
Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution



Abstract
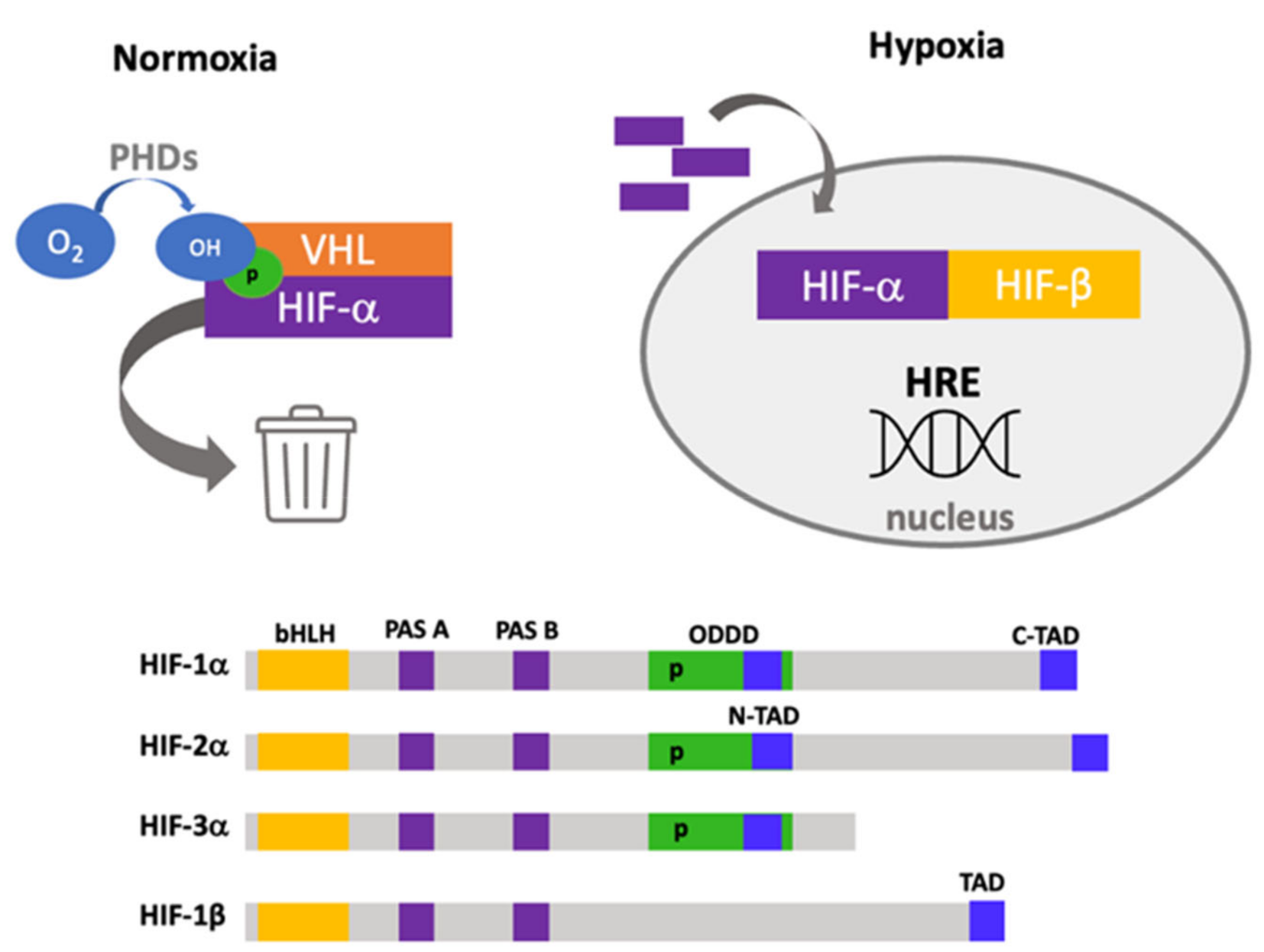
1. Hypoxia Adaptation and Signaling
Structure and Functional Domains of Hypoxia-Inducible Factors
- Basic helix–loop–helix (bHLH). This is a basic region followed by two alpha-helices separated by a variable loop region, and this region confers the ability to bind DNA as homodimers or heterodimers, which is found in other transcription factors [13].
- PAS domains. PAS-A and PAS-B domains also build a stable heterodimer that enables robust DNA binding. In the PAS-B structure, a central β-sheet provides a hydrophobic surface for HIF-1β that is conserved among the HIFα isoforms [14].
- Oxygen-dependent degradation domain (ODDD). This domain is responsible for degradation by the ubiquitin–proteome system, located ~200 amino acids in the central region and has two functional domains: N-terminal (NODD) and the C-terminal (CODD) [15]. The hydroxylation of NODD is more sensitive to hypoxia than CODD; thus, it is possible to block them differentially [16].
