
Novel Nested-Seq Approach for SARS-CoV-2 Real-Time Epidemiology and In-Depth Mutational Profiling in Wastewater

 ,
,  ,
, 
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
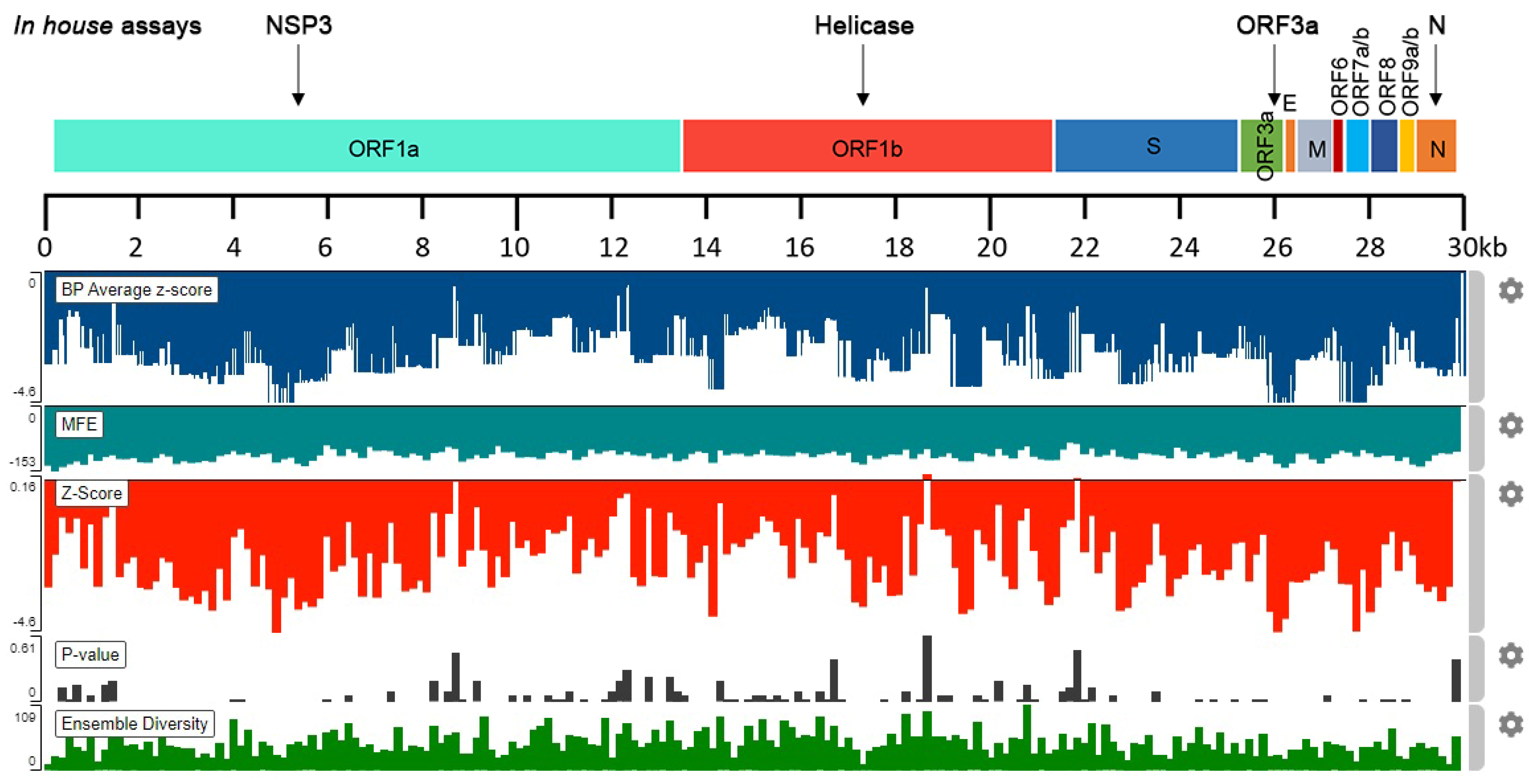
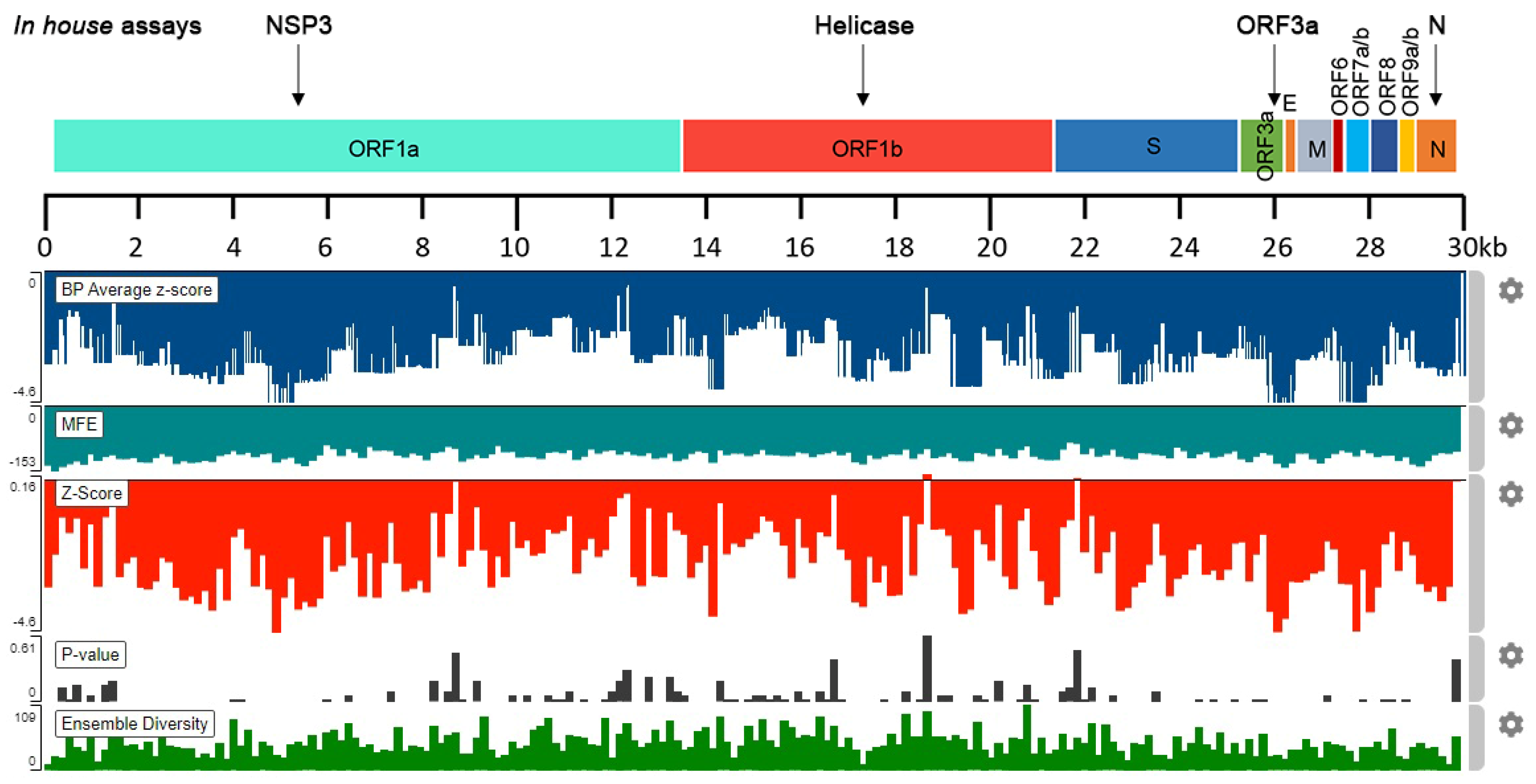
2.1. In Silico Analysis of SARS-CoV-2 RNA Stability
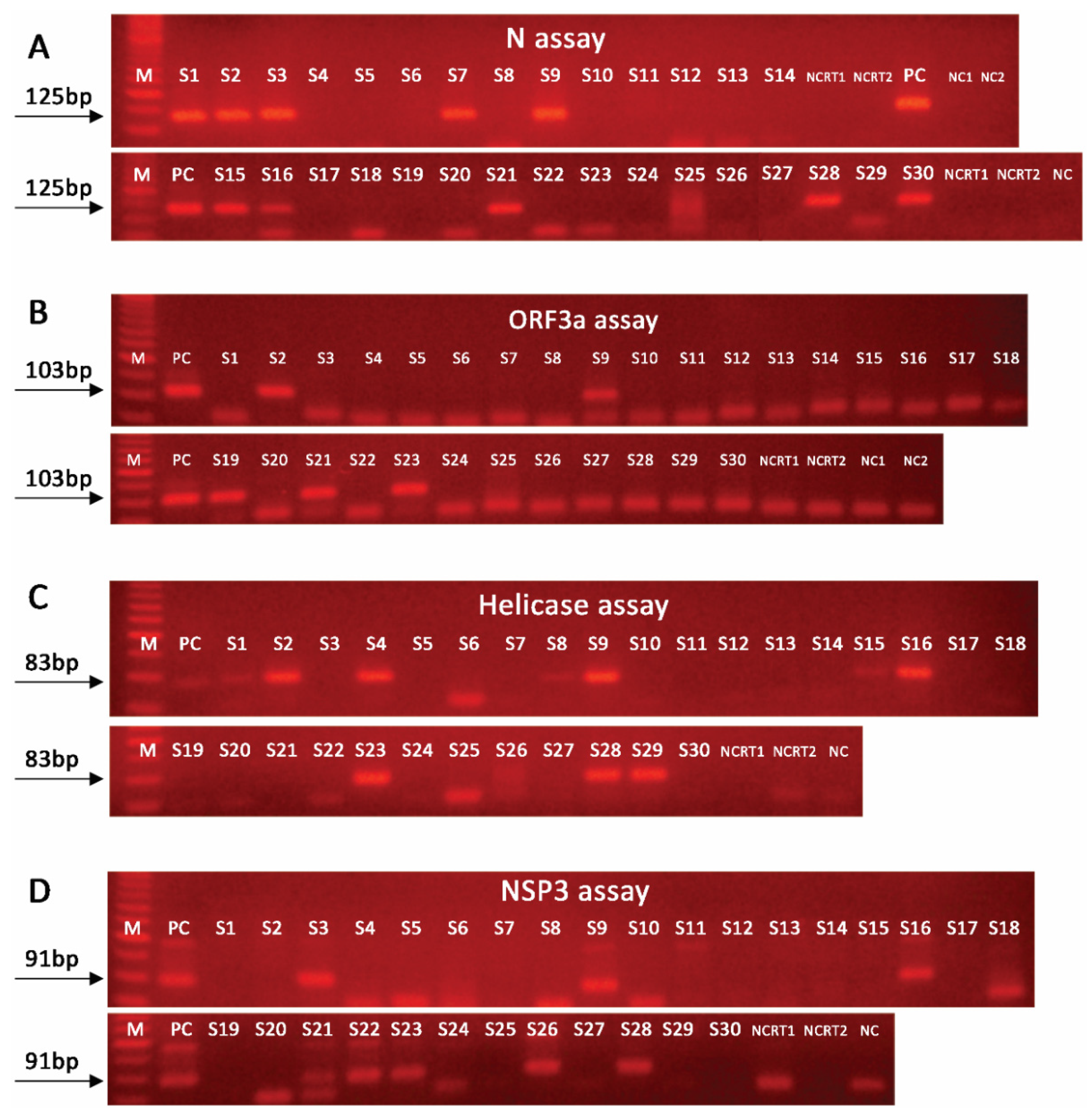
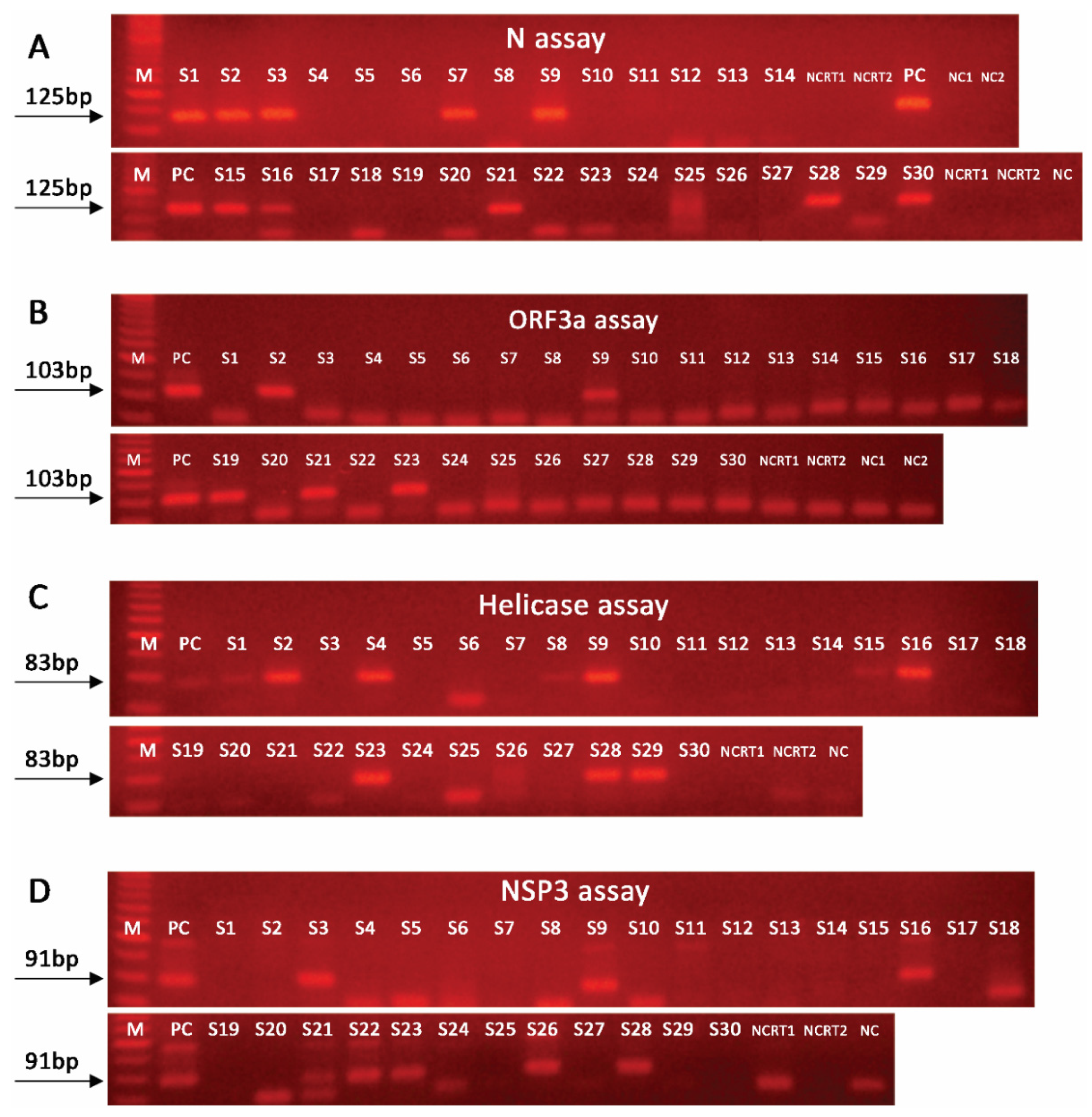
2.2. Nested PCR Approach Significantly Improves SARS-CoV-2 Detection Sensitivity
2.3. Effective SARS-CoV-2 Detection in Wastewater Requires Amplification of Multiple Targets
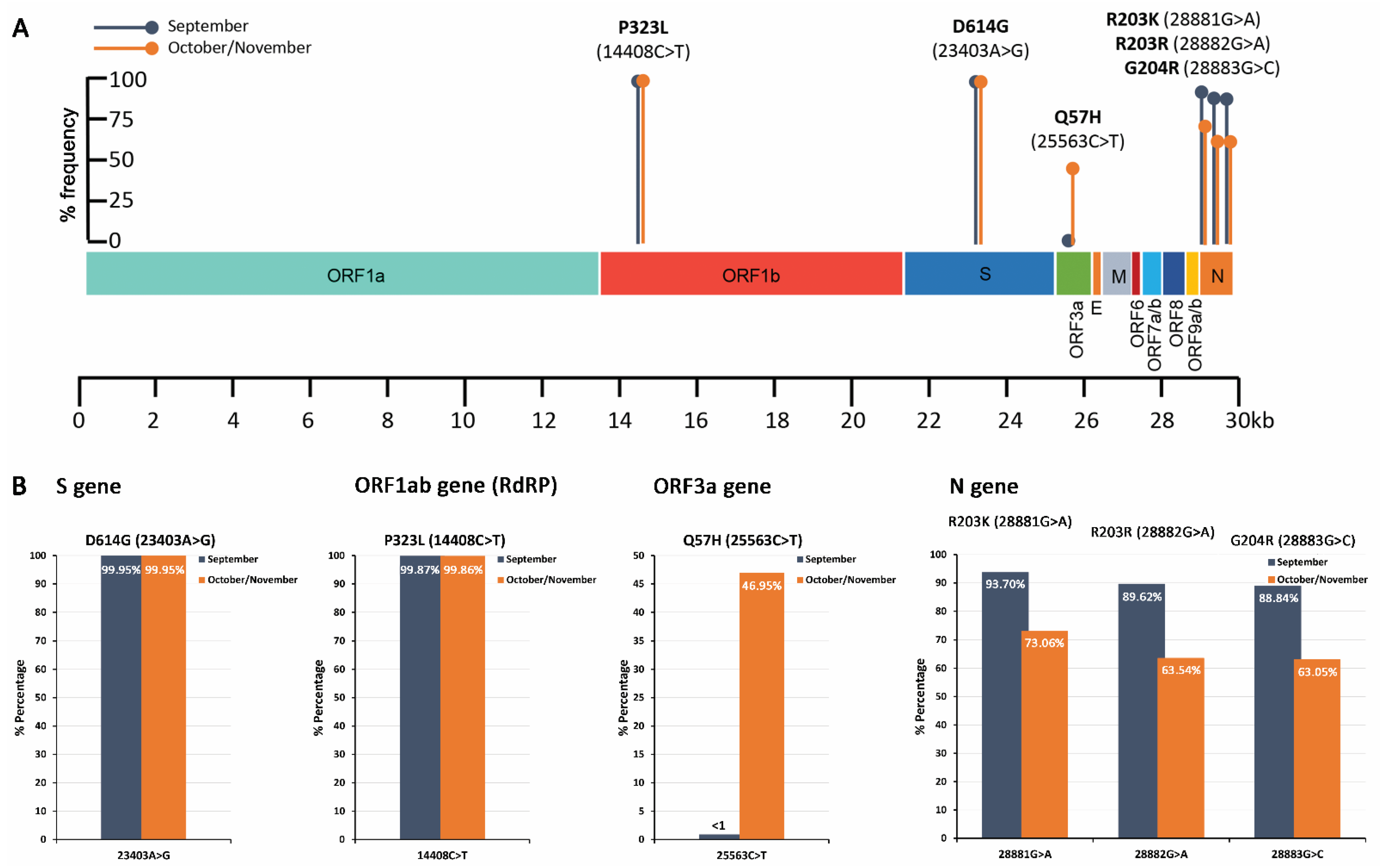
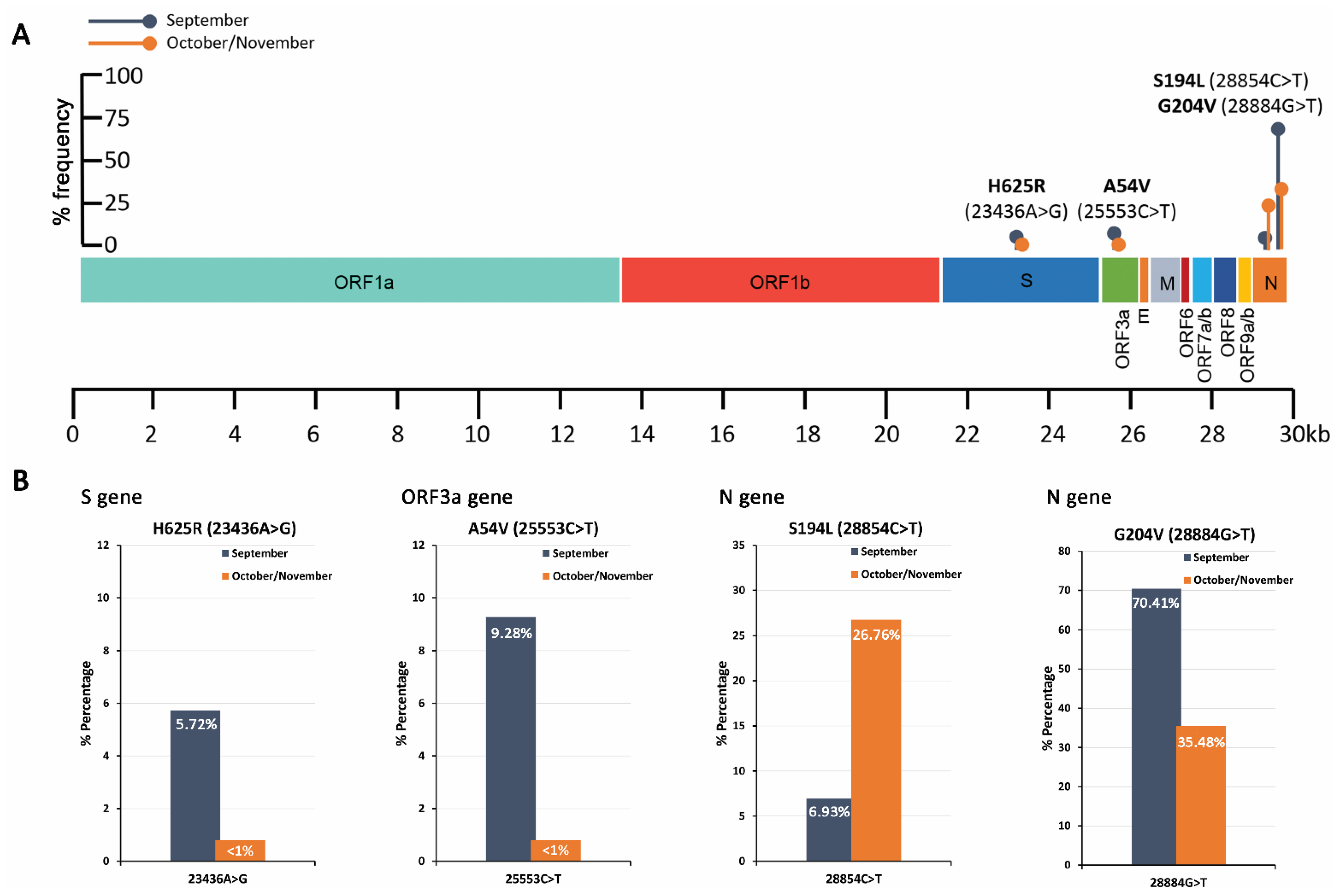
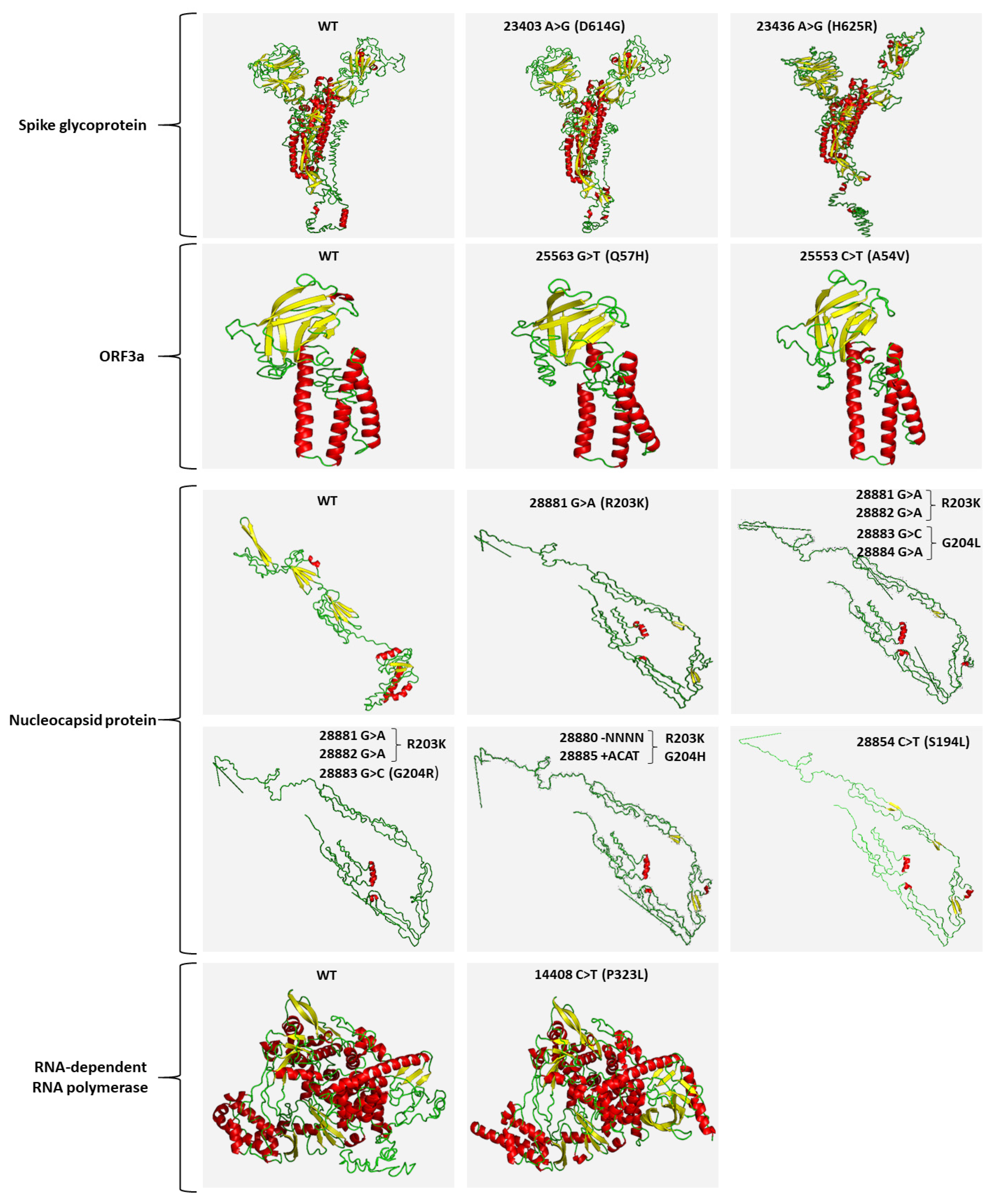
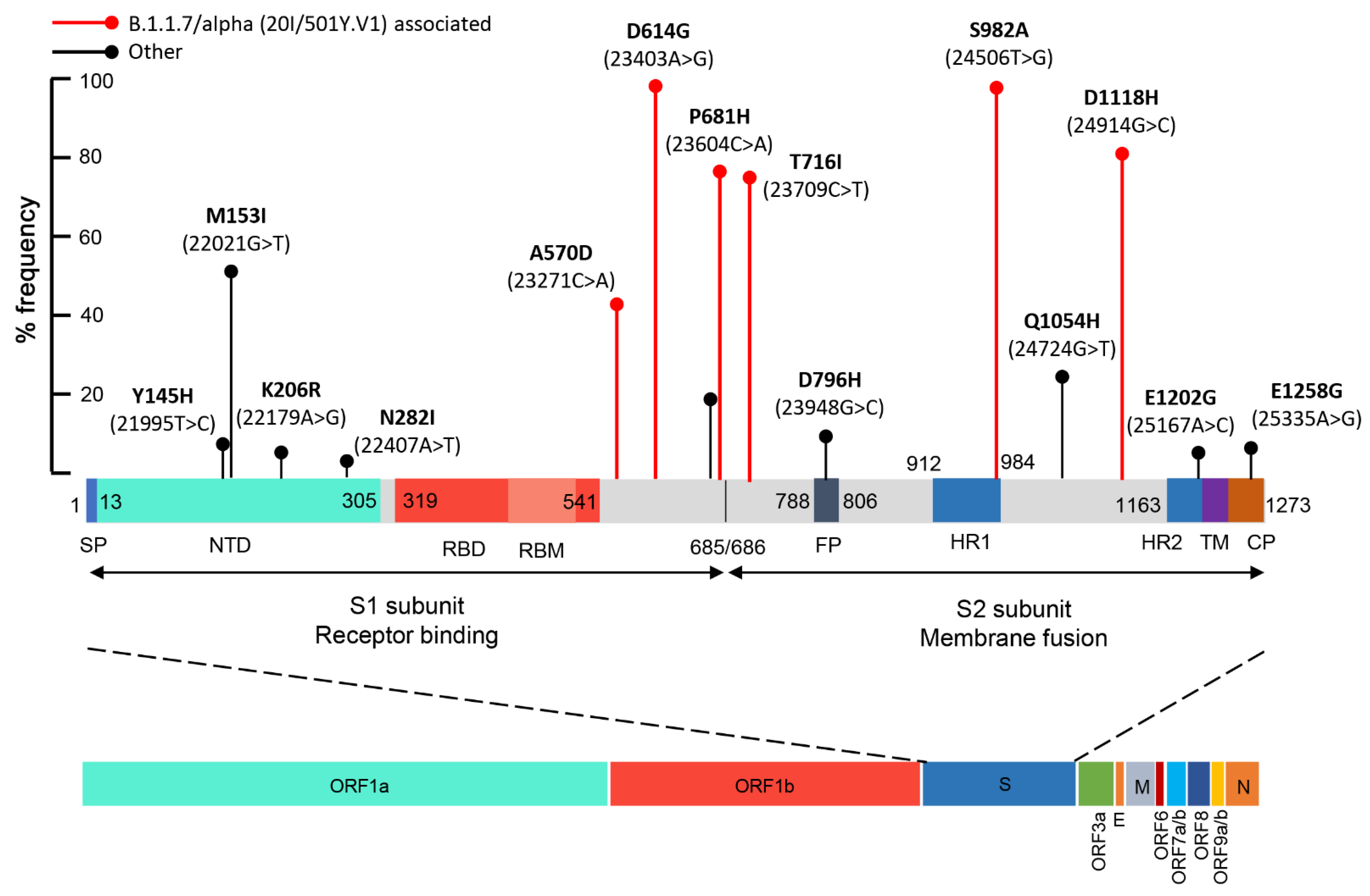
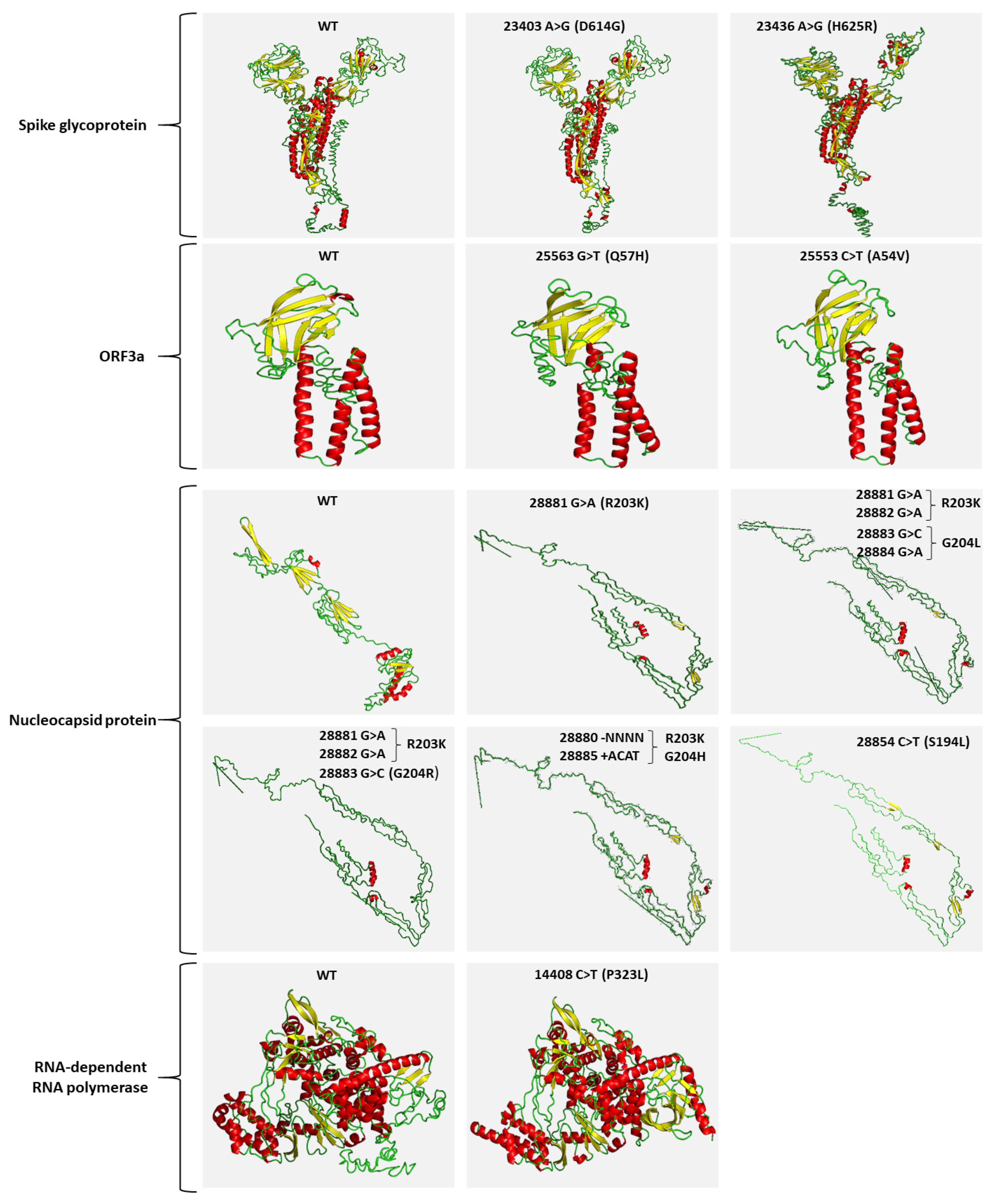
2.4. Mutational Analysis of SARS-CoV-2 in Wastewater Using Targeted DNA-seq
3. Discussion
4. Materials and Methods
4.1. SARS-CoV-2 RNA Stability Analysis
4.2. Wastewater Sampling
4.3. Sample Concentration and RNA Extraction
4.4. First-Strand cDNA Synthesis
4.5. Detection of SARS-CoV-2
4.6. Targeted DNA-seq for the Mutational Analysis of SARS-CoV-2 in Wastewater
4.7. Bioinformatics Analysis
4.8. In Silico 3D Protein Folding Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; Kastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J. Biomed. Sci. 2021, 28, 9. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e1289. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, F.; Cui, J.; Peng, Z.; Chang, Z.; Lai, S.; Chen, Q.; Wang, L.; Gao, G.F.; Feng, Z. Comprehensive large-scale nucleic acid-testing strategies support China’s sustained containment of COVID-19. Nat. Med. 2021, 27, 740–742. [Google Scholar] [CrossRef]
