
Decreased Brain pH and Pathophysiology in Schizophrenia


Abstract
:1. Introduction
2. Decreased Brain pH in Patients with Schizophrenia
3. Mitochondria Dysfunction in Schizophrenia
4. Effects of Low Brain pH on Neuronal Activity in Schizophrenia
4.1. Dopaminergic and Glutamatergic Systems
4.2. Noradrenergic System
5. Possible Involvement of pH-Regulating Proteins in Schizophrenia
5.1. NHEs
5.1.1. NHE1 and NHE3
5.1.2. NHE5
5.1.3. NHE6 and NHE9
5.2. NCBTs
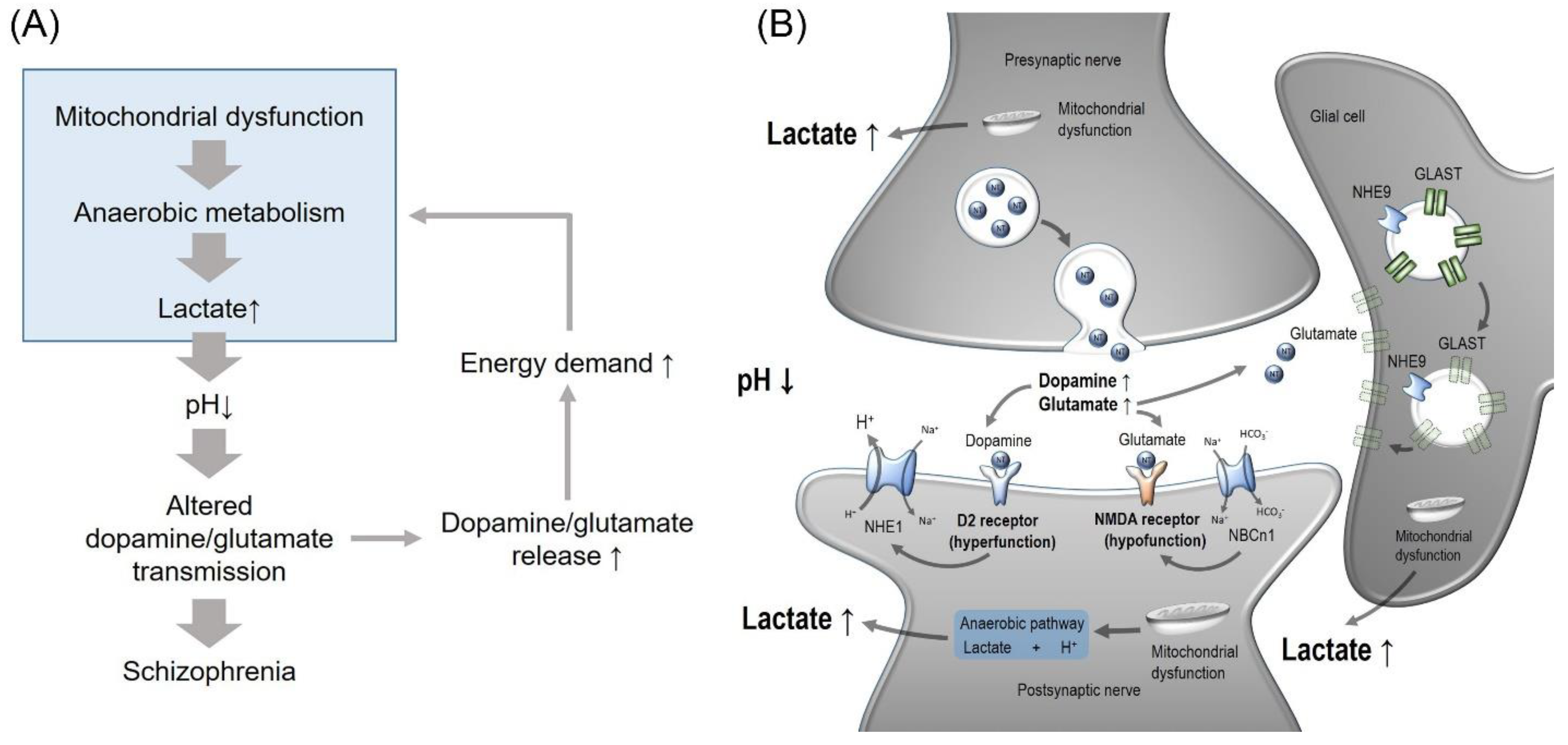
6. Model for pH Involvement in Schizophrenia
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.G.; Steiner, J.; Bogerts, B.; Braun, K.; Jankowski, Z.; Kumaratilake, J.; et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: Old fashioned, but still in vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef]
- Shen, L.H.; Liao, M.H.; Tseng, Y.C. Recent advances in imaging of dopaminergic neurons for evaluation of neuropsychiatric disorders. J. Biomed. Biotechnol. 2012, 2012, 259349. [Google Scholar] [CrossRef] [Green Version]
- Stahl, S.M.; Buckley, P.F. Negative symptoms of schizophrenia: A problem that will not go away. Acta Psychiatr. Scand. 2007, 115, 4–11. [Google Scholar] [CrossRef]
- Du, F.; Cooper, A.J.; Thida, T.; Sehovic, S.; Lukas, S.E.; Cohen, B.M.; Zhang, X.; Ongur, D. In vivo evidence for cerebral bioenergetic abnormalities in schizophrenia measured using 31P magnetization transfer spectroscopy. JAMA Psychiatry 2014, 71, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halim, N.D.; Lipska, B.K.; Hyde, T.M.; Deep-Soboslay, A.; Saylor, E.M.; Herman, M.M.; Thakar, J.; Verma, A.; Kleinman, J.E. Increased lactate levels and reduced pH in postmortem brains of schizophrenics: Medication confounds. J. Neurosci. Methods 2008, 169, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Pruett, B.S.; Meador-Woodruff, J.H. Evidence for altered energy metabolism, increased lactate, and decreased pH in schizophrenia brain: A focused review and meta-analysis of human postmortem and magnetic resonance spectroscopy studies. Schizophr. Res. 2020, 223, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Schulze, E.; Weiss, E.; Weiss, F.; Bohme, H.J.; Hofmann, E. An improved method for adrenalectomy of suckling rats. The influence of thrombin treatment and deoxycorticosterone substitution on survival and on hepatic and renal enzyme activities. Biomed. Biochim. Acta 1986, 45, 1049–1056. [Google Scholar]
- Altar, C.A.; Jurata, L.W.; Charles, V.; Lemire, A.; Liu, P.; Bukhman, Y.; Young, T.A.; Bullard, J.; Yokoe, H.; Webster, M.J.; et al. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol. Psychiatry 2005, 58, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Clay, H.B.; Sillivan, S.; Konradi, C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int. J. Dev. Neurosci. 2011, 29, 311–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [Green Version]
- Valiente-Palleja, A.; Torrell, H.; Alonso, Y.; Vilella, E.; Muntane, G.; Martorell, L. Increased blood lactate levels during exercise and mitochondrial DNA alterations converge on mitochondrial dysfunction in schizophrenia. Schizophr. Res. 2020, 220, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chesler, M. Regulation and modulation of pH in the brain. Physiol. Rev. 2003, 83, 1183–1221. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.S.; Yang, H.S.; He, P.; Kim, E.; Rajbhandari, I.; Yun, C.C.; Choi, I. Sodium/bicarbonate cotransporter NBCn1/slc4a7 increases cytotoxicity in magnesium depletion in primary cultures of hippocampal neurons. Eur. J. Neurosci. 2009, 29, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, S.; Ruusuvuori, E.; Sipila, S.T.; Haapanen, A.; Damkier, H.H.; Kurth, I.; Hentschke, M.; Schweizer, M.; Rudhard, Y.; Laatikainen, L.M.; et al. Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc. Natl. Acad. Sci. USA 2008, 105, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffin, V.A.; Salameh, A.I.; Boron, W.F.; Parker, M.D. Intracellular pH regulation by acid-base transporters in mammalian neurons. Front. Physiol. 2014, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Carney, K.E.; Falgoust, L.; Pan, J.W.; Sun, D.; Zhang, Z. Emerging roles of Na+/H+ exchangers in epilepsy and developmental brain disorders. Prog. Neurobiol. 2016, 138–140, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Sinning, A.; Hubner, C.A. Minireview: pH and synaptic transmission. FEBS Lett. 2013, 587, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Soto, E.; Ortega-Ramirez, A.; Vega, R. Protons as Messengers of Intercellular Communication in the Nervous System. Front. Cell Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Mozrzymas, J.W.; Zarnowska, E.D.; Pytel, M.; Mercik, K. Modulation of GABA(A) receptors by hydrogen ions reveals synaptic GABA transient and a crucial role of the desensitization process. J. Neurosci. 2003, 23, 7981–7992. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Gonzalez-Islas, C.E.; Kang, Y.; Li, J.M.; Choi, I. Deletion of the Na/HCO3 Transporter NBCn1 Protects Hippocampal Neurons from NMDA-induced Seizures and Neurotoxicity in Mice. Sci. Rep. 2019, 9, 15981. [Google Scholar] [CrossRef] [Green Version]
