
SerpinB10, a Serine Protease Inhibitor, Is Implicated in UV-Induced Cellular Response

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
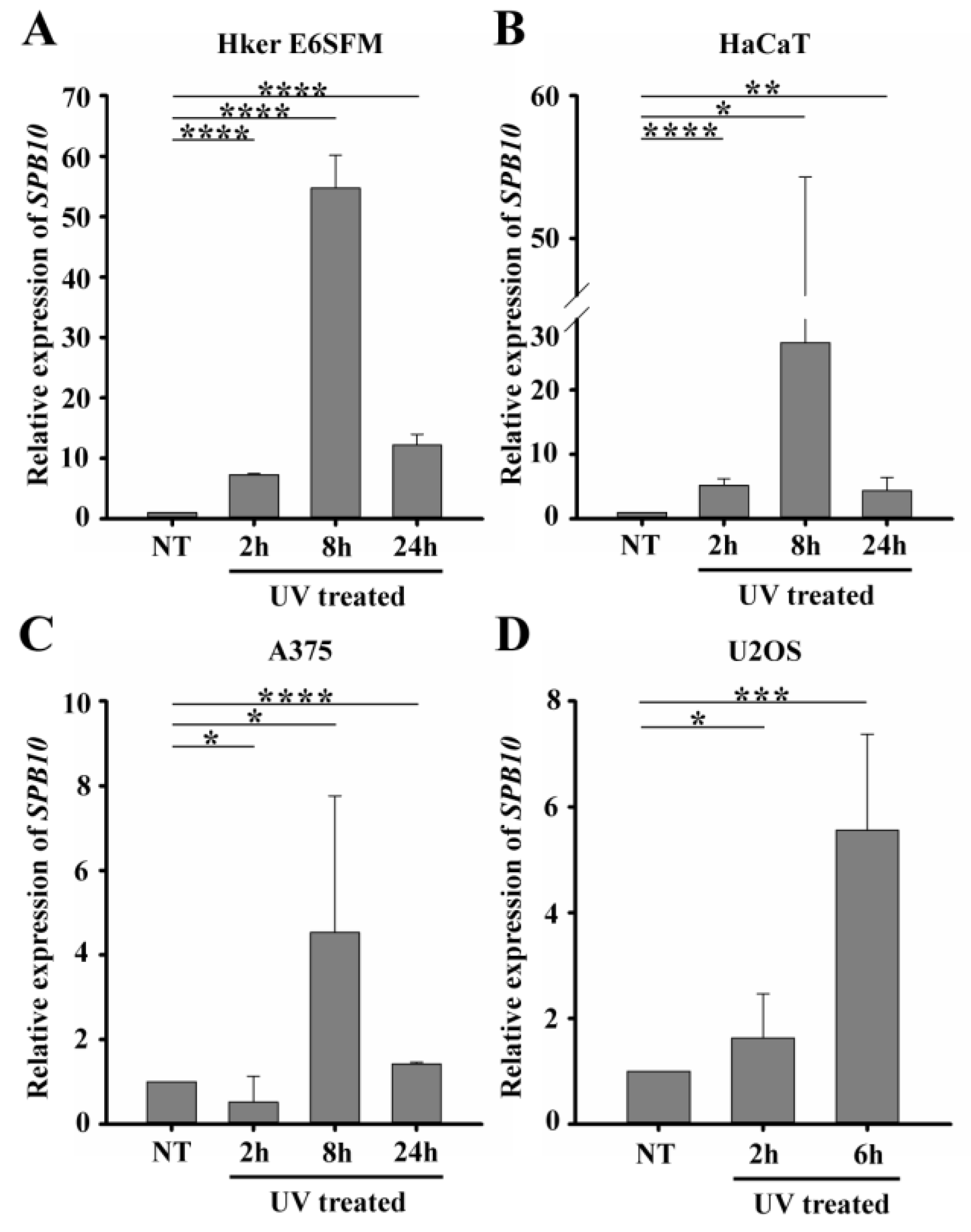
2.1. Expression of Several Members of the SerpinB Superfamily, Including SPB10, Is Increased Following UV Irradiation
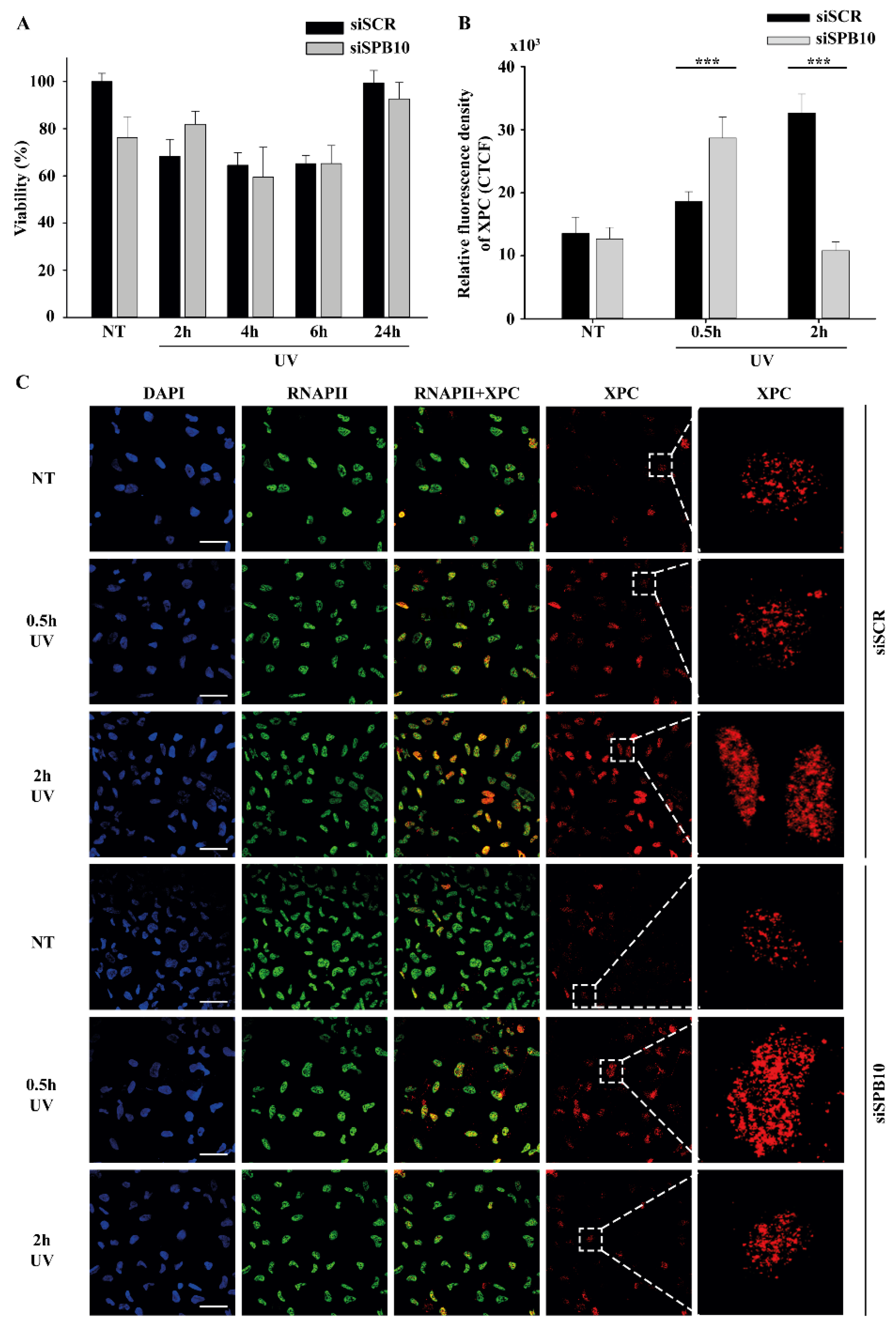
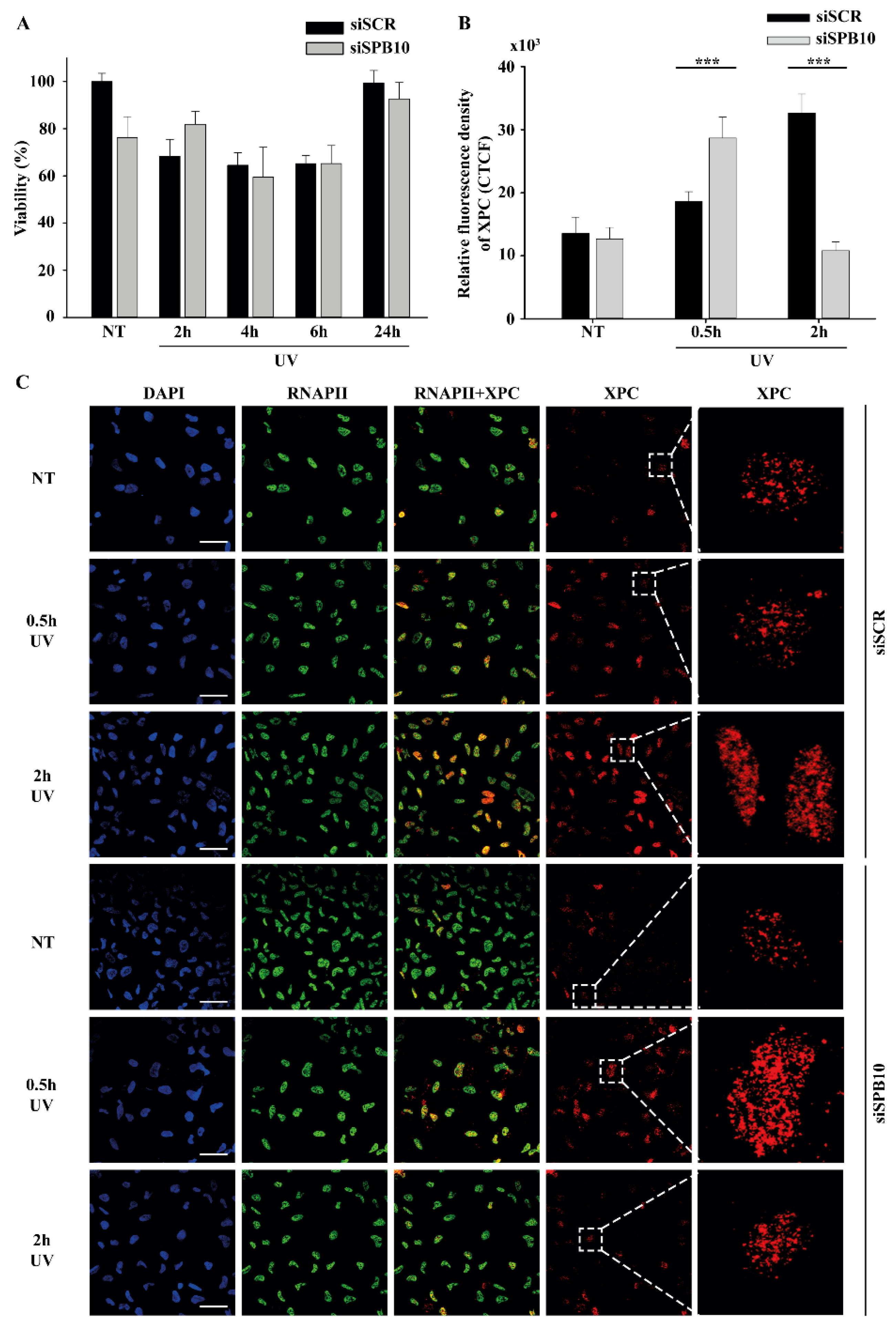
2.2. SPB10 Is Dispensable for Cell Survival but Influences DNA Repair Kinetics in Interphase Cells upon UV Irradiation
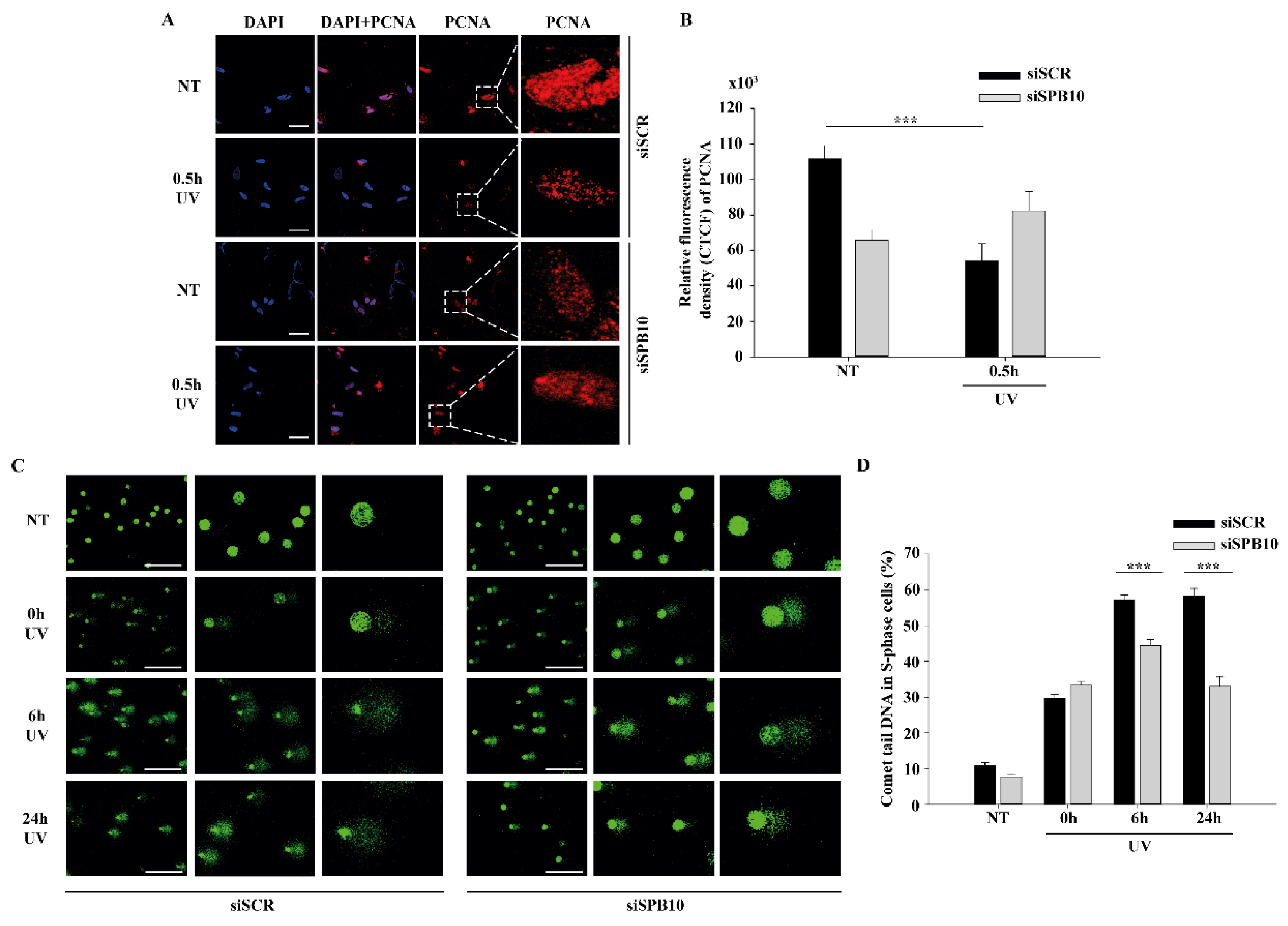
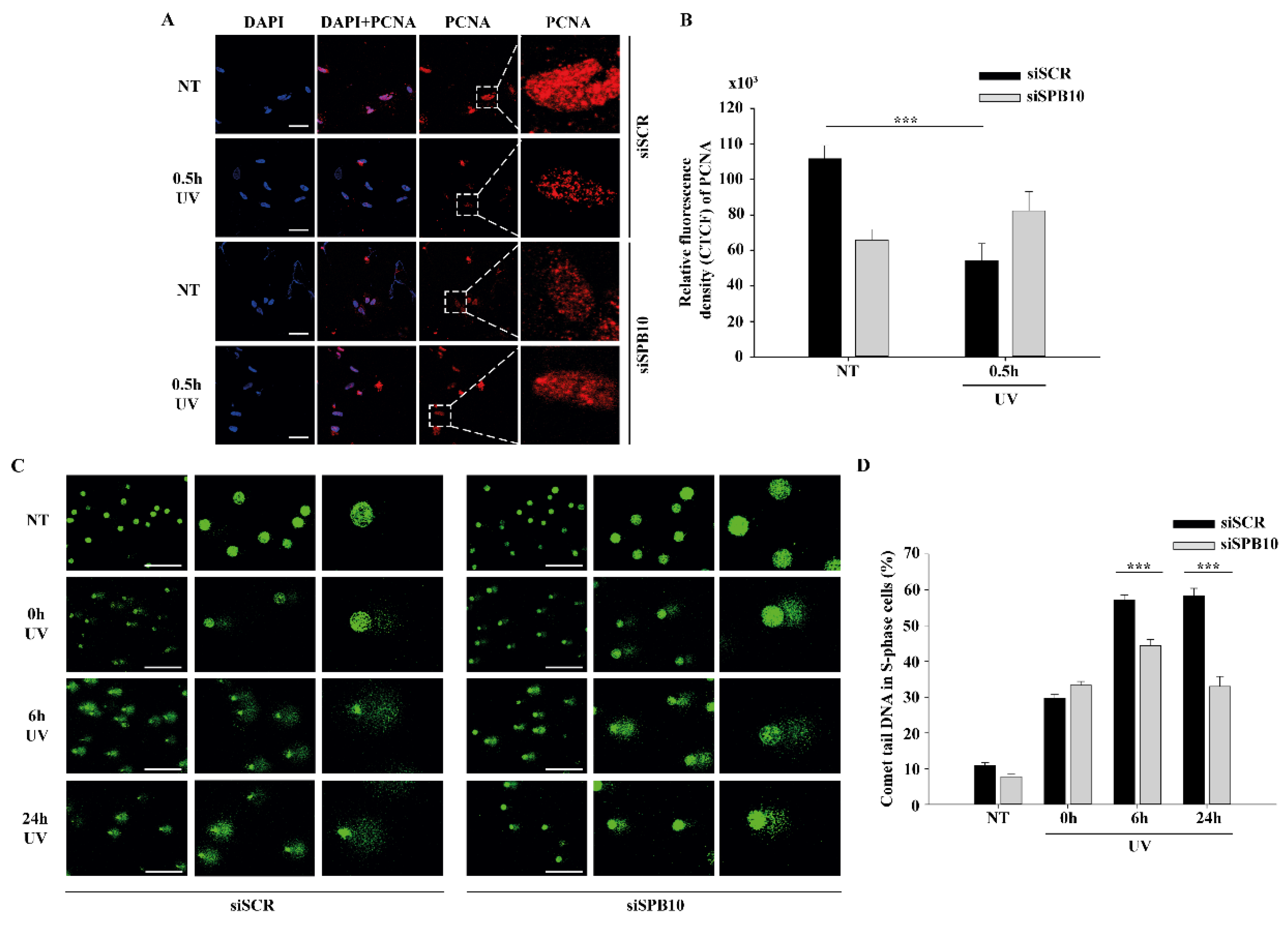
2.3. SPB10 Influences DNA Repair Kinetics in S-Phase Cells upon UV Irradiation
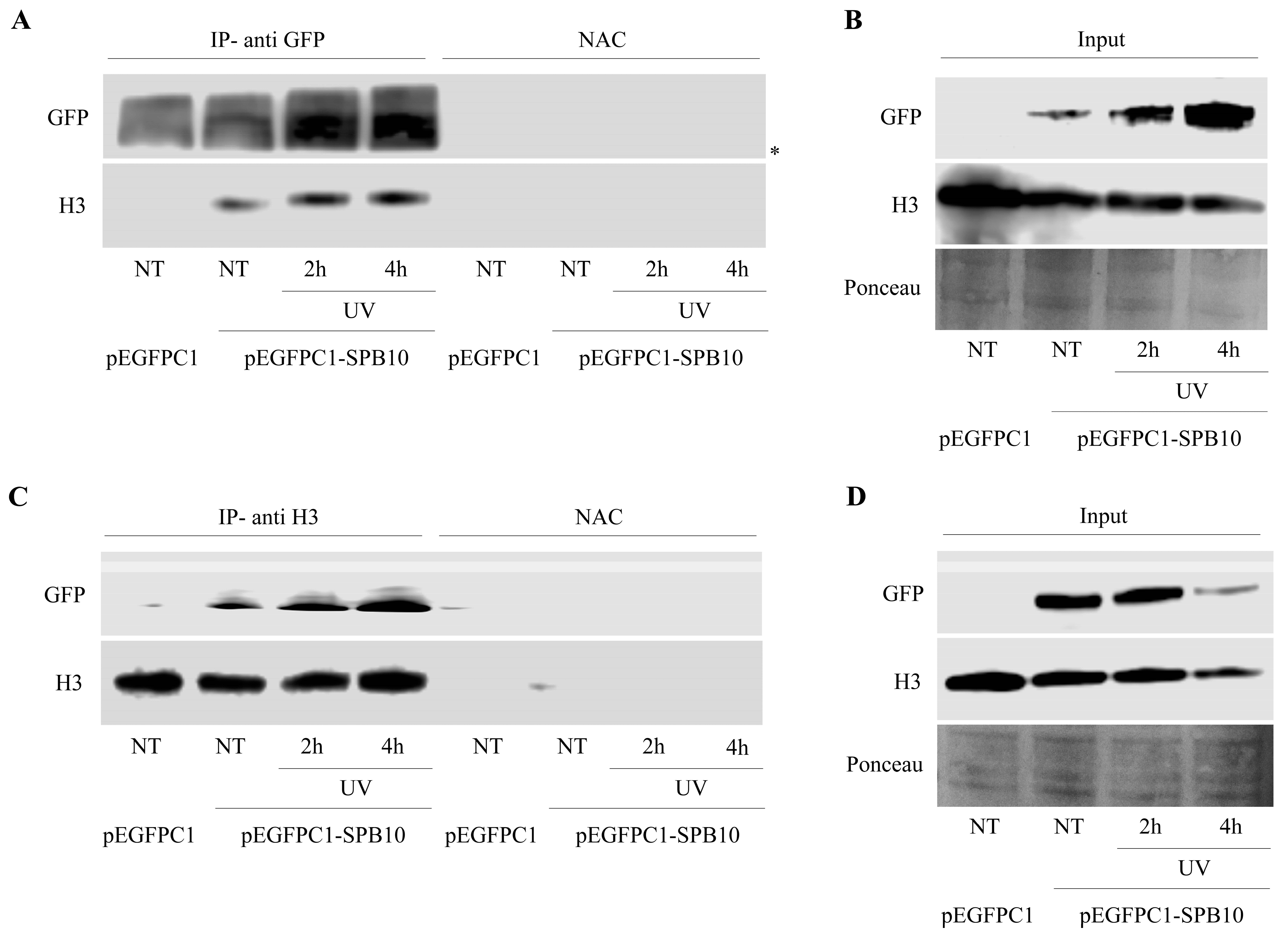
2.4. SPB10 Is Associated with Chromatin
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Media and Culturing Conditions
4.2. UV Irradiation
4.3. Microarray Experiment
4.4. RNA Extraction, Reverse Transcription, and qRT-PCR
4.5. Western Blot
4.6. Cytoskeletal (CSK) Immunocytochemistry
4.7. Transfection
4.8. Co-Immunoprecipitation
4.9. Viability Assay
4.10. siRNA Transfection
4.11. Post-Replication Repair (PRR) Comet Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-4 PP | 6-4 pirimidine-pirimidon photoproduct |
| BrdU | bromodeoxyuridine |
| CPD | cyclobutane-pyrimidine dimers |
| co-IP | co-immunoprecipitation |
| DMEM | Dulbecco’s Modified Eagle Medium |
| GFP | green fluorescent protein |