- Kuguyo, O.; Kengne, A.P.; Dandara, C. Singapore COVID-19 Pandemic Response as a Successful Model Framework for Low-Resource Health Care Settings in Africa? OMICS 2020, 24, 470–478. [Google Scholar] [CrossRef]
- Thomaidis, N.S.; Gago-Ferrero, P.; Ort, C.; Maragou, N.C.; Alygizakis, N.A.; Borova, V.L.; Dasenaki, M.E. Reflection of Socioeconomic Changes in Wastewater: Licit and Illicit Drug Use Patterns. Environ. Sci. Technol. 2016, 50, 10065–10072. [Google Scholar] [CrossRef]
- Gonzalez-Marino, I.; Baz-Lomba, J.A.; Alygizakis, N.A.; Andres-Costa, M.J.; Bade, R.; Bannwarth, A.; Barron, L.P.; Been, F.; Benaglia, L.; Berset, J.D.; et al. Spatio-temporal assessment of illicit drug use at large scale: Evidence from 7 years of international wastewater monitoring. Addiction 2020, 115, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.L.; McElroy, K.A.; Armas, F.; Imakaev, M.; Gu, X.; Duvallet, C.; Chandra, F.; Chen, H.; Leifels, M.; Mendola, S.; et al. Quantitative detection of SARS-CoV-2 B.1.1.7 variant in wastewater by allele-specific RT-qPCR. medRxiv 2021. [Google Scholar] [CrossRef]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Surya, K.; Buyukyoruk, M.; Cicha, C.; Vanderwood, K.K.; Wilkinson, R.; Wiedenheft, B. Temporal Detection and Phylogenetic Assessment of SARS-CoV-2 in Municipal Wastewater. Cell Rep. Med. 2020, 1, 100098. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Kantor, R.S.; Olm, M.R.; Whitney, O.N.; Al-Shayeb, B.; Lou, Y.C.; Flamholz, A.; Kennedy, L.C.; Greenwald, H.; Hinkle, A.; et al. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. mBio 2021, 12, e02703-20. [Google Scholar] [CrossRef] [PubMed]
- Alygizakis, N.; Markou, A.N.; Rousis, N.I.; Galani, A.; Avgeris, M.; Adamopoulos, P.G.; Scorilas, A.; Lianidou, E.S.; Paraskevis, D.; Tsiodras, S.; et al. Analytical methodologies for the detection of SARS-CoV-2 in wastewater: Protocols and future perspectives. Trends Analyt. Chem. 2020, 134, 116125. [Google Scholar] [CrossRef]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J. 2020, 39, e105938. [Google Scholar] [CrossRef] [PubMed]
- Wurtzer, S.; Marechal, V.; Mouchel, J.M.; Maday, Y.; Teyssou, R.; Richard, E.; Almayrac, J.L.; Moulin, L. Evaluation of lockdown effect on SARS-CoV-2 dynamics through viral genome quantification in waste water, Greater Paris, France, 5 March to 23 April 2020. Eurosurveillance 2020, 25, 2000776. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Farkas, K.; Hillary, L.S.; Thorpe, J.; Walker, D.I.; Lowther, J.A.; McDonald, J.E.; Malham, S.K.; Jones, D.L. Concentration and Quantification of SARS-CoV-2 RNA in Wastewater Using Polyethylene Glycol-Based Concentration and qRT-PCR. Methods Protoc. 2021, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, G.; Iaconelli, M.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Bonadonna, L.; Lucentini, L.; Suffredini, E. First detection of SARS-CoV-2 in untreated wastewaters in Italy. Sci. Total Environ. 2020, 736, 139652. [Google Scholar] [CrossRef] [PubMed]