- Nuclear localization signal (NLS). α and β subunits have two nuclear localization sequences in N- and C-terminal domains [19]. Direct interaction of HIFs with importins 1α, 3α, 5α and 7α has been described, and this interaction depends on a nuclear localization signal within the C-terminal region [20]. Furthermore, an additional interaction site has been reported with importins 4 and 7, which is more efficient than NLS interaction [21].
- The nuclear export signal (NES), located in amino acids 616–658, has been reported to be regulated by MAPK phosphorylation [22].
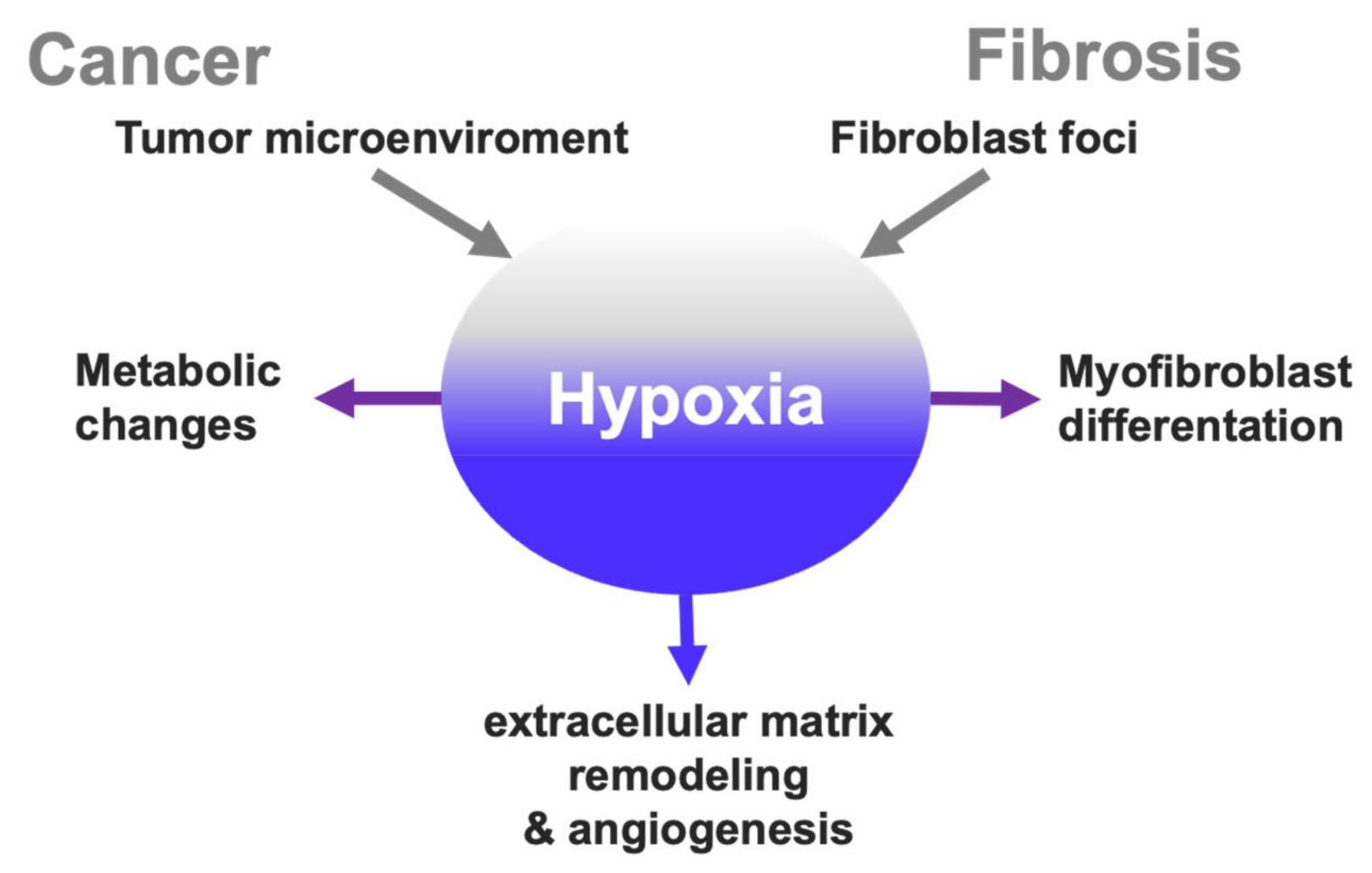
2. Cancer Hallmarks Associated with Hypoxia
2.1. Hypoxic Microenvironment in Tumors
2.2. Hypoxia-Induced Metabolic Changes
2.3. Integration of HIFs Signaling in Lung Cancer
2.4. Interconnected Pathways with Hypoxia
2.5. Hypoxia and Extracellular Matrix Remodeling
3. Hypoxia Signaling Overlap in Cancer and Fibrosis?
3.1. Hypoxia in the Pathophysiology of Idiopathic Pulmonary Fibrosis
3.2. Effects of Hypoxia on the Alveolar Epithelium
3.3. Profibrotic Feedback Loops Related to Hypoxia
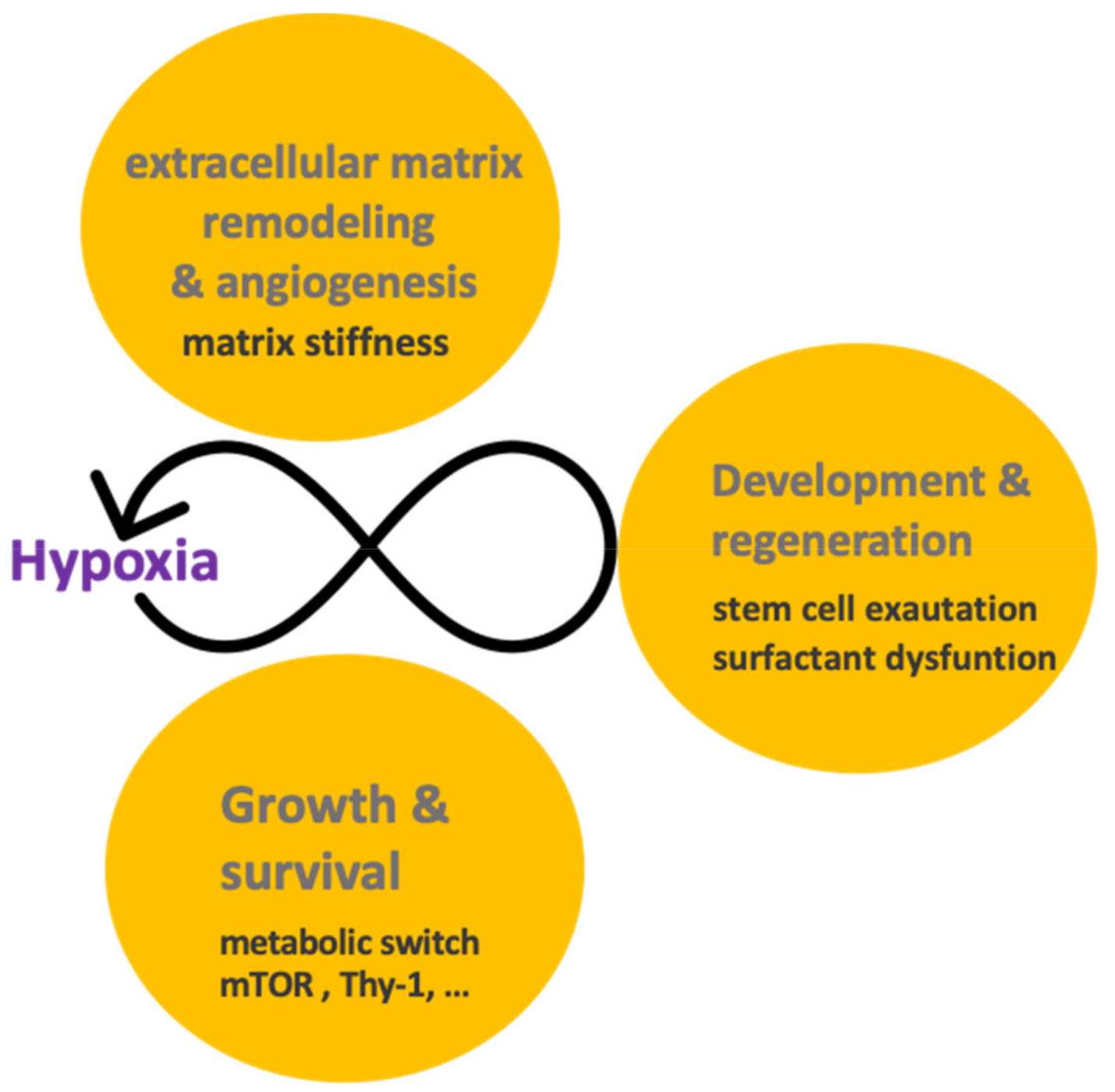
4. Nothing in Hypoxia Signaling Makes Sense except in Light of Regeneration and Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef] [PubMed]
- Olenyuk, B.Z.; Zhang, G.-J.; Klco, J.M.; Nickols, N.G.; Kaelin, W.G., Jr.; Dervan, P.B. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc. Natl. Acad. Sci. USA 2004, 101, 16768–16773. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef]
- Tian, H.; Mcknight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Erbel, P.J.A.; Card, P.B.; Karakuzu, O.; Bruick, R.K.; Gardner, K.H. Structural basis for PAS domain heterodimerization in the basic helix--loop--helix-PAS transcription factor hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2003, 100, 15504–15509. [Google Scholar] [CrossRef]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef]
- Chowdhury, R.; Leung, I.K.H.; Tian, Y.-M.; Abboud, M.I.; Ge, W.; Domene, C.; Cantrelle, F.-X.; Landrieu, I.; Hardy, A.P.; Pugh, C.W.; et al. Structural basis for oxygen degradation domain selectivity of the HIF prolyl hydroxylases. Nat. Commun. 2016, 7, 12673. [Google Scholar] [CrossRef]
- Ruas, J.L.; Poellinger, L.; Pereira, T. Functional analysis of hypoxia-inducible factor-1 alpha-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J. Biol. Chem. 2002, 277, 38723–38730. [Google Scholar] [CrossRef]
- Hu, C.-J.; Iyer, S.; Sataur, A.; Covello, K.L.; Chodosh, L.A.; Simon, M.C. Differential Regulation of the Transcriptional Activities of Hypoxia-Inducible Factor 1 Alpha (HIF-1α) and HIF-2α in Stem Cells. Mol. Cell. Biol. 2006, 26, 3514–3526. [Google Scholar] [CrossRef] [PubMed]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of theCBP/p300 coactivator by the hypoxia-induciblefactor-1α. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Köhler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin alpha/beta pathway. Biochim. Biophys. Acta 2008, 1783, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Chachami, G.; Paraskeva, E.; Mingot, J.M.; Braliou, G.G.; Görlich, D.; Simos, G. Transport of hypoxia-inducible factor HIF-1α into the nucleus involves importins 4 and 7. Biochem. Biophys. Res. Commun. 2009, 390, 235–240. [Google Scholar] [CrossRef]