| NER | Nucleotide Excision Repair |
| NT | non-treated (basal condition) |
| PCNA | proliferating cell nuclear antigen |
| PRR comet assay | post-replication repair comet assay |
| qPCR | quantitative polymerase chain reaction |
| siSCR | non-targeting scrambled siRNA |
| SPB | SerpinB |
| SPB2 | SerpinB2 |
| SPB10 | SerpinB10 (Bomapin) |
| SPB13 | SerpinB13 |
| TLS DNA Polymerase | translesion DNA polymerase |
| TPCK | N-tosyl-L-phenylalanine chloromethyl ketone |
| XPC | Xeroderma Pigmentosum C |
References
- Epstein, J.H. Photocarcinogenesis, skin cancer, and aging. J. Am. Acad. Dermatol. 1983, 9, 487–502. [Google Scholar] [CrossRef]
- de Gruijl, F.R.; van Kranen, H.J.; Mullenders, L.H. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol. B 2001, 63, 19–27. [Google Scholar] [CrossRef]
- Kielbassa, C.; Roza, L.; Epe, B. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 1997, 18, 811–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rünger, T.M.; Kappes, U.P. Mechanisms of mutation formation with long-wave ultraviolet light (UVA). Photodermatol. Photoimmunol. Photomed. 2008, 24, 2–10. [Google Scholar] [CrossRef]
- de Gruijl, F.R. Photocarcinogenesis: UVA vs UVB. Methods Enzymol. 2000, 319, 359–366. [Google Scholar]