- Haramoto, E.; Malla, B.; Thakali, O.; Kitajima, M. First environmental surveillance for the presence of SARS-CoV-2 RNA in wastewater and river water in Japan. Sci. Total Environ. 2020, 737, 140405. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, G.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Iaconelli, M.; Bonadonna, L.; Lucentini, L.; Suffredini, E. SARS-CoV-2 has been circulating in northern Italy since December 2019: Evidence from environmental monitoring. Sci. Total Environ. 2021, 750, 141711. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Klapsa, D.; Wilton, T.; Zambon, M.; Bentley, E.; Bujaki, E.; Fritzsche, M.; Mate, R.; Majumdar, M. Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic. Viruses 2020, 12, 1144. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Sevajol, M.; Subissi, L.; Decroly, E.; Canard, B.; Imbert, I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 2014, 194, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2020, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2020, 184, 64–75. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schafer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, A.; Selisko, B.; Le, N.T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Andrews, R.J.; Roche, J.; Moss, W.N. ScanFold: An approach for genome-wide discovery of local RNA structural elements-applications to Zika virus and HIV. PeerJ 2018, 6, e6136. [Google Scholar] [CrossRef] [Green Version]
- Galani, A.; Aalizadeh, R.; Kostakis, M.; Markou, A.; Alygizakis, N.; Lytras, T.; Adamopoulos, P.G.; Peccia, J.; Thompson, D.C.; Kontou, A.; et al. SARS-CoV-2 wastewater surveillance data can predict hospitalizations and ICU admissions. Sci. Total Environ. 2021. (Under Review). [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | CDC N1-based | N | NSP3 | Helicase | ORF3a |
|---|---|---|---|---|---|
| S1 | 36.2 | 27.0 | - | - | - |
| S2 | - | 28.0 | - | 26.2 | 26.8 |
| S3 | 34.3 | 27.2 | 26.3 | - | - |
| S4 | - | - | - | 26.6 | - |
| S7 | - | 28.0 | - | - | - |
| S9 | - | 26.5 | 26.4 | 26.7 | 27.3 |
| S15 | 35.2 | 28.9 | - | - | - |
| S16 | - | 26.7 | 25.6 | 25.9 | - |
| S19 | - | - | - | - | 28.0 |
| S20 | - | - | 24.9 | - | - |
| S21 | - | 27.1 | 25.7 | - | 28.0 |
| S22 | - | - | 25.4 | - | - |
| S23 | - | - | 30.9 | 25.1 | 28.5 |
| S26 | - | - | 26.1 | - | - |
| S28 | - | 29.0 | 26.1 | 26.3 | - |
| S29 | 36.7 | - | - | 25.9 | - |
| S30 | 35.9 | 28.2 | - | - | - |
| Positive | 5/30 | 10/30 | 9/30 | 7/30 | 5/30 |
| % Sensitivity | 29.4% | 58.8% | 52.9% | 41.2% | 29.4% |
| Assay Combinations % Sensitivity | |||||
| N and NSP3 | 82.4% | ||||
| N, NSP3 and helicase | 94.1% | ||||
| “-”: Not Detected | |||||
| September 2020 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Position | Ref. Base | Alt. Base | Alt. Freq. (%) | Total Depth | Ref. Codon | Ref. AA | Alt. Codon | Alt. AA |
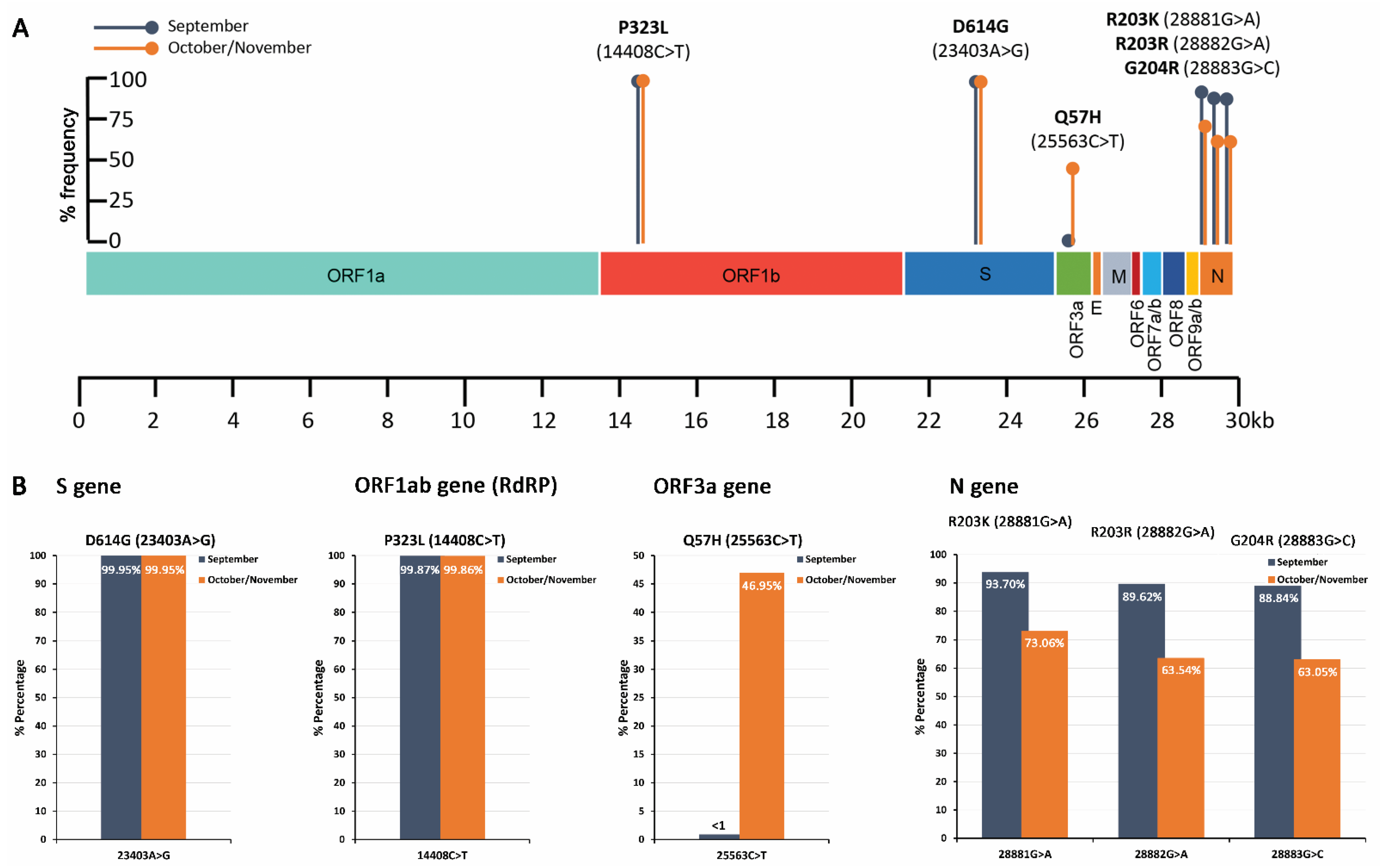
| P323L (14408C>T) | C | T | 99.87 | 240,183 | CCT | P | CTT | L |
| D614G (23403A>G) | A | G | 99.95 | 236,549 | GAT | D | GGT | G |