- Schank, J.R.; Lee, S.; Gonzalez-Islas, C.E.; Nennig, S.E.; Fulenwider, H.D.; Chang, J.; Li, J.M.; Kim, Y.; Jeffers, L.A.; Chung, J.; et al. Increased Alcohol Consumption in Mice Lacking Sodium Bicarbonate Transporter NBCn1. Sci. Rep. 2020, 10, 11017. [Google Scholar] [CrossRef]
- Sinning, A.; Liebmann, L.; Kougioumtzes, A.; Westermann, M.; Bruehl, C.; Hubner, C.A. Synaptic glutamate release is modulated by the Na+ -driven Cl−/HCO3− exchanger Slc4a8. J. Neurosci. 2011, 31, 7300–7311. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.; Politis, M.; Rabiner, E.A.; Middleton, L.T. Novel PET Biomarkers to Disentangle Molecular Pathways across Age-Related Neurodegenerative Diseases. Cells 2020, 9, 2581. [Google Scholar] [CrossRef]
- Galijašević, M.; Steiger, R.; Regodić, M.; Waibel, M.; Sommer, P.J.D.; Grams, A.E.; Singewald, N.; Gizewski, E.R. Brain Energy Metabolism in Two States of Mind Measured by Phosphorous Magnetic Resonance Spectroscopy. Front. Hum. Neurosci. 2021, 15, 686433. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; Thomas, N.; Scarr, E.; Udawela, M. Evidence for impaired glucose metabolism in the striatum, obtained postmortem, from some subjects with schizophrenia. Transl. Psychiatry 2016, 6, e949. [Google Scholar] [CrossRef] [Green Version]
- Hagihara, H.; Catts, V.S.; Katayama, Y.; Shoji, H.; Takagi, T.; Huang, F.L.; Nakao, A.; Mori, Y.; Huang, K.P.; Ishii, S.; et al. Decreased Brain pH as a Shared Endophenotype of Psychiatric Disorders. Neuropsychopharmacology 2018, 43, 459–468. [Google Scholar] [CrossRef] [Green Version]
- das Neves Duarte, J.M.; Kulak, A.; Gholam-Razaee, M.M.; Cuenod, M.; Gruetter, R.; Do, K.Q. N-acetylcysteine normalizes neurochemical changes in the glutathione-deficient schizophrenia mouse model during development. Biol. Psychiatry 2012, 71, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, C.R.; Mielnik, C.A.; Funk, A.; O’Donovan, S.M.; Bentea, E.; Pletnikov, M.; Ramsey, A.J.; Wen, Z.; Rowland, L.M.; McCullumsmith, R.E. Measurement of lactate levels in postmortem brain, iPSCs, and animal models of schizophrenia. Sci. Rep. 2019, 9, 5087. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Z.; Vawter, M.P.; Walsh, D.M.; Tomita, H.; Evans, S.J.; Choudary, P.V.; Lopez, J.F.; Avelar, A.; Shokoohi, V.; Chung, T.; et al. Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum. Mol. Genet. 2004, 13, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, H.; Vawter, M.P.; Walsh, D.M.; Evans, S.J.; Choudary, P.V.; Li, J.; Overman, K.M.; Atz, M.E.; Myers, R.M.; Jones, E.G.; et al. Effect of agonal and postmortem factors on gene expression profile: Quality control in microarray analyses of postmortem human brain. Biol. Psychiatry 2004, 55, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Vawter, M.P.; Tomita, H.; Meng, F.; Bolstad, B.; Li, J.; Evans, S.; Choudary, P.; Atz, M.; Shao, L.; Neal, C.; et al. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: Implications for brain disorders. Mol. Psychiatry 2006, 11, 663–679. [Google Scholar] [CrossRef] [Green Version]
- Glavina, T.; Mrass, D.; Dodig, T.; Glavina, G.; Pranic, S.; Uglesic, B. Blood lactate levels in patients receiving first- or second- generation antipsychotics. Croat. Med. J. 2011, 52, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmorsy, E.; Shahda, M.; Mahmoud, E.-H.M.; Rakha, S.A.; Shoaib, M. Blood lactate levels as a biomarker of antipsychotic side effects in patients with schizophrenia. J. Psychopharmacol. 2016, 30, 63–68. [Google Scholar] [CrossRef]
- Goh, S.; Dong, Z.; Zhang, Y.; DiMauro, S.; Peterson, B.S. Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder: Evidence from brain imaging. JAMA Psychiatry 2014, 71, 665–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavelier, L.; Jazin, E.E.; Eriksson, I.; Prince, J.; Bave, U.; Oreland, L.; Gyllensten, U. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics 1995, 29, 217–224. [Google Scholar] [CrossRef]
- Inuwa, I.M.; Peet, M.; Williams, M.A. QSAR modeling and transmission electron microscopy stereology of altered mitochondrial ultrastructure of white blood cells in patients diagnosed as schizophrenic and treated with antipsychotic drugs. Biotech. Histochem. 2005, 80, 133–137. [Google Scholar] [CrossRef]