- Kabuyama, Y.; Homma, M.K.; Kurosaki, T.; Homma, Y. Early signaling events induced by 280-nm UV irradiation. Eur. J. Biochem. 2002, 269, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Pustovalova, Y.; MacIejewski, M.W.; Korzhnev, D.M. NMR mapping of PCNA interaction with translesion synthesis DNA polymerase Rev1 mediated by Rev1-BRCT domain. J. Mol. Biol. 2013, 425, 3091–3105. [Google Scholar] [CrossRef]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Kosarek-Stancel, J.N.; Tang, T.S.; Friedberg, E.C. Y-family DNA polymerases in mammalian cells. Cell. Mol. Life Sci. 2009, 66, 2363–2381. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, A.A.; Korzhnev, D.M. The Rev1-Polζ translesion synthesis mutasome: Structure, interactions and inhibition. In Enzymes; Academic Press: Cambridge, MA, USA, 2019; Volume 45, pp. 139–181. ISBN 9780128173961. [Google Scholar]
- Acharya, N.; Patel, S.K.; Sahu, S.R.; Kumari, P. “PIPs” in DNA polymerase: PCNA interaction affairs. Biochem. Soc. Trans. 2020, 48, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Lehmann, A.R.; Fuchs, R.P.P. Trading Places: How do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 2005, 18, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Johnson, R.E.; Haracska, L.; Prakash, L.; Prakash, S.; Benkovic, S.J. Regulation of polymerase exchange between Polη and Polδ by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 5361–5366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorano, D.; El Etri, J.; Franchet, C.; Hoffmann, J.-S. Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. Int. J. Mol. Sci. 2021, 22, 3924. [Google Scholar] [CrossRef]