| H625R (23436A>G) | A | G | 5.72 | 249,444 | CAT | H | CGT | R |
| A54V (25553C>T) | C | T | 9.28 | 240,436 | GCT | A | GTT | V |
| S194L (28854C>T) | C | T | 6.93 | 307,007 | TCA | S | TTA | L |
| 28880 | A | −NNNN | 7.95 | 337,302 | NA | NA | NA | NA |
| R203K (28881G>A) | G | A | 93.70 | 264,387 | AGG | R | AAG | K |
| R203 (28882G>A) | G | A | 89.62 | 255,837 | AGG | R | AGA | R |
| G204R (28883G>C) | G | C | 88.84 | 269,981 | GGA | G | CGA | R |
| G204V (28884G>T) | G | T | 70.41 | 263,960 | GGA | G | GTA | V |
| 28885 | A | +ACAT | 3.09 | 340,323 | NA | NA | NA | NA |
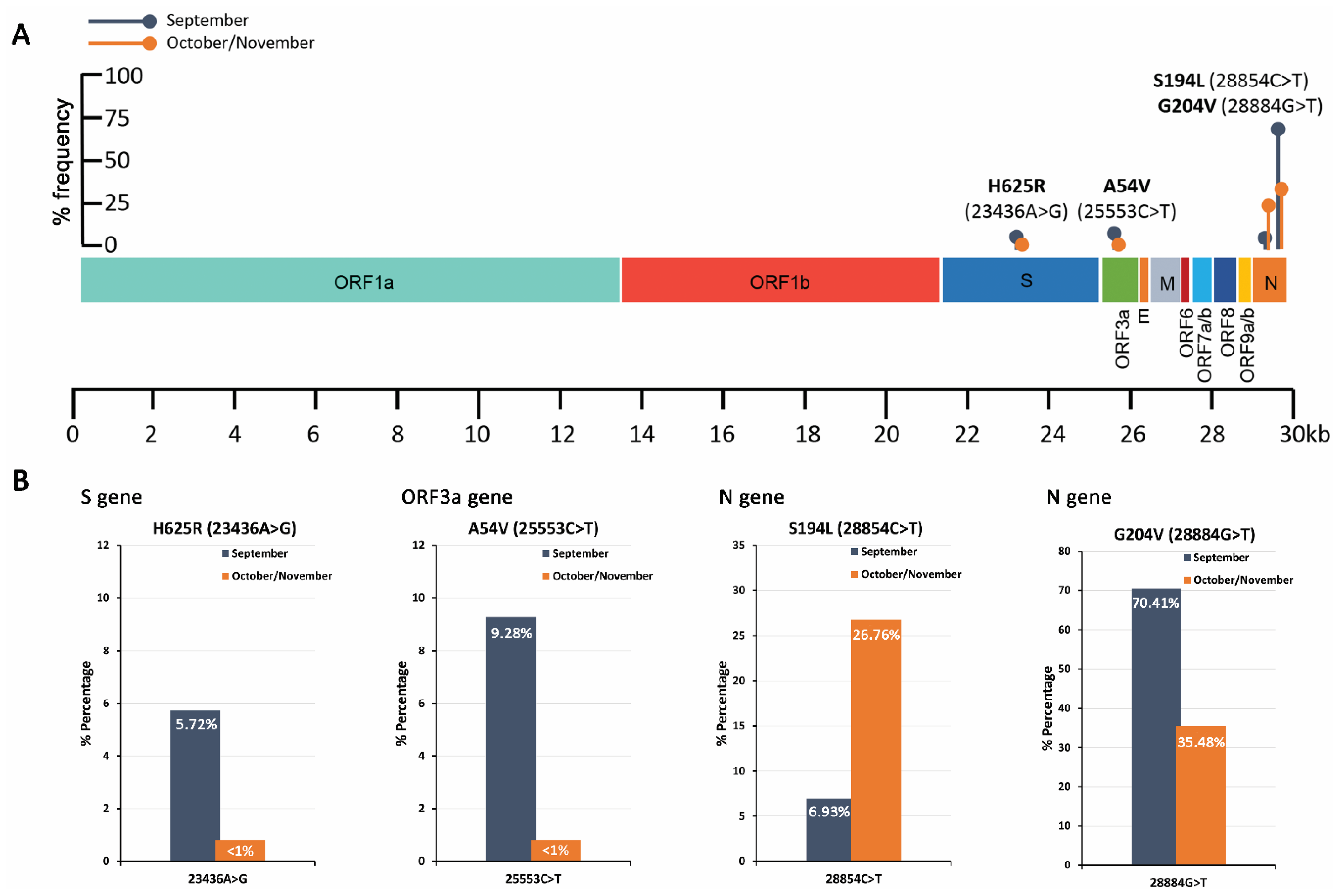
| October/November 2020 | ||||||||
| Position | Ref. Base | Alt. Base | Alt. Freq.(%) | Total Depth | Ref. Codon | Ref. AA | Alt. Codon | Alt. AA |
| P323L (14408C>T) | C | T | 99.86 | 249,568 | CCT | P | CTT | L |
| D614G (23403A>G) | A | G | 99.95 | 563,339 | GAT | D | GGT | G |
| Q57H (25563G>T) | G | T | 46.95 | 342,018 | CAG | Q | CAT | H |
| S194L (28854C>T) | C | T | 26.76 | 507,486 | TCA | S | TTA | L |
| 28881_28884del | A | −NNNN | 5.08 | 597,906 | NA | NA | NA | NA |
| R203K (28881G>A) | G | A | 73.06 | 453,201 | AGG | R | AAG | K |
| R203 (28882G>A) | G | A | 63.54 | 463,095 | AGG | R | AGA | R |
| G204R (28883G>C) | G | C | 63.05 | 490,594 | GGA | G | CGA | R |
| G204V (28884G>T) | G | T | 35.48 | 429,127 | GGA | G | GTA | V |
| 28885_28886insACAT | A | +ACAT | 3.50 | 599,861 | NA | NA | NA | NA |
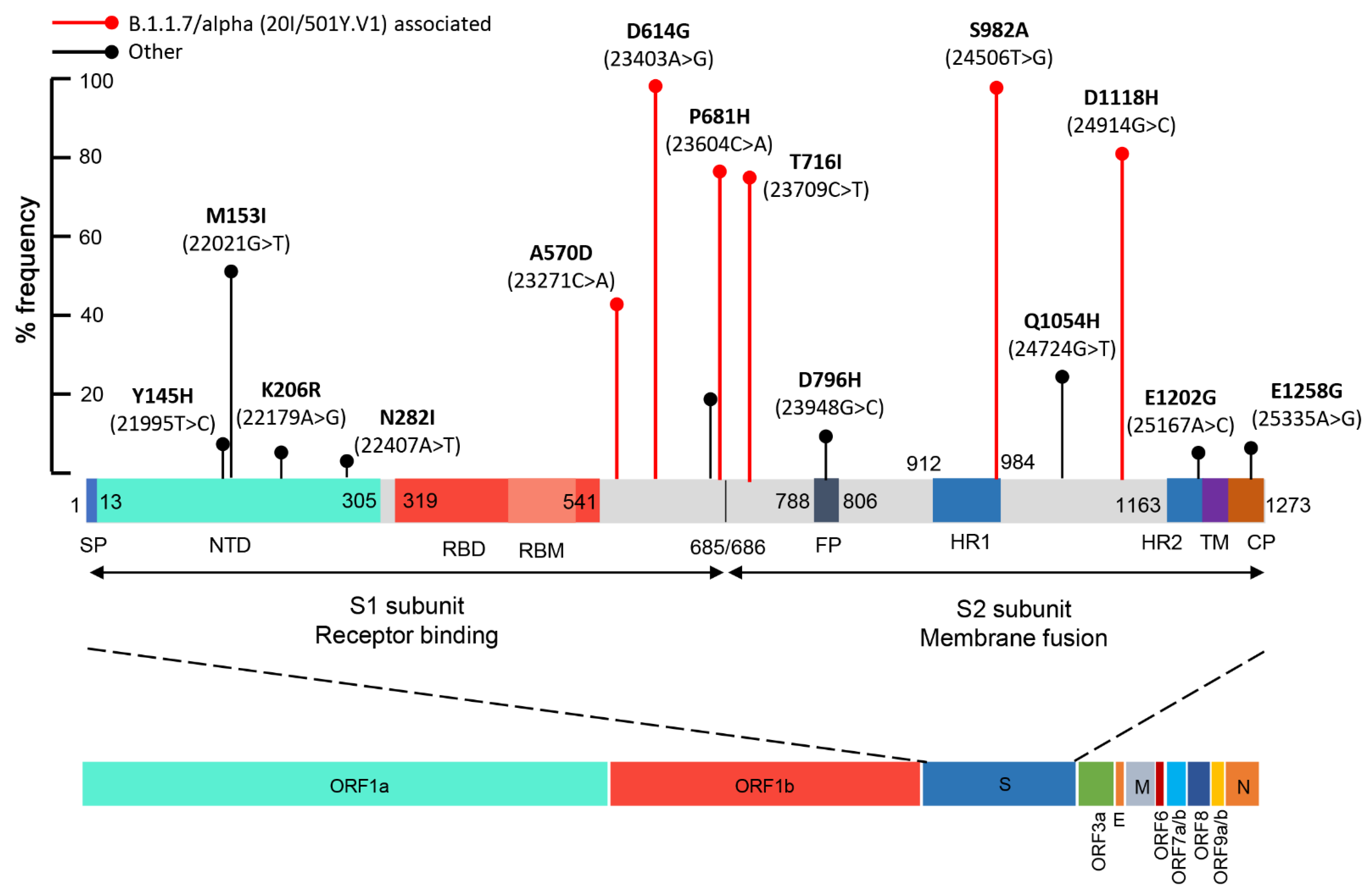
| Position | Ref. Base | Alt. Base | Alt. Freq. (%) | Total Depth | Ref. Codon | Ref. AA | Alt. Codon | Alt. AA |
|---|---|---|---|---|---|---|---|---|
| Y144 (21994T>C) | T | C | 6.75 | 116,465 | TAT | Y | TAC | Y |
| Y145H (21995T>C) | T | C | 8.75 | 100,235 | TAC | Y | CAC | H |
| M153I (22021G>T) | G | T | 52.41 | 114,991 | ATG | M | ATT | I |
| K206R (22179A>G) | A | G | 6.51 | 71,676 | AAG | K | AGG | R |
| N282I (22407A>T) | A | T | 4.12 | 153,621 | AAT | N | ATT | I |
| A570D (23271C>A) | C | A | 44.41 | 286,530 | GCT | A | GAT | D |
| D614G (23403A>G) | A | G | 99.95 | 135,792 | GAT | D | GGT | G |
| C649 (23509T>C) | T | C | 3.12 | 103,991 | TGT | C | TGC | C |
| S659 (23539A>G) | A | G | 17.97 | 263,236 | TCA | S | TCG | S |
| Q675H (23587G>T) | G | T | 20.12 | 154,225 | CAG | Q | CAT | H |
| P681H (23604C>A) | C | A | 78.56 | 150,675 | CCT | P | CAT | H |
| T716I (23709C>T) | C | T | 77.62 | 152,037 | ACA | T | ATA | I |
| D796H (23948G>C) | G | C | 11.16 | 147,600 | GAT | D | CAT | H |
| I934 (24364T>C) | T | C | 3.56 | 174,369 | ATT | I | ATC | I |
| L959 (24437T>C) | T | C | 5.09 | 156,278 | TTA | L | CTA | L |
| S982A (24506T>G) | T | G | 99.72 | 178,323 | TCA | S | GCA | A |
| Q1054H (24724G>T) | G | T | 25.88 | 175,445 | CAG | Q | CAT | H |
| D1118H (24914G>C) | G | C | 82.96 | 176,660 | GAC | D | CAC | H |
| F1156 (25030T>C) | T | C | 3.07 | 94,796 | TTT | F | TTC | F |
| E1202G (25167A>G) | A | G | 6.56 | 77,473 | GAA | E | GGA | G |
| E1258G (25335A>G) | A | G | 7.80 | 253,421 | GAA | E | GGA | G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avgeris, M.; Adamopoulos, P.G.; Galani, A.; Xagorari, M.; Gourgiotis, D.; Trougakos, I.P.; Voulgaris, N.; Dimopoulos, M.-A.; Thomaidis, N.S.; Scorilas, A. Novel Nested-Seq Approach for SARS-CoV-2 Real-Time Epidemiology and In-Depth Mutational Profiling in Wastewater. Int. J. Mol. Sci. 2021, 22, 8498. https://doi.org/10.3390/ijms22168498
Avgeris M, Adamopoulos PG, Galani A, Xagorari M, Gourgiotis D, Trougakos IP, Voulgaris N, Dimopoulos M-A, Thomaidis NS, Scorilas A. Novel Nested-Seq Approach for SARS-CoV-2 Real-Time Epidemiology and In-Depth Mutational Profiling in Wastewater. International Journal of Molecular Sciences. 2021; 22(16):8498. https://doi.org/10.3390/ijms22168498
Chicago/Turabian StyleAvgeris, Margaritis, Panagiotis G. Adamopoulos, Aikaterini Galani, Marieta Xagorari, Dimitrios Gourgiotis, Ioannis P. Trougakos, Nikolaos Voulgaris, Meletios-Athanasios Dimopoulos, Nikolaos S. Thomaidis, and Andreas Scorilas. 2021. "Novel Nested-Seq Approach for SARS-CoV-2 Real-Time Epidemiology and In-Depth Mutational Profiling in Wastewater" International Journal of Molecular Sciences 22, no. 16: 8498. https://doi.org/10.3390/ijms22168498
APA StyleAvgeris, M., Adamopoulos, P. G., Galani, A., Xagorari, M., Gourgiotis, D., Trougakos, I. P., Voulgaris, N., Dimopoulos, M.-A., Thomaidis, N. S., & Scorilas, A. (2021). Novel Nested-Seq Approach for SARS-CoV-2 Real-Time Epidemiology and In-Depth Mutational Profiling in Wastewater. International Journal of Molecular Sciences, 22(16), 8498. https://doi.org/10.3390/ijms22168498