- Karry, R.; Klein, E.; Ben Shachar, D. Mitochondrial complex I subunits expression is altered in schizophrenia: A postmortem study. Biol. Psychiatry 2004, 55, 676–684. [Google Scholar] [CrossRef]
- Kung, L.; Roberts, R.C. Mitochondrial pathology in human schizophrenic striatum: A postmortem ultrastructural study. Synapse 1999, 31, 67–75. [Google Scholar] [CrossRef]
- Maurer, I.; Zierz, S.; Moller, H. Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr. Res. 2001, 48, 125–136. [Google Scholar] [CrossRef]
- Uranova, N.; Bonartsev, P.; Brusov, O.; Morozova, M.; Rachmanova, V.; Orlovskaya, D. The ultrastructure of lymphocytes in schizophrenia. World J. Biol. Psychiatry 2007, 8, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Uranova, N.; Orlovskaya, D.; Vikhreva, O.; Zimina, I.; Kolomeets, N.; Vostrikov, V.; Rachmanova, V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res. Bull. 2001, 55, 597–610. [Google Scholar] [CrossRef]
- Deicken, R.F.; Calabrese, G.; Merrin, E.L.; Vinogradov, S.; Fein, G.; Weiner, M.W. Asymmetry of temporal lobe phosphorous metabolism in schizophrenia: A 31phosphorous magnetic resonance spectroscopic imaging study. Biol. Psychiatry 1995, 38, 279–286. [Google Scholar] [CrossRef]
- Deicken, R.F.; Merrin, E.L.; Floyd, T.C.; Weiner, M.W. Correlation between left frontal phospholipids and Wisconsin Card Sort Test performance in schizophrenia. Schizophr. Res. 1995, 14, 177–181. [Google Scholar] [CrossRef]
- Fujimoto, T.; Nakano, T.; Takano, T.; Hokazono, Y.; Asakura, T.; Tsuji, T. Study of chronic schizophrenics using 31P magnetic resonance chemical shift imaging. Acta Psychiatr. Scand. 1992, 86, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.; Rzanny, R.; Riehemann, S.; Volz, H.P.; Schmidt, B.; Gerhard, U.J.; Filz, C.; Schonberg, A.; Mentzel, H.J.; Kaiser, W.A.; et al. Cerebral phosphate metabolism in first-degree relatives of patients with schizophrenia. Am. J. Psychiatry 2001, 158, 958–960. [Google Scholar] [CrossRef]
- Pettegrew, J.W.; Keshavan, M.S.; Panchalingam, K.; Strychor, S.; Kaplan, D.B.; Tretta, M.G.; Allen, M. Alterations in brain high-energy phosphate and membrane phospholipid metabolism in first-episode, drug-naive schizophrenics. A pilot study of the dorsal prefrontal cortex by in vivo phosphorus 31 nuclear magnetic resonance spectroscopy. Arch. Gen. Psychiatry 1991, 48, 563–568. [Google Scholar] [CrossRef]
- Stanley, J.A.; Williamson, P.C.; Drost, D.J.; Carr, T.J.; Rylett, R.J.; Morrison-Stewart, S.; Thompson, R.T. Membrane phospholipid metabolism and schizophrenia: An in vivo 31P-MR spectroscopy study. Schizophr. Res. 1994, 13, 209–215. [Google Scholar] [CrossRef]
- Regenold, W.T.; Pratt, M.; Nekkalapu, S.; Shapiro, P.S.; Kristian, T.; Fiskum, G. Mitochondrial detachment of hexokinase 1 in mood and psychotic disorders: Implications for brain energy metabolism and neurotrophic signaling. J. Psychiatr. Res. 2012, 46, 95–104. [Google Scholar] [CrossRef]
- Rowland, L.M.; Pradhan, S.; Korenic, S.; Wijtenburg, S.A.; Hong, L.E.; Edden, R.A.; Barker, P.B. Elevated brain lactate in schizophrenia: A 7 T magnetic resonance spectroscopy study. Transl. Psychiatry 2016, 6, e967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, B.; La, Y.; Gao, L.; Zhu, H.; Tian, N.; Zhang, M.; Yang, Y.; Zhao, X.; Tang, R.; Ma, G.; et al. A comparative proteomics analysis of rat mitochondria from the cerebral cortex and hippocampus in response to antipsychotic medications. J. Proteome Res. 2009, 8, 3633–3641. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Rego, A.C.; Cardoso, S.M.; Borges, F.; Swerdlow, R.H.; Macedo, T.; de Oliveira, C.R. Mitochondrial dysfunction and caspase activation in rat cortical neurons treated with cocaine or amphetamine. Brain Res. 2006, 1089, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Chen, W.; Andreazza, A.C. The Potential Role of the NLRP3 Inflammasome as a Link between Mitochondrial Complex I Dysfunction and Inflammation in Bipolar Disorder. Neural Plast. 2015, 2015, 408136. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Amar, S.; Shamir, A.; Ovadia, O.; Blanaru, M.; Reshef, A.; Kremer, I.; Rietschel, M.; Schulze, T.G.; Maier, W.; Belmaker, R.H.; et al. Mitochondrial DNA HV lineage increases the susceptibility to schizophrenia among Israeli Arabs. Schizophr. Res. 2007, 94, 354–358. [Google Scholar] [CrossRef]
- Munakata, K.; Iwamoto, K.; Bundo, M.; Kato, T. Mitochondrial DNA 3243A>G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol. Psychiatry 2005, 57, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Nishigaki, Y.; Kong, Q.P.; Fuku, N.; Kojima, S.; Iwata, N.; Ozaki, N.; Tanaka, M. Analysis of mitochondrial DNA variants in Japanese patients with schizophrenia. Mitochondrion 2009, 9, 385–393. [Google Scholar] [CrossRef]