- Guilliam, T.A.; Yeeles, J.T.P. Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat. Struct. Mol. Biol. 2020, 27, 450–460. [Google Scholar] [CrossRef]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Ujfaludi, Z.; Tuzesi, A.; Majoros, H.; Rothler, B.; Pankotai, T.; Boros, I.M. Coordinated activation of a cluster of MMP genes in response to UVB radiation. Sci. Rep. 2018, 8, 2660. [Google Scholar] [CrossRef]
- Majoros, H.; Ujfaludi, Z.; Borsos, B.N.; Hudacsek, V.V.; Nagy, Z.; Coin, F.; Buzas, K.; Kovács, I.; Bíró, T.; Boros, I.M.; et al. SerpinB2 is involved in cellular response upon UV irradiation. Sci. Rep. 2019, 9, 2753. [Google Scholar] [CrossRef]
- Polyanka, H.; Szabo, K.; Tax, G.; Tubak, V.; Kusz, E.; Ujfaludi, Z.; Boros, I.; Bata-Csorgo, Z.; Kemény, L.; Szell, M. Primary characterization of a novel HPV-E6 oncogene immortalized keratinocyte cell line. J. Investig. Dermatol. 2018, 131, 70. [Google Scholar]
- Szlavicz, E.; Szabo, K.; Groma, G.; Bata-Csorgo, Z.; Pagani, F.; Kemeny, L.; Szell, M. Splicing factors differentially expressed in psoriasis alter mRNA maturation of disease-associated EDA+ fibronectin. Mol. Cell. Biochem. 2017, 436, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Silverman, G.A.; Whisstock, J.C.; Askew, D.J.; Pak, S.C.; Luke, C.J.; Cataltepe, S.; Irving, J.A.; Bird, P.I. Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell. Mol. Life Sci. 2004, 61, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.S.; Bleakley, P.; Woodrow, G.C.; Doe, W.F. Inhibition of cancer cell urokinase plasminogen activator by its specific inhibitor PAI-2 and subsequent effects on extracellular matrix degradation. Cancer Res. 1990, 50, 4676–4684. [Google Scholar]
- Sheng, S.; Truong, B.; Fredrickson, D.; Wu, R.; Pardee, A.B.; Sager, R. Tissue-type plasminogen activator is a target of the tumor suppressor gene maspin. Proc. Natl. Acad. Sci. USA 1998, 95, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Li, X.; He, Q.; Zhang, L.; Song, K.; Yang, X.; He, Q.; Wang, Y.; Hong, X.; Ma, J.; et al. TRIM21-SERPINB5 AIDS GMPS repression to protect nasopharyngeal carcinoma cells from radiation-induced apoptosis. J. Biomed. Sci. 2020, 27, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Y.; Zhang, K.; Feng, Y.; Yi, L.; Liang, Y.; Wu, W.; Zhao, J.; Zhang, Z.; Xu, Y.; Hu, Q.; et al. Epithelial serpinb10, a novel marker of airway eosinophilia in asthma, contributes to allergic airway inflammation. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L245–L254. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.W.; Atwell, B.J.; Roberts, T.H. Serpin genes AtSRP2 and AtSRP3 are required for normal growth sensitivity to a DNA alkylating agent in Arabidopsis. BMC Plant Biol. 2009, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, H.-H.; Chen, Y.-C.; Jhan, J.-R.; Lin, J.