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1α by MAPK. J. Biol. Chem. 2008, 283, 27620–27627. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C. Idiopathic pulmonary fibrosis: An altered fibroblast proliferation linked to cancer biology. Proc. Am. Thorac. Soc. 2012, 9, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic pulmonary fibrosis and lung cancer: Mechanisms and molecular targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Kenny, P.A.; Bissell, M.J. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J. Cell. Biochem. 2007, 101, 830–839. [Google Scholar] [CrossRef]
- Rubio, K.; Castillo-Negrete, R.; Barreto, G. Non-coding RNAs and nuclear architecture during epithelial-mesenchymal transition in lung cancer and idiopathic pulmonary fibrosis. Cell. Signal. 2020, 70, 109593. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Martinive, P.; Defresne, F.; Bouzin, C.; Saliez, J.; Lair, F.; Grégoire, V.; Michiels, C.; Dessy, C.; Feron, O. Preconditioning of the tumor vasculature and tumor cells by intermittent hypoxia: Implications for anticancer therapies. Cancer Res. 2006, 66, 11736–11744. [Google Scholar] [CrossRef] [PubMed]
- Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Review cyclic hypoxia: An update on its characteristics, methods to measure it and biological implications in cancer. Cancers 2021, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Gozal, D.; Farré, R.; Nieto, F.J. Obstructive sleep apnea and cancer: Epidemiologic links and theoretical biological constructs. Sleep Med. Rev. 2016, 27, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, V.K.; Mohanam, I.; Rao, J.S.; Mohanam, S. Intermittent hypoxia regulates stem-like characteristics and differentiation of neuroblastoma cells. PLoS ONE 2012, 7, e30905. [Google Scholar] [CrossRef]
- Hao, S.; Zhu, X.; Liu, Z.; Wu, X.; Li, S.; Jiang, P.; Jiang, L. Chronic intermittent hypoxia promoted lung cancer stem cell-like properties via enhancing Bach1 expression. Respir. Res. 2021, 22, 58. [Google Scholar] [CrossRef] [PubMed]
- Louie, E.; Nik, S.; Chen, J.-S.; Schmidt, M.; Song, B.; Pacson, C.; Chen, X.F.; Park, S.; Ju, J.; Chen, E.I. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. 2010, 12, R94. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Martínez-García, M.Á.; Díaz-García, E.; Jaureguizar, A.; Campos-Rodríguez, F.; Sánchez-de-la-Torre, M.; Nagore, E.; Martorell-Calatayud, A.; Blasco, L.H.; Pastor, E.; et al. Obesity attenuates the effect of sleep apnea on active TGF-ß1 levels and tumor aggressiveness in patients with melanoma. Sci. Rep. 2020, 10, 15528. [Google Scholar] [CrossRef]
- Nanduri, J.; Wang, N.; Yuan, G.; Khan, S.A.; Souvannakitti, D.; Peng, Y.J.; Kumar, G.K.; Garcia, J.A.; Prabhakar, N.R. Intermittent hypoxia degrades HIF-2α via calpains resulting in oxidative stress: Implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. USA 2009, 106, 1199–1204. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Peng, Y.J.; Nanduri, J. Hypoxia-inducible factors and obstructive sleep apnea. J. Clin. Invest. 2020, 130, 5042–5051. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial Complex III stabilize hypoxia-inducible factor-1α during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, A.P.J.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis Activates Rag-mTORC1 Signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Cervantes-Madrid, D.; Romero, Y.; Dueñas-González, A. Reviving Lonidamine and 6-diazo-5-oxo-l-norleucine to be used in combination for metabolic cancer therapy. Biomed Res. Int. 2015, 2015, 690492. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Bracken, C.P.; Fedele, A.O.; Linke, S.; Balrak, W.; Lisy, K.; Whitelaw, M.L.; Peet, D.J. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J. Biol. Chem. 2006, 281, 22575–22585. [Google Scholar] [CrossRef] [PubMed]
- Petrella, B.L.; Lohi, J.; Brinckerhoff, C.E. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2α in von Hippel-Lindau renal cell carcinoma. Oncogene 2005, 24, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, C.; Zaborowska, Z.; Kurreck, J.; Erdmann, V.A.; Frei, U.; Wiesener, M.; Eckardt, K. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: Erythropoietin is a HIF-2α target gene in Hep3B and Kelly cells. FASEB J. 2004, 18, 1462–1464. [Google Scholar] [CrossRef]
- Sato, M.; Tanaka, T.; Maemura, K.; Uchiyama, T.; Sato, H.; Maeno, T.; Suga, T.; Iso, T.; Ohyama, Y.; Arai, M.; et al. The PAI-1 gene as a direct target of endothelial PAS domain protein-1 in adenocarcinoma A549 cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Compernolle, V.; Brusselmans, K.; Acker, T.; Hoet, P.; Tjwa, M.; Beck, H.; Plaisance, S.; Dor, Y.; Keshet, E.; Lupu, F.; et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002, 8, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Koukourakis, M.I.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.C.; Harris, A.L. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br. J. Cancer 2001, 85, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, A.; Heikkilä, M.; Rautavuoma, K.; Hirsilä, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3α is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int. J. Biochem. Cell Biol. 2010, 42, 1189–1200. [Google Scholar] [CrossRef]
- Heikkilä, M.; Pasanen, A.; Kivirikko, K.I.; Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3α variants in the hypoxia response. Cell. Mol. Life Sci. 2011, 68, 3885–3901. [Google Scholar] [CrossRef]
- Romero, Y.; Castillejos-López, M.; Romero-Garciá, S.; Aguayo, A.S.; Herrera, I.; Garcia-Martin, M.O.; Torres-Espíndola, L.M.; Negrete-Garciá, M.C.; Olvera, A.C.; Huerta-Cruz, J.C.; et al. Antitumor Therapy under Hypoxic Microenvironment by the Combination of 2-Methoxyestradiol and Sodium Dichloroacetate on Human Non-Small-Cell Lung Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 3176375. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chang, C.C.; Teng, S.C.; Wu, K.J. Interplay between HDAC3 and WDR5 Is Essential for Hypoxia-Induced Epithelial-Mesenchymal Transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Swinson, D.E.B.; Jones, J.L.; Cox, G.; Richardson, D.; Harris, A.L.; O’Byrne, K.J. Hypoxia-inducible factor-1α in non small cell lung cancer: Relation to growth factor, protease and apoptosis pathways. Int. J. Cancer 2004, 111, 43–50. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; DeBerardinis, R.J.; Thompson, C.B. Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 2005, 6, 439–448. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Wouters, B.G. “Translating” tumor hypoxia: Unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol. Cancer Res. 2006, 4, 423–436. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Kozlova, N.; Mennerich, D.; Samoylenko, A.; Dimova, E.Y.; Koivunen, P.; Biterova, E.; Richter, K.; Hassinen, A.; Kellokumpu, S.; Manninen, A.; et al. The pro-oncogenic adaptor CIN85 acts as an inhibitory binding partner of hypoxia-inducible factor prolyl hydroxylase 2. Cancer Res. 2019, 79, 4042–4056. [Google Scholar] [CrossRef]
- Li, D.; Shimamura, T.; Ji, H.; Chen, L.; Haringsma, H.J.; McNamara, K.; Liang, M.C.; Perera, S.A.; Zaghlul, S.; Borgman, C.L.; et al. Bronchial and Peripheral Murine Lung Carcinomas Induced by T790M-L858R Mutant EGFR Respond to HKI-272 and Rapamycin Combination Therapy. Cancer Cell 2007, 12, 81–93. [Google Scholar] [CrossRef]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Kondo, K.; Klco, J.; Nakamura, E.; Lechpammer, M.; Kaelin, W.G. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002, 1, 237–246. [Google Scholar] [CrossRef]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. Landmark Ed. 2010, 15, 166–179. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Giulia, B.; David, A.T. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2020, 53, 1689–1699. [Google Scholar]
- Barbazan, J.; Pérez-González, C.; Gómez-González, M.; Dedenon, M.; Richon, S.; Latorre, E.; Serra, M.; Mariani, P.; Descroix, S.; Sens, P.; et al. Cancer-associated fibroblasts actively compress cancer cells and modulate mechanotransduction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Miyoshi, A.; Kitajima, Y.; Ide, T.; Ohtaka, K.; Nagasawa, H.; Uto, Y.; Hori, H.; Miyazaki, K. Hypoxia accelerates cancer invasion of hepatoma cells by upregulating MMP expression in an HIF-1α-independent manner. Int. J. Oncol. 2006, 29, 1533–1539. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lukacova, S.; Khalil, A.A.; Overgaard, J.; Alsner, J.; Horsman, M.R. Relationship between radiobiological hypoxia in a C3H mouse mammary carcinoma and osteopontin levels in mouse serum. Int. J. Radiat. Biol. 2005, 81, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Le Jeune, I.; Gribbin, J.; West, J.; Smith, C.; Cullinan, P.; Hubbard, R. The incidence of cancer in patients with idiopathic pulmonary fibrosis and sarcoidosis in the UK. Respir. Med. 2007, 101, 2534–2540. [Google Scholar] [CrossRef]
- Hubbard, R.; Venn, A.; Lewis, S.; Britton, J. Lung cancer and cryptogenic fibrosing alveolitis: A population-based cohort study. Am. J. Respir. Crit. Care Med. 2000, 161, 5–8. [Google Scholar] [CrossRef]
- Selman, M.; TE, K., Jr.; Pardo, A.; King, J.; Pardo, A. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- King, T.E.; Pardo, A.; Selman, M.M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 2020, 66, 109482. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis: An integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Romero, Y.; Pardo, A. Aging and IPF: What Is the Link? In BT—Idiopathic Pulmonary Fibrosis: A Comprehensive Clinical Guide; Meyer, K.C., Nathan, S.D., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 259–279. ISBN 978-1-62703-682-5. [Google Scholar]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Sørensen, B.S.; Horsman, M.R. Tumor Hypoxia: Impact on Radiation Therapy and Molecular Pathways. Front. Oncol. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008, 5. [Google Scholar] [CrossRef]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2, 891–903. [Google Scholar] [CrossRef]
- Sørensen, B.S.; Alsner, J.; Overgaard, J.; Horsman, M.R. Hypoxia induced expression of endogenous markers in vitro is highly influenced by pH. Radiother. Oncol. 2007, 83, 362–366. [Google Scholar] [CrossRef]
- Placido, L.; Romero, Y.; Maldonado, M.; Toscano-Marquez, F.; Ramírez, R.; Calyeca, J.; Mora, A.L.; Selman, M.; Pardo, A. Loss of mt1-mmp in alveolar epithelial cells exacerbates pulmonary fibrosis. Int. J. Mol. Sci. 2021, 22, 2923. [Google Scholar] [CrossRef] [PubMed]
- Oblander, S.A.; Zhou, Z.; Gálvez, B.G.; Starcher, B.; Shannon, J.M.; Durbeej, M.; Arroyo, A.G.; Tryggvason, K.; Apte, S.S. Distinctive functions of membrane type 1 matrix-metalloprotease (MT1-MMP or MMP-14) in lung and submandibular gland development are independent of its role in pro-MMP-2 activation. Dev. Biol. 2005, 277, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Clerici, C.; Planès, C. Gene regulation in the adaptive process to hypoxia in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L267–L274. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Eul, B.G.; Hänze, J.; Savai, R.; Grimminger, F.; Seeger, W.; Rose, F. Role of hypoxia-inducible factor-1alpha in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 395–403. [Google Scholar] [CrossRef]
- Jain, M.; Sznajder, J.I. Effects of hypoxia on the alveolar epithelium. Proc. Am. Thorac. Soc. 2005, 2, 202–205. [Google Scholar] [CrossRef]
- Autilio, C.; Pérez-Gil, J. Understanding the principle biophysics concepts of pulmonary surfactant in health and disease. Arch. Dis. Child. Fetal Neonatal Ed. 2019, 104, F443–F451. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, E.; Boden, C.; Echaide, M.; Perez-Gil, J.; Kolb, M.; Gauldie, J.; Maus, U.A.; Ochs, M.; Knudsen, L. Surfactant dysfunction during overexpression of TGF-β1 precedes profibrotic lung remodeling in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1260–L1271. [Google Scholar] [CrossRef] [PubMed]
- Ruwisch, J.; Sehlmeyer, K.; Roldan, N.; Garcia-Alvarez, B.; Perez-Gil, J.; Weaver, T.E.; Ochs, M.; Knudsen, L.; Lopez-Rodriguez, E. Air space distension precedes spontaneous fibrotic remodeling and impaired cholesterol metabolism in the absence of surfactant protein C. Am. J. Respir. Cell Mol. Biol. 2020, 62, 466–478. [Google Scholar] [CrossRef]
- Vazquez-De-Lara, L.G.; Tlatelpa-Romero, B.; Romero, Y.; Fernández-Tamayo, N.; Vazquez-De-Lara, F.; Justo-Janeiro, J.M.; Garcia-Carrasco, M.; Paredes, R.D.L.R.; Cisneros-Lira, J.G.; Mendoza-Milla, C.; et al. Phosphatidylethanolamine induces an antifibrotic phenotype in normal human lung fibroblasts and ameliorates bleomycin-induced lung fibrosis in mice. Int. J. Mol. Sci. 2018, 19, 2758. [Google Scholar] [CrossRef]
- Uhal, B.D.; Joshi, I.; Hughes, W.F.; Ramos, C.; Pardo, A.; Selman, M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 275, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1α in disease pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Shochet, G.E.; Bardenstein-Wald, B.; McElroy, M.; Kukuy, A.; Surber, M.; Edelstein, E.; Pertzov, B.; Kramer, M.R.; Shitrit, D. Hypoxia inducible factor 1a supports a pro-fibrotic phenotype loop in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2021, 22, 3331. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, G.; Hagood, J.S.; Sanders, Y.; Ramírez, R.; Becerril, C.; Segura, L. Absence of Thy-1 results in TGF-β induced MMP-9 expression and confers a profibrotic phenotype to human lung fibroblasts. Lab. Invest. 2011, 91, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Neary, R.; Levendale, A.; Watson, C.J.; Baugh, J.A. Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir. Res. 2012, 13, 74. [Google Scholar] [CrossRef] [PubMed]
- Hagood, J.S.; Prabhakaran, P.; Kumbla, P.; Salazar, L.; MacEwen, M.W.; Barker, T.H. Loss of fibroblast Thy-1 expression correlates with lung fibrogenesis. Am. J. Pathol. 2005, 167, 365–379. [Google Scholar] [CrossRef]
- Rege, T.A.; Hagood, J.S. Thy-1 as a regulator of cell-cell and cell-matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis. FASEB J. 2006, 20, 1045–1054. [Google Scholar] [CrossRef]
- Aquino-Gálvez, A.; González-Ávila, G.; Jiménez-Sánchez, L.L.; Maldonado-Martínez, H.A.; Cisneros, J.; Toscano-Marquez, F.; Castillejos-López, M.; Torres-Espíndola, L.M.; Velázquez-Cruz, R.; Rodríguez, V.H.O.; et al. Dysregulated expression of hypoxia-inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir. Res. 2019, 20, 130. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Fibroageing: An ageing pathological feature driven by dysregulated extracellular matrix-cell mechanobiology. Ageing Res. Rev. 2021, 70, 101393. [Google Scholar] [CrossRef]
- Cool, C.D.; Groshong, S.D.; Rai, P.R.; Henson, P.M.; Stewart, J.S.; Brown, K.K. Fibroblast foci are not discrete sites of lung injury or repair: The fibroblast reticulum. Am. J. Respir. Crit. Care Med. 2006, 174, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur. Respir. Rev. 2013, 22, 265–272. [Google Scholar] [CrossRef]
- Braun, R.K.; Broytman, O.; Braun, F.M.; Brinkman, J.A.; Clithero, A.; Modi, D.; Pegelow, D.F.; Eldridge, M.; Teodorescu, M. Chronic intermittent hypoxia worsens bleomycin-induced lung fibrosis in rats. Respir. Physiol. Neurobiol. 2018, 256, 97–108. [Google Scholar] [CrossRef]
- Lancaster, L.H.; Mason, W.R.; Parnell, J.A.; Rice, T.W.; Loyd, J.E.; Milstone, A.P.; Collard, H.R.; Malow, B.A. Obstructive Sleep Apnea Is Common in Idiopathic Pulmonary Fibrosis. Chest 2009, 136, 772–778. [Google Scholar] [CrossRef]
- Schiza, S.; Mermigkis, C.; Margaritopoulos, G.A.; Daniil, Z.; Harari, S.; Poletti, V.; Renzoni, E.A.; Torre, O.; Visca, D.; Bouloukaki, I.; et al. Idiopathic pulmonary fibrosis and sleep disorders: No longer strangers in the night. Eur. Respir. Rev. 2015, 24, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Nigdelioglu, R.; Hamanaka, R.B.; Meliton, A.Y.; O’Leary, E.; Witt, L.J.; Cho, T.; Sun, K.; Bonham, C.; Wu, D.; Woods, P.S.; et al. Transforming Growth Factor (TGF)-β Promotes de Novo Serine Synthesis for Collagen Production. J. Biol. Chem. 2016, 291, 27239–27251. [Google Scholar] [CrossRef]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Gopu, V.; Fan, L.; Shetty, R.S.; Nagaraja, M.R.; Shetty, S. Caveolin-1 scaffolding domain peptide regulates glucose metabolism in lung fibrosis. JCI Insight 2020, 5, e137969. [Google Scholar] [CrossRef]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.-M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Moon, J.-S.; Lee, C.-M.; Choi, A.M.K.; Stout-Delgado, H.W. Glucose Transporter 1-Dependent Glycolysis Is Increased during Aging-Related Lung Fibrosis, and Phloretin Inhibits Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 521–531. [Google Scholar] [CrossRef]
- Han, S.; Chandel, N.S. Lessons from Cancer Metabolism for Pulmonary Arterial Hypertension and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 1–38. [Google Scholar] [CrossRef]
- Herrera, I.; Cisneros, J.; Maldonado, M.; Ramírez, R.; Ortiz-Quintero, B.; Anso, E. Matrix metalloproteinase (MMP)-1 induces lung alveolar epithelial cell migration and proliferation, protects from apoptosis, and represses mitochondrial oxygen consumption. J. Biol. Chem. 2013, 288, 25964–25975. [Google Scholar] [CrossRef] [PubMed]
- Mohyeldin, A.; Garzón-Muvdi, T.; Quiñones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.H.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.K.; Gotts, J.E.; et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 2017, 19, 904–914. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Kato, T.; Furu, M.; Nasu, A.; Kajita, Y.; Mitsui, H.; Ueda, M.; Aoyama, T.; Nakayama, T.; Nakamura, T.; et al. Mesenchymal stem cells cultured under hypoxia escape from senescence via down-regulation of p16 and extracellular signal regulated kinase. Biochem. Biophys. Res. Commun. 2010, 391, 1471–1476. [Google Scholar] [CrossRef]
- Lan, Y.W.; Choo, K.B.; Chen, C.M.; Hung, T.H.; Chen, Y.B.; Hsieh, C.H.; Kuo, H.P.; Chong, K.Y. Hypoxia-preconditioned mesenchymal stem cells attenuate bleomycin-induced pulmonary fibrosis. Stem Cell Res. Ther. 2015, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef]
- Villaseñor-Altamirano, A.B.; Moretto, M.; Maldonado, M.; Zayas-Del Moral, A.; Munguía-Reyes, A.; Romero, Y.; García-Sotelo, J.S.; Aguilar, L.A.; Aldana-Assad, O.; Engelen, K.; et al. PulmonDB: A curated lung disease gene expression database. Sci. Rep. 2020, 10, 514. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Scadden, D.T. The stem-cell niche as an entity of action. Nature 2006, 441, 1075–1079. [Google Scholar] [CrossRef]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef]
- Parmar, K.; Mauch, P.; Vergilio, J.-A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed]
- Jarecki, J.; Johnson, E.; Krasnow, M.A. Oxygen regulation of airway branching in Drosophila is mediated by branchless FGF. Cell 1999, 99, 211–220. [Google Scholar] [CrossRef]
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kapere Ochieng, J.; Kempen, M.B.; Munck, A.B.; Swagemakers, S.; van Ijcken, W.; Grosveld, F.; Tibboel, D.; Rottier, R.J. Hypoxia inducible factor 3α plays a critical role in alveolarization and distal epithelial cell differentiation during mouse lung development. PLoS ONE 2013, 8, e57695. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef] [PubMed]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Chu, Q.; Jiang, K.; Li, J.; Tang, N. The Strength of Mechanical Forces Determines the Differentiation of Alveolar Epithelial Cells. Dev. Cell 2018, 44, 297–312.e5. [Google Scholar] [CrossRef]
- Becerril, C.; Montaño, M.; Cisneros, J.; Mendoza-Milla, C.; Pardo, A.; Ortiz-Quintero, B.; Selman, M.; Ramos, C. Mesenchymal-epithelial transition in fibroblasts of human normal lungs and interstitial lung diseases. Biomolecules 2021, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Hallmark | Cause or Effect | Associated with Fibrosis | References |
|---|---|---|---|
| Tumor microenvironment | Cause | --- | [28,52,53,54,60,65,66,68,82,86,87] |
| Metabolic switch | Effect | + | [30,31,42,43,44,45,51,59,62] |
| Cyclic hypoxia | Cause | + | [32,33,34,35,36,37,38,39] |
| Cancer stem cell | Effect | + | [34,35,36] |
| Cancer-associated fibroblasts | Effect | +++ | [67,82,83,84] |
| ECM remodeling | Effect/Cause | +++ | [55,67,83] |
| Hallmark | Cause or Effect | Associated with Cancer | References |
|---|---|---|---|
| Fibroblast foci | Cause | --- | [114,115,117,120,129,130] |
| Metabolic switch | Effect | +++ | [129,131,132,133,134] |
| Cyclic hypoxia | Cause | + | [124,125] |
| Fibroblast activation/differentiation | Effect | + | [115,117,120,129,131,132] |
| Stem cell | Effect | ++ | [135,136,137,138,139] |
| ECM remodeling | Effect/Cause | + | [115,117,120] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero, Y.; Aquino-Gálvez, A. Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution. Int. J. Mol. Sci. 2021, 22, 8335. https://doi.org/10.3390/ijms22158335
Romero Y, Aquino-Gálvez A. Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution. International Journal of Molecular Sciences. 2021; 22(15):8335. https://doi.org/10.3390/ijms22158335
Chicago/Turabian StyleRomero, Yair, and Arnoldo Aquino-Gálvez. 2021. "Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution" International Journal of Molecular Sciences 22, no. 15: 8335. https://doi.org/10.3390/ijms22158335
APA StyleRomero, Y., & Aquino-Gálvez, A. (2021). Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution. International Journal of Molecular Sciences, 22(15), 8335. https://doi.org/10.3390/ijms22158335