- Reale, M.; Costantini, E.; Greig, N.H. Cytokine Imbalance in Schizophrenia. From Research to Clinic: Potential Implications for Treatment. Front. Psychiatry 2021, 12, 536257. [Google Scholar] [CrossRef]
- Korf, J.; Gramsbergen, J.B. Timing of potential and metabolic brain energy. J. Neurochem. 2007, 103, 1697–1708. [Google Scholar] [CrossRef]
- Stojanov, D.; Korf, J.; de Jonge, P.; Popov, G. The possibility of evidence-based psychiatry: Depression as a case. Clin. Epigenetics 2011, 2, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Barrier, L.; Barc, S.; Fauconneau, B.; Pontcharraud, R.; Kelani, A.; Bestel, E.; Page, G. Evidence that acidosis alters the high-affinity dopamine uptake in rat striatal slices and synaptosomes by different mechanisms partially related to oxidative damage. Neurochem. Int. 2003, 42, 27–34. [Google Scholar] [CrossRef]
- Remblier, C.; Jolimay, N.; Wahl, A.; Pariat, C.; Piriou, A.; Huguet, F. Extracellular dopamine and catabolites in rat striatum during lactic acid perfusion as determined by in vivo microdialysis. Brain Res. 1998, 804, 224–230. [Google Scholar] [CrossRef]
- Berman, S.B.; Zigmond, M.J.; Hastings, T.G. Modification of dopamine transporter function: Effect of reactive oxygen species and dopamine. J. Neurochem. 1996, 67, 593–600. [Google Scholar] [CrossRef]
- Bindoli, A.; Rigobello, M.P.; Deeble, D.J. Biochemical and toxicological properties of the oxidation products of catecholamines. Free Radic. Biol. Med. 1992, 13, 391–405. [Google Scholar] [CrossRef]
- Klegeris, A.; Korkina, L.G.; Greenfield, S.A. Autoxidation of dopamine: A comparison of luminescent and spectrophotometric detection in basic solutions. Free Radic. Biol. Med. 1995, 18, 215–222. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamate and schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar] [CrossRef]
- Labrie, V.; Roder, J.C. The involvement of the NMDA receptor D-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci. Biobehav. Rev. 2010, 34, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; D’Souza, D.C.; Mathalon, D.; Perry, E.; Belger, A.; Hoffman, R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: Toward a paradigm shift in medication development. Psychopharmacology 2003, 169, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Pinals, D.A.; Adler, C.M.; Elman, I.; Clifton, A.; Pickar, D.; Breier, A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gil, X.; Babot, Z.; Amargos-Bosch, M.; Sunol, C.; Artigas, F.; Adell, A. Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 2007, 32, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorrain, D.S.; Baccei, C.S.; Bristow, L.J.; Anderson, J.J.; Varney, M.A. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 2003, 117, 697–706. [Google Scholar] [CrossRef]
- Lorrain, D.S.; Schaffhauser, H.; Campbell, U.C.; Baccei, C.S.; Correa, L.D.; Rowe, B.; Rodriguez, D.E.; Anderson, J.J.; Varney, M.A.; Pinkerton, A.B.; et al. Group II mGlu receptor activation suppresses norepinephrine release in the ventral hippocampus and locomotor responses to acute ketamine challenge. Neuropsychopharmacology 2003, 28, 1622–1632. [Google Scholar] [CrossRef] [Green Version]
- Egerton, A.; Broberg, B.V.; Van Haren, N.; Merritt, K.; Barker, G.J.; Lythgoe, D.J.; Perez-Iglesias, R.; Baandrup, L.; During, S.W.; Sendt, K.V.; et al. Response to initial antipsychotic treatment in first episode psychosis is related to anterior cingulate glutamate levels: A multicentre (1)H-MRS study (OPTiMiSE). Mol. Psychiatry 2018, 23, 2145–2155. [Google Scholar] [CrossRef]
- Egerton, A.; Brugger, S.; Raffin, M.; Barker, G.J.; Lythgoe, D.J.; McGuire, P.K.; Stone, J.M. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology 2012, 37, 2515–2521. [Google Scholar] [CrossRef] [PubMed]
- Merritt, K.; Egerton, A.; Kempton, M.J.; Taylor, M.J.; McGuire, P.K. Nature of Glutamate Alterations in Schizophrenia: A Meta-analysis of Proton Magnetic Resonance Spectroscopy Studies. JAMA Psychiatry 2016, 73, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Mouchlianitis, E.; Bloomfield, M.A.; Law, V.; Beck, K.; Selvaraj, S.; Rasquinha, N.; Waldman, A.; Turkheimer, F.E.; Egerton, A.; Stone, J.; et al. Treatment-Resistant Schizophrenia Patients Show Elevated Anterior Cingulate Cortex Glutamate Compared to Treatment-Responsive. Schizophr. Bull. 2016, 42, 744–752. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Cull-Candy, S.G. Pharmacological properties and H+ sensitivity of excitatory amino acid receptor channels in rat cerebellar granule neurones. J. Physiol. 1991, 433, 727–763. [Google Scholar] [CrossRef] [Green Version]