-J. The serine protease inhibitor serpinB2 binds and stabilizes p21 in senescent cells. J. Cell Sci. 2017, 130, 3272–3281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Ding, Y.; Wu, M.; Chen, Y.; Lyu, X.; Lu, J.; Wang, H.; Teng, L. Integrated genomic analysis identifies a genetic mutation model predicting response to immune checkpoint inhibitors in melanoma. Cancer Med. 2020, 9, 8498–8518. [Google Scholar] [CrossRef]
- Przygodzka, P.; Ramstedt, B.; Tengel, T.; Larsson, G.; Wilczynska, M. Bomapin is a redox-sensitive nuclear serpin that affects responsiveness of myeloid progenitor cells to growth environment. BMC Cell Biol. 2010, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Chou, R.-H.; Wen, H.-C.; Liang, W.-G.; Lin, S.-C.; Yuan, H.-W.; Wu, C.-W.; Chang, W.-S.W. Suppression of the invasion and migration of cancer cells by SERPINB family genes and their derived peptides. Oncol. Rep. 2011, 27, 238–245. [Google Scholar] [CrossRef]
- Shioji, G.; Ezura, Y.; Nakajima, T.; Ohgaki, K.; Fujiwara, H.; Kubota, Y.; Ichikawa, T.; Inoue, K.; Shuin, T.; Habuchi, T.; et al. Nucleotide variations in genes encoding plasminogen activator inhibitor-2 and serine proteinase inhibitor B10 associated with prostate cancer. J. Hum. Genet. 2005, 50, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garmyn, M.; Young, A.R.; Miller, S.A. Mechanisms of and variables affecting UVR photoadaptation in human skin. Photochem. Photobiol. Sci. 2018, 17, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- López-Camarillo, C.; Aréchaga Ocampo, E.; López Casamichana, M.; Pérez-Plasencia, C.; Álvarez-Sánchez, E.; Marchat, L.A. Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis. Int. J. Mol. Sci. 2011, 13, 142–172. [Google Scholar] [CrossRef]
- Kirillova, I.; Chaisson, M.; Fausto, N. Tumor necrosis factor induces DNA replication in hepatic cells through nuclear factor κB activation. Cell Growth Differ. 1999, 10, 819–828. [Google Scholar]
- Schleef, R.R.; Chuang, T.L. Protease Inhibitor 10 Inhibits Tumor Necrosis Factor α-induced Cell Death. J. Biol. Chem. 2000, 275, 26385–26389. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Machida, Y.; Palani, S.; Caulfield, T.R.; Radisky, E.S.; Kaufmann, S.H.; Machida, Y.J. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 2020, 11, 1318. [Google Scholar] [CrossRef]
- Borsos, B.N.; Majoros, H.; Pankotai, T. Ubiquitylation-Mediated Fine-Tuning of DNA Double-Strand Break Repair. Cancers 2020, 12, 1617. [Google Scholar] [CrossRef]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiroli, F.; Vissers, J.H.A.; Van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatti, M.; Pinato, S.; Maspero, E.; Soffientini, P.; Polo, S.; Penengo, L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 2012, 11, 2538–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.R.; Wang, K.; Smith, J.B.; Heslin, M.J.; Diasio, R.B. Quantitation of Dihydropyrimidine Dehydrogenase Expression by Real-Time Reverse Transcription Polymerase Chain Reaction. Anal. Biochem. 2000, 278, 175–184. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majoros, H.; Borsos, B.N.; Ujfaludi, Z.; Páhi, Z.G.; Mórocz, M.; Haracska, L.; Boros, I.M.; Pankotai, T. SerpinB10, a Serine Protease Inhibitor, Is Implicated in UV-Induced Cellular Response. Int. J. Mol. Sci. 2021, 22, 8500. https://doi.org/10.3390/ijms22168500
Majoros H, Borsos BN, Ujfaludi Z, Páhi ZG, Mórocz M, Haracska L, Boros IM, Pankotai T. SerpinB10, a Serine Protease Inhibitor, Is Implicated in UV-Induced Cellular Response. International Journal of Molecular Sciences. 2021; 22(16):8500. https://doi.org/10.3390/ijms22168500
Chicago/Turabian StyleMajoros, Hajnalka, Barbara N. Borsos, Zsuzsanna Ujfaludi, Zoltán G. Páhi, Mónika Mórocz, Lajos Haracska, Imre Miklós Boros, and Tibor Pankotai. 2021. "SerpinB10, a Serine Protease Inhibitor, Is Implicated in UV-Induced Cellular Response" International Journal of Molecular Sciences 22, no. 16: 8500. https://doi.org/10.3390/ijms22168500
APA StyleMajoros, H., Borsos, B. N., Ujfaludi, Z., Páhi, Z. G., Mórocz, M., Haracska, L., Boros, I. M., & Pankotai, T. (2021). SerpinB10, a Serine Protease Inhibitor, Is Implicated in UV-Induced Cellular Response. International Journal of Molecular Sciences, 22(16), 8500. https://doi.org/10.3390/ijms22168500