
Signaling Pathways Regulated by UBR Box-Containing E3 Ligases

 ,
, 
Abstract
:1. Introduction
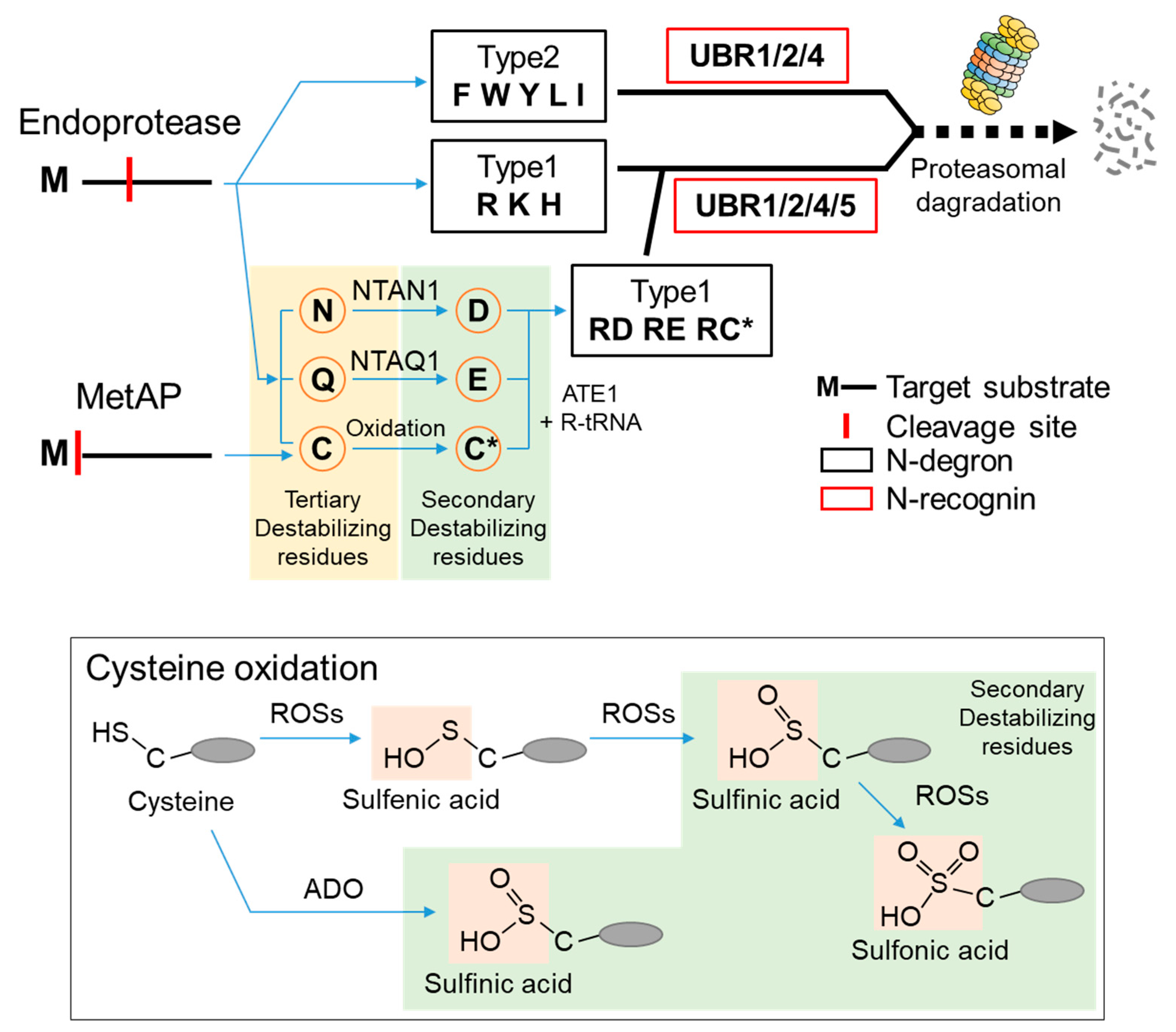
2. N-Degrons and the UBR Box E3 Ligases
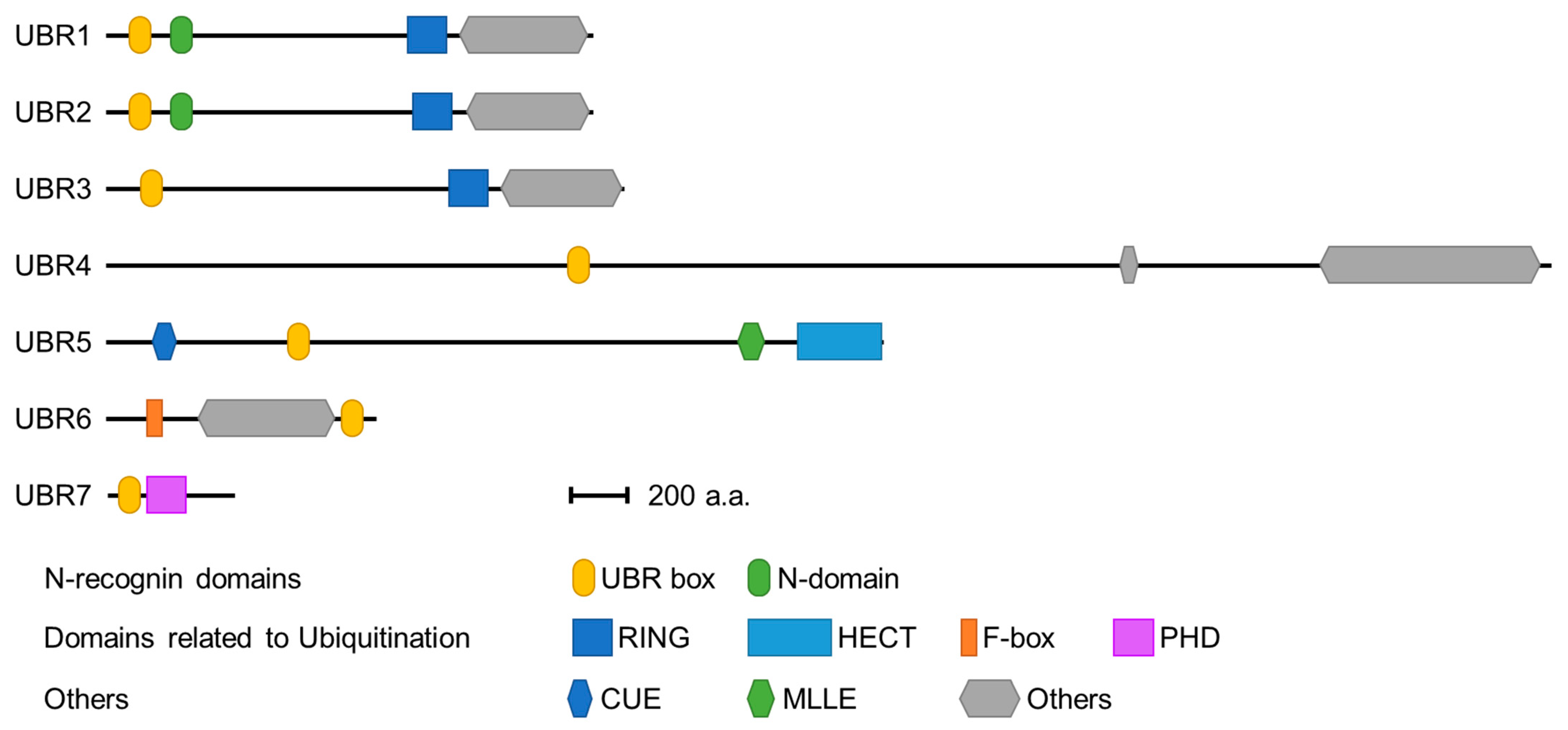
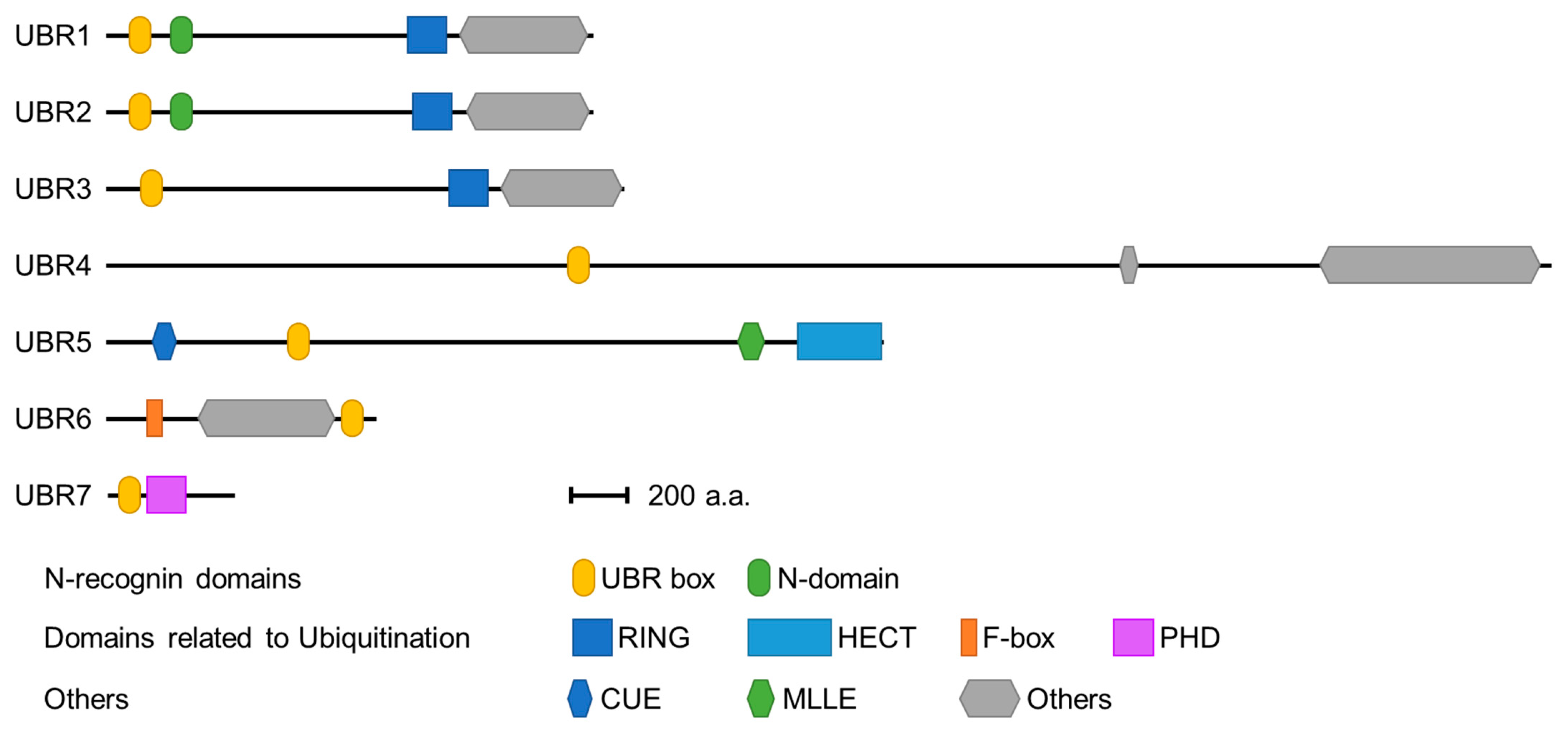
3. The Structure of the Family of UBR Box Proteins
3.1. UBR Box Protein Domains Associated with E3 Ubiquitin Ligases
3.2. Substrate Recognition Domains of UBR Box-Containing N-Recognins
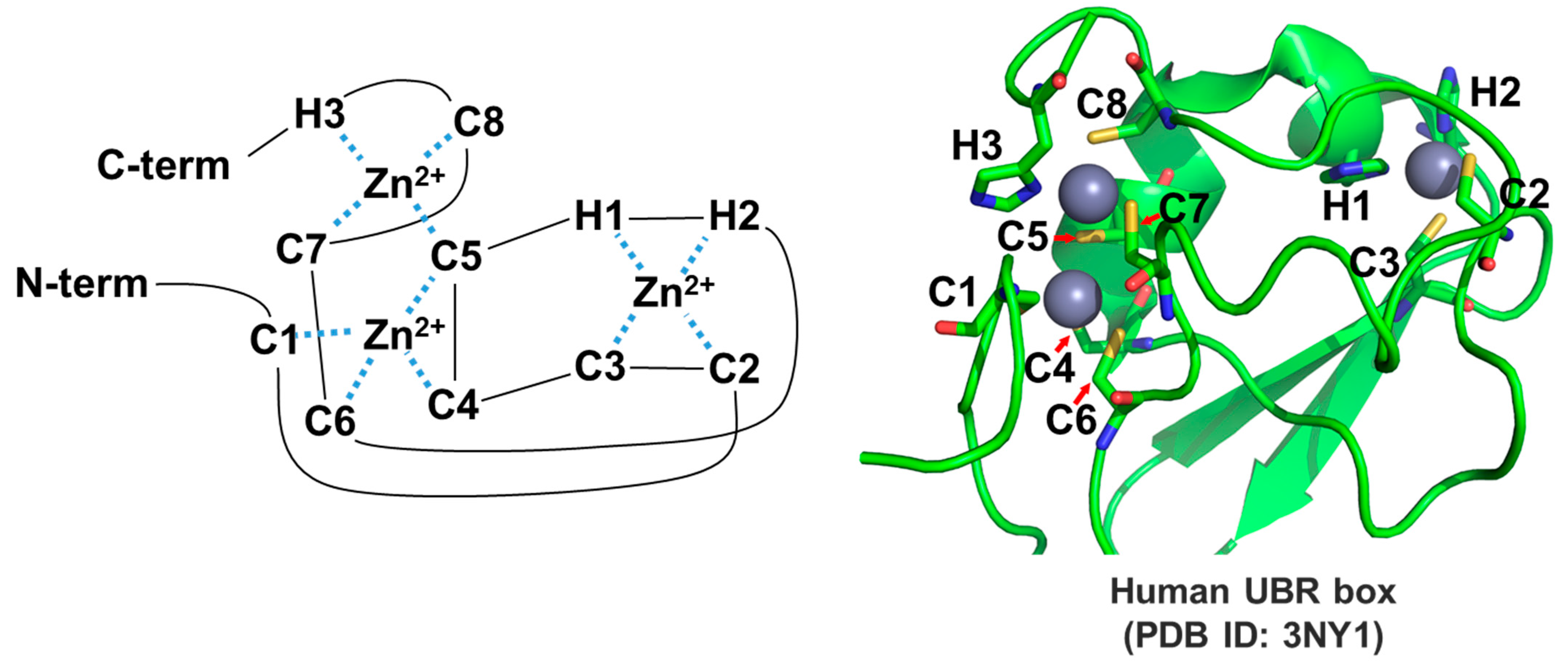
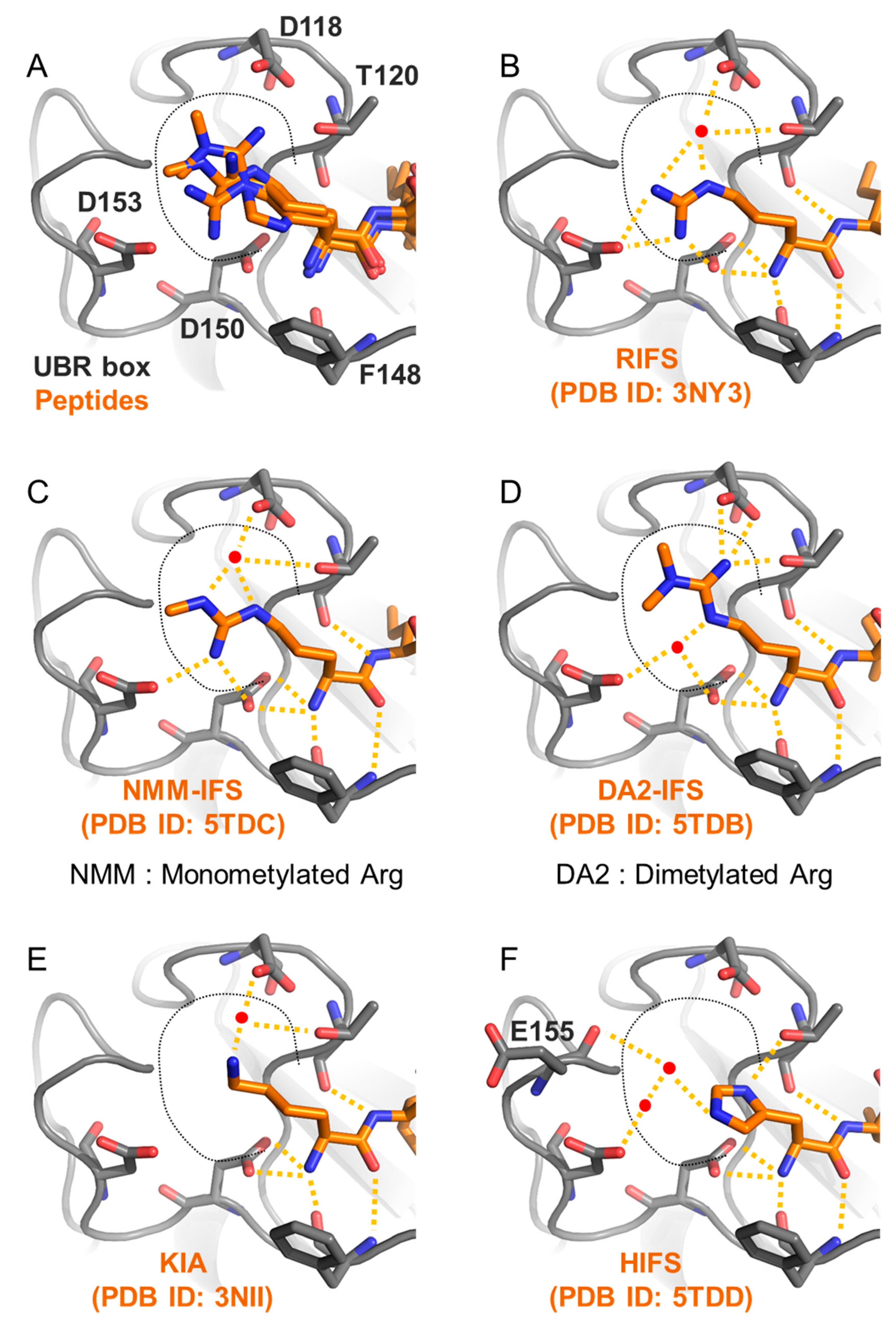
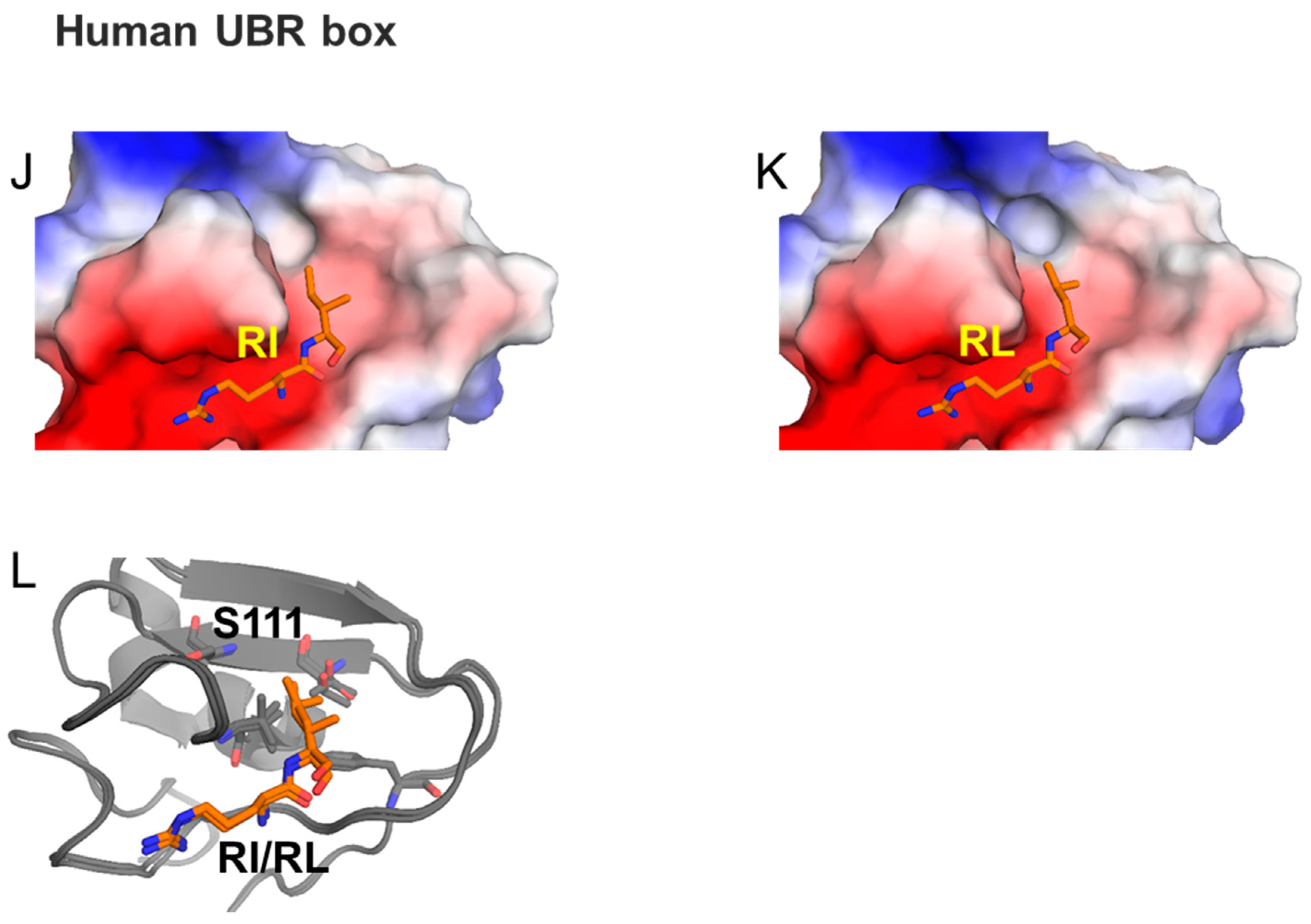
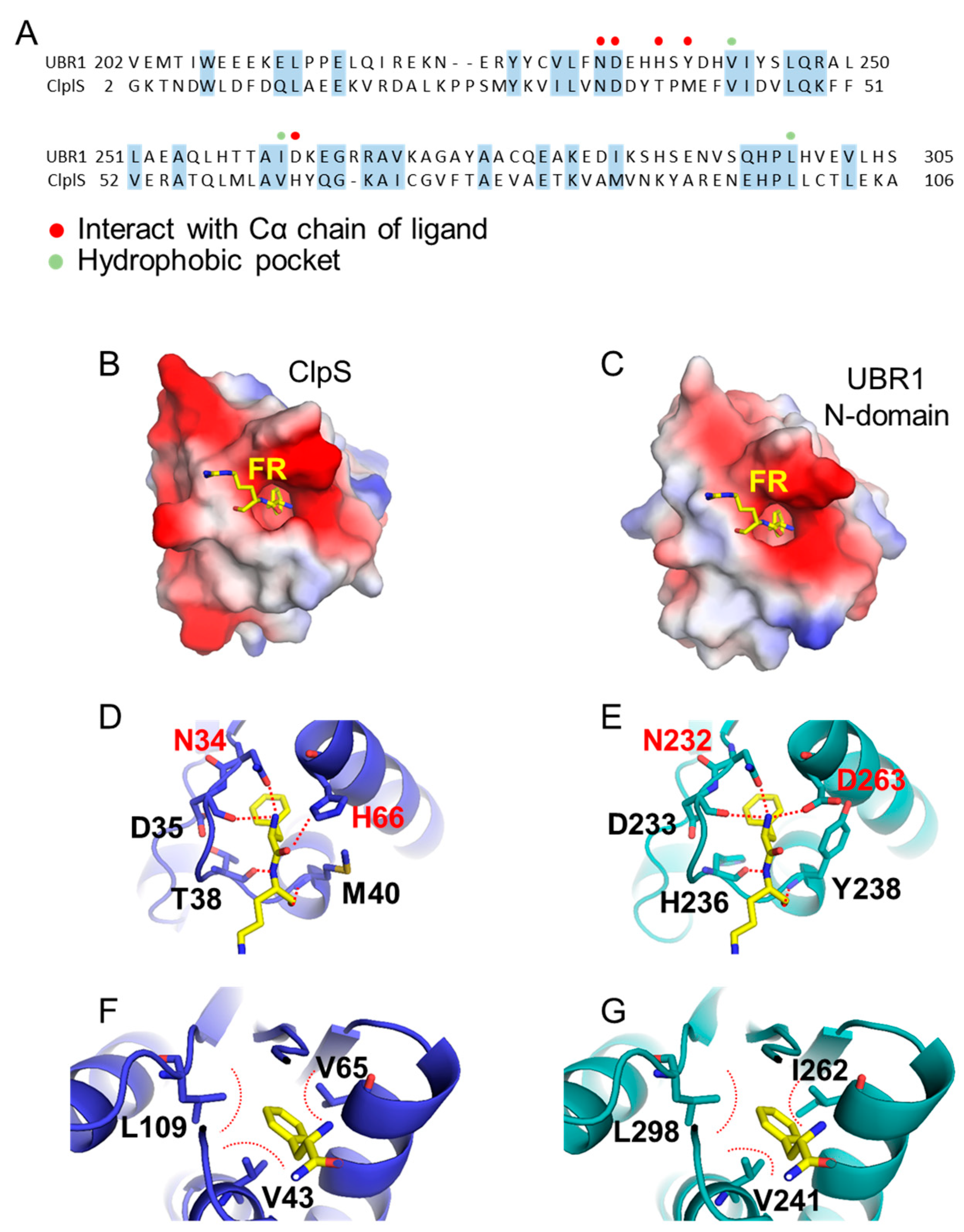
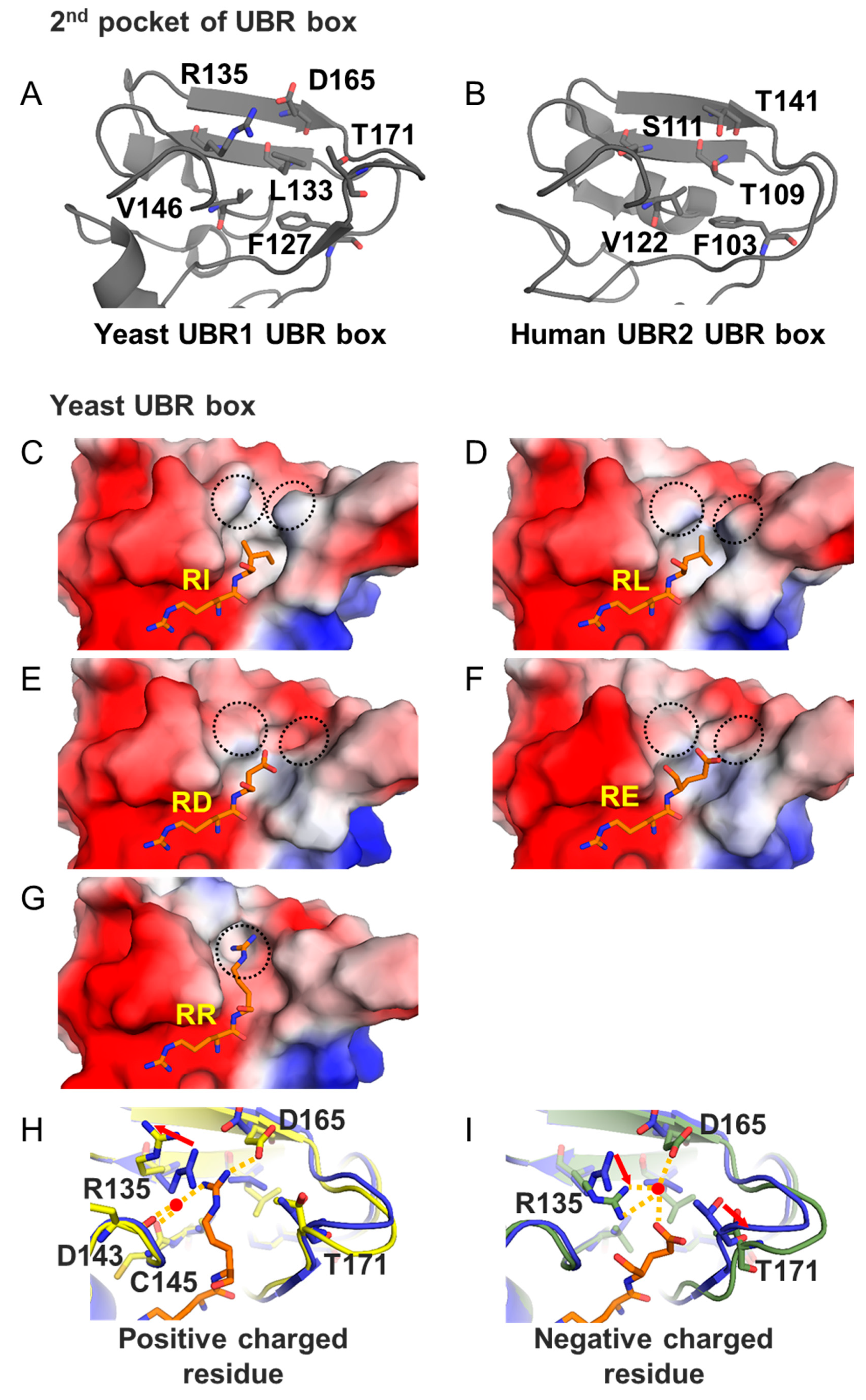
3.2.1. UBR Box