- Mony, L.; Kew, J.N.; Gunthorpe, M.J.; Paoletti, P. Allosteric modulators of NR2B-containing NMDA receptors: Molecular mechanisms and therapeutic potential. Br. J. Pharm. 2009, 157, 1301–1317. [Google Scholar] [CrossRef] [Green Version]
- Bondy, B.; Ackenheil, M.; Birzle, W.; Elbers, R.; Fröhler, M. Catecholamines and their receptors in blood: Evidence for alterations in schizophrenia. Biol. Psychiatry 1984, 19, 1377–1393. [Google Scholar]
- Kemali, D.; Del Vecchio, M.; Maj, M. Increased noradrenaline levels in CSF and plasma of schizophrenic patients. Biol. Psychiatry 1982, 17, 711–717. [Google Scholar]
- Yamamoto, K.; Hornykiewicz, O. Proposal for a noradrenaline hypothesis of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 913–922. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, D.; Marques, D.A.; Bernuci, M.P.; Leite, C.M.; Araújo-Lopes, R.; Anselmo-Franci, J.; Bícego, K.C.; Szawka, R.E.; Gargaglioni, L.H. Role of sex hormones in hypercapnia-induced activation of the locus coeruleus in female and male rats. Neuroscience 2016, 313, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, N.; Zhang, X.; Tadepalli, J.S.; Yu, L.; Gai, H.; Petit, J.; Pamulapati, R.T.; Jin, X.; Jiang, C. Involvement of TRP channels in the CO₂ chemosensitivity of locus coeruleus neurons. J. Neurophysiol. 2011, 105, 2791–2801. [Google Scholar] [CrossRef]
- Devoto, P.; Flore, G.; Saba, P.; Fà, M.; Gessa, G.L. Stimulation of the locus coeruleus elicits noradrenaline and dopamine release in the medial prefrontal and parietal cortex. J. Neurochem. 2005, 92, 368–374. [Google Scholar] [CrossRef]
- Devoto, P.; Flore, G. On the origin of cortical dopamine: Is it a co-transmitter in noradrenergic neurons? Curr. Neuropharmacol. 2006, 4, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.C.; Greene, R.W. CNS dopamine transmission mediated by noradrenergic innervation. J. Neurosci. 2012, 32, 6072–6080. [Google Scholar] [CrossRef] [Green Version]
- Devoto, P.; Flore, G.; Saba, P.; Scheggi, S.; Mulas, G.; Gambarana, C.; Spiga, S.; Gessa, G.L. Noradrenergic terminals are the primary source of α(2)-adrenoceptor mediated dopamine release in the medial prefrontal cortex. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 90, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Devoto, P.; Sagheddu, C.; Santoni, M.; Flore, G.; Saba, P.; Pistis, M.; Gessa, G.L. Noradrenergic Source of Dopamine Assessed by Microdialysis in the Medial Prefrontal Cortex. Front. Pharm. 2020, 11, 588160. [Google Scholar] [CrossRef] [PubMed]
- Severson, C.A.; Wang, W.; Pieribone, V.A.; Dohle, C.I.; Richerson, G.B. Midbrain serotonergic neurons are central pH chemoreceptors. Nat. Neurosci. 2003, 6, 1139–1140. [Google Scholar] [CrossRef]
- Donowitz, M.; Ming Tse, C.; Fuster, D. SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol. Asp. Med. 2013, 34, 236–251. [Google Scholar] [CrossRef] [Green Version]
- Obara, M.; Szeliga, M.; Albrecht, J. Regulation of pH in the mammalian central nervous system under normal and pathological conditions: Facts and hypotheses. Neurochem. Int. 2008, 52, 905–919. [Google Scholar] [CrossRef]
- Halestrap, A.P. The SLC16 gene family–structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef]
- Orlowski, J.; Grinstein, S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflug. Arch. 2004, 447, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A.; Di Sole, F.; Moe, O.W. Na+/H+ exchangers: Physiology and link to hypertension and organ ischemia. Curr. Opin. Nephrol. Hypertens. 2005, 14, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, J.; Grinstein, S. Emerging roles of alkali cation/proton exchangers in organellar homeostasis. Curr. Opin. Cell Biol. 2007, 19, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, D.G.; Alexander, R.T. Traditional and emerging roles for the SLC9 Na+/H+ exchangers. Pflug. Arch. 2014, 466, 61–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, C.L.; Drong, R.F.; Riley, D.T.; Gill, G.S.; Slightom, J.L.; Huff, R.M. D4 dopamine receptor-mediated signaling events determined in transfected Chinese hamster ovary cells. J. Biol. Chem. 1994, 269, 11813–11819. [Google Scholar] [CrossRef]
- Neve, K.A.; Kozlowski, M.R.; Rosser, M.P. Dopamine D2 receptor stimulation of Na+/H+ exchange assessed by quantification of extracellular acidification. J. Biol. Chem. 1992, 267, 25748–25753. [Google Scholar] [CrossRef]
- Felder, C.C.; Campbell, T.; Albrecht, F.; Jose, P.A. Dopamine inhibits Na+-H+ exchanger activity in renal BBMV by stimulation of adenylate cyclase. Am. J. Physiol. 1990, 259, F297–F303. [Google Scholar] [CrossRef]
- Gomes, P.; Soares-da-Silva, P. Dopamine acutely decreases type 3 Na+/H+ exchanger activity in renal OK cells through the activation of protein kinases A and C signalling cascades. Eur. J. Pharm. 2004, 488, 51–59. [Google Scholar] [CrossRef]
- Gomes, P.; Vieira-Coelho, M.A.; Soares-Da-Silva, P. Ouabain-insensitive acidification by dopamine in renal OK cells: Primary control of the Na+/H+ exchanger. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R10–R18. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Oh, S.; Zizak, M.; Steplock, D.; Tsao, S.; Tse, C.M.; Weinman, E.J.; Donowitz, M. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc. Natl. Acad. Sci. USA 1997, 94, 3010–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aperia, A.; Bertorello, A.; Seri, I. Dopamine causes inhibition of Na+-K+-ATPase activity in rat proximal convoluted tubule segments. Am. J. Physiol. 1987, 252, F39–F45. [Google Scholar] [CrossRef]
- Kunimi, M.; Seki, G.; Hara, C.; Taniguchi, S.; Uwatoko, S.; Goto, A.; Kimura, S.; Fujita, T. Dopamine inhibits renal Na+:HCO3− cotransporter in rabbits and normotensive rats but not in spontaneously hypertensive rats. Kidney Int. 2000, 57, 534–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, M.A.; Simon, J.N.; Ma, R.; Loonat, A.A.; Crabtree, M.J.; Paterson, D.J.; Fahlman, R.P.; Casadei, B.; Fliegel, L.; Swietach, P. Nitric oxide modulates cardiomyocyte pH control through a biphasic effect on sodium/hydrogen exchanger-1. Cardiovasc. Res. 2020, 116, 1958–1971. [Google Scholar] [CrossRef]
- Diering, G.H.; Mills, F.; Bamji, S.X.; Numata, M. Regulation of dendritic spine growth through activity-dependent recruitment of the brain-enriched Na+/H+ exchanger NHE5. Mol. Biol. Cell 2011, 22, 2246–2257. [Google Scholar] [CrossRef]
- Sin, W.C.; Moniz, D.M.; Ozog, M.A.; Tyler, J.E.; Numata, M.; Church, J. Regulation of early neurite morphogenesis by the Na+/H+ exchanger NHE1. J. Neurosci. 2009, 29, 8946–8959. [Google Scholar] [CrossRef]
- Pizzonia, J.H.; Ransom, B.R.; Pappas, C.A. Characterization of Na+/H+ exchange activity in cultured rat hippocampal astrocytes. J. Neurosci. Res. 1996, 44, 191–198. [Google Scholar] [CrossRef]
- Ma, E.; Haddad, G.G. Expression and localization of Na+/H+ exchangers in rat central nervous system. Neuroscience 1997, 79, 591–603. [Google Scholar] [CrossRef]
- Kiwull-Schone, H.; Kiwull, P.; Frede, S.; Wiemann, M. Role of brainstem sodium/proton exchanger 3 for breathing control during chronic acid base imbalance. Am. J. Respir. Crit. Care Med. 2007, 176, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Wiemann, M.; Frede, S.; Bingmann, D.; Kiwull, P.; Kiwull-Schone, H. Sodium/Proton exchanger 3 in the medulla oblongata and set point of breathing control. Am. J. Respir. Crit. Care Med. 2005, 172, 244–249. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.M.; Schreiner, C.M.; Schultheis, P.J.; Miller, M.L.; Evans, R.L.; Vorhees, C.V.; Shull, G.E.; Scott, W.J. Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am. J. Physiol. 1999, 276, C788–C795. [Google Scholar] [CrossRef]
- Gu, X.Q.; Yao, H.; Haddad, G.G. Increased neuronal excitability and seizures in the Na+/H+ exchanger null mutant mouse. Am. J. Physiol. Cell Physiol. 2001, 281, C496–C503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Zhao, P.; Xue, J.; Gu, X.Q.; Sun, X.; Yao, H.; Haddad, G.G. Na+ channel expression and neuronal function in the Na+/H+ exchanger 1 null mutant mouse. J. Neurophysiol. 2003, 89, 229–236. [Google Scholar] [CrossRef]
- Chen, X.; Wang, X.; Tang, L.; Wang, J.; Shen, C.; Liu, J.; Lu, S.; Zhang, H.; Kuang, Y.; Fei, J.; et al. Nhe5 deficiency enhances learning and memory via upregulating Bdnf/TrkB signaling in mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, C.; Monteggia, L.M. BDNF—A key transducer of antidepressant effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Fang, H.; Zhang, L.; Hu, M.; He, S.; Li, H.; Zhu, H. Reversible Changes in BDNF Expression in MK-801-Induced Hippocampal Astrocytes Through NMDAR/PI3K/ERK Signaling. Front. Cell Neurosci. 2021, 15, 672136. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Brenè, S.; Mathé, A.A. BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry 2005, 10, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Bergen, S.E.; Nguyen, Q.L.; Xu, B.; Monteggia, L.M.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 2005, 25, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weickert, C.S.; Hyde, T.M.; Lipska, B.K.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2003, 8, 592–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, P.F.; Pillai, A.; Evans, D.; Stirewalt, E.; Mahadik, S. Brain derived neurotropic factor in first-episode psychosis. Schizophr. Res. 2007, 91, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Palomino, A.; Vallejo-Illarramendi, A.; González-Pinto, A.; Aldama, A.; González-Gómez, C.; Mosquera, F.; González-García, G.; Matute, C. Decreased levels of plasma BDNF in first-episode schizophrenia and bipolar disorder patients. Schizophr. Res. 2006, 86, 321–322. [Google Scholar] [CrossRef]
- Tan, Y.L.; Zhou, D.F.; Cao, L.Y.; Zou, Y.Z.; Zhang, X.Y. Decreased BDNF in serum of patients with chronic schizophrenia on long-term treatment with antipsychotics. Neurosci. Lett. 2005, 382, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, K.; Asama, K.; Watanabe, Y.; Muratake, T.; Takahashi, M.; Someya, T.; Nawa, H. Decreased levels of brain-derived neurotrophic factor in serum of chronic schizophrenic patients. Psychiatry Res. 2002, 110, 249–257. [Google Scholar] [CrossRef]
- Nakamura, N.; Tanaka, S.; Teko, Y.; Mitsui, K.; Kanazawa, H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J. Biol. Chem. 2005, 280, 1561–1572. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, K.C.; Prasad, H.; Rao, R. An inside job: How endosomal Na+/H+ exchangers link to autism and neurological disease. Front. Cell. Neurosci. 2014, 8, 172. [Google Scholar] [CrossRef] [Green Version]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef]
- Christianson, A.L.; Stevenson, R.E.; van der Meyden, C.H.; Pelser, J.; Theron, F.W.; van Rensburg, P.L.; Chandler, M.; Schwartz, C.E. X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J. Med. Genet. 1999, 36, 759–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilfillan, G.D.; Selmer, K.K.; Roxrud, I.; Smith, R.; Kyllerman, M.; Eiklid, K.; Kroken, M.; Mattingsdal, M.; Egeland, T.; Stenmark, H.; et al. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am. J. Hum. Genet. 2008, 82, 1003–1010. [Google Scholar] [CrossRef] [Green Version]
- Garbern, J.Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M.; Feldman, G.; Norris, J.W.; Friez, M.J.; Schwartz, C.E.; Stevenson, R.; Sima, A.A. A mutation affecting the sodium/proton exchanger, SLC9A6, causes mental retardation with tau deposition. Brain 2010, 133, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Piton, A.; Gauthier, J.; Hamdan, F.F.; Lafrenière, R.G.; Yang, Y.; Henrion, E.; Laurent, S.; Noreau, A.; Thibodeau, P.; Karemera, L.; et al. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry 2011, 16, 867–880. [Google Scholar] [CrossRef]
- Ouyang, Q.; Lizarraga, S.B.; Schmidt, M.; Yang, U.; Gong, J.; Ellisor, D.; Kauer, J.A.; Morrow, E.M. Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 2013, 80, 97–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Silva, M.G.; Elliott, K.; Dahl, H.H.; Fitzpatrick, E.; Wilcox, S.; Delatycki, M.; Williamson, R.; Efron, D.; Lynch, M.; Forrest, S. Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. J. Med. Genet. 2003, 40, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwede, M.; Garbett, K.; Mirnics, K.; Geschwind, D.H.; Morrow, E.M. Genes for endosomal NHE6 and NHE9 are misregulated in autism brains. Mol. Psychiatry 2014, 19, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Vink, J.M.; Smit, A.B.; de Geus, E.J.; Sullivan, P.; Willemsen, G.; Hottenga, J.J.; Smit, J.H.; Hoogendijk, W.J.; Zitman, F.G.; Peltonen, L.; et al. Genome-wide association study of smoking initiation and current smoking. Am. J. Hum. Genet. 2009, 84, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Zayats, T.; Athanasiu, L.; Sonderby, I.; Djurovic, S.; Westlye, L.T.; Tamnes, C.K.; Fladby, T.; Aase, H.; Zeiner, P.; Reichborn-Kjennerud, T.; et al. Genome-wide analysis of attention deficit hyperactivity disorder in Norway. PloS ONE 2015, 10, e0122501. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Faraone, S.V.; Zhang-James, Y. Autism spectrum disorder traits in Slc9a9 knock-out mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 363–376. [Google Scholar] [CrossRef]
- Beydoun, R.; Hamood, M.A.; Gomez Zubieta, D.M.; Kondapalli, K.C. Na+/H+ Exchanger 9 Regulates Iron Mobilization at the Blood-Brain Barrier in Response to Iron Starvation. J. Biol. Chem. 2017, 292, 4293–4301. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, K.C.; Hack, A.; Schushan, M.; Landau, M.; Ben-Tal, N.; Rao, R. Functional evaluation of autism-associated mutations in NHE9. Nat. Commun. 2013, 4, 2510. [Google Scholar] [CrossRef] [Green Version]
- McQueen, G.; Sendt, K.V.; Gillespie, A.; Avila, A.; Lally, J.; Vallianatou, K.; Chang, N.; Ferreira, D.; Borgan, F.; Howes, O.D.; et al. Changes in Brain Glutamate on Switching to Clozapine in Treatment-Resistant Schizophrenia. Schizophr. Bull. 2021, 47, 662–671. [Google Scholar] [CrossRef]
- Choi, I. SLC4A transporters. Curr. Top. Membr. 2012, 70, 77–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, M.F.; Chen, A.P.; Parker, M.D.; Boron, W.F. The SLC4 family of bicarbonate (HCO3−) transporters. Mol. Asp. Med. 2013, 34, 159–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Rajbhandari, I.; Yang, H.S.; Lee, S.; Cucoranu, D.; Cooper, D.S.; Klein, J.D.; Sands, J.M.; Choi, I. Neuronal expression of sodium/bicarbonate cotransporter NBCn1 (SLC4A7) and its response to chronic metabolic acidosis. Am. J. Physiol. Cell Physiol. 2010, 298, C1018–C1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burette, A.C.; Weinberg, R.J.; Sassani, P.; Abuladze, N.; Kao, L.; Kurtz, I. The sodium-driven chloride/bicarbonate exchanger in presynaptic terminals. J. Comp. Neurol. 2012, 520, 1481–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yang, H.S.; Kim, E.; Ju, E.J.; Kwon, M.H.; Dudley, R.K.; Smith, Y.; Yun, C.C.; Choi, I. PSD-95 interacts with NBCn1 and enhances channel-like activity without affecting Na/HCO3 cotransport. Cell. Physiol. Biochem. 2012, 30, 1444–1455. [Google Scholar] [CrossRef]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Glass, M.J.; Robinson, D.C.; Waters, E.; Pickel, V.M. Deletion of the NMDA-NR1 receptor subunit gene in the mouse nucleus accumbens attenuates apomorphine-induced dopamine D1 receptor trafficking and acoustic startle behavior. Synapse 2013, 67, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laviolette, S.R. Dopamine modulation of emotional processing in cortical and subcortical neural circuits: Evidence for a final common pathway in schizophrenia? Schizophr. Bull. 2007, 33, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotoyama, H.; Namba, H.; Kobayashi, Y.; Hasegawa, T.; Watanabe, D.; Nakatsukasa, E.; Sakimura, K.; Furuyashiki, T.; Nawa, H. Resting-state dopaminergic cell firing in the ventral tegmental area negatively regulates affiliative social interactions in a developmental animal model of schizophrenia. Transl. Psychiatry 2021, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Bubenikova-Valesova, V.; Svoboda, J.; Horacek, J.; Vales, K. The effect of a full agonist/antagonist of the D1 receptor on locomotor activity, sensorimotor gating and cognitive function in dizocilpine-treated rats. Int. J. Neuropsychopharmacol. 2009, 12, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, A.; Fujita, Y.; Iyo, M.; Hashimoto, K. Effects of sodium benzoate on pre-pulse inhibition deficits and hyperlocomotion in mice after administration of phencyclidine. Acta Neuropsychiatr. 2015, 27, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Lee, Y.; Oh, H.K.; Lee, H.E.; Lee, Y.; Ko, S.Y.; Kim, B.; Cheong, J.H.; Shin, C.Y.; Ryu, J.H. Oleanolic acid attenuates MK-801-induced schizophrenia-like behaviors in mice. Neuropharmacology 2014, 86, 49–56. [Google Scholar] [CrossRef]

{kind=link}
| Protein | Type | Function | Reference |
|---|---|---|---|
| Na+/H+ exchanger | NHE1–9 | Exchange extracellular Na+ for intracellular H+ | [95] |
| Na+-coupled HCO3− transporter | NBCe1, e2, NBCn1, n2/NCBE | Cotransport Na+ and HCO3− into cells | [15,95] |
| NDCBE | Move Na+ and HCO3− into cells in exchange for intracellular Cl− | [17] | |
| Cl−/HCO3− exchanger | AE1–3 | Exchange intracellular HCO3− for extracellular Cl− | [95] |
| Vacuolar type H-ATPase | V-ATPase | Move H+ from cytoplasm into vesicles or extracellular space by using ATP | [95] |
| Monocarboxylate transporter | MCT1, 2, 4 | Cotransport H+ and monocarboxylate anion (such as lactate, pyruvate, acetoacetate, and/or β-hydroxybutyrate) | [95,96] |
| Carbonic anhydrase | CA1–15 | Catalyze the inter conversion of CO2 and H2O to HCO3− and H+ | [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-J.; Choi, I.; Leem, K.-H. Decreased Brain pH and Pathophysiology in Schizophrenia. Int. J. Mol. Sci. 2021, 22, 8358. https://doi.org/10.3390/ijms22168358
Park H-J, Choi I, Leem K-H. Decreased Brain pH and Pathophysiology in Schizophrenia. International Journal of Molecular Sciences. 2021; 22(16):8358. https://doi.org/10.3390/ijms22168358
Chicago/Turabian StylePark, Hae-Jeong, Inyeong Choi, and Kang-Hyun Leem. 2021. "Decreased Brain pH and Pathophysiology in Schizophrenia" International Journal of Molecular Sciences 22, no. 16: 8358. https://doi.org/10.3390/ijms22168358
APA StylePark, H.-J., Choi, I., & Leem, K.-H. (2021). Decreased Brain pH and Pathophysiology in Schizophrenia. International Journal of Molecular Sciences, 22(16), 8358. https://doi.org/10.3390/ijms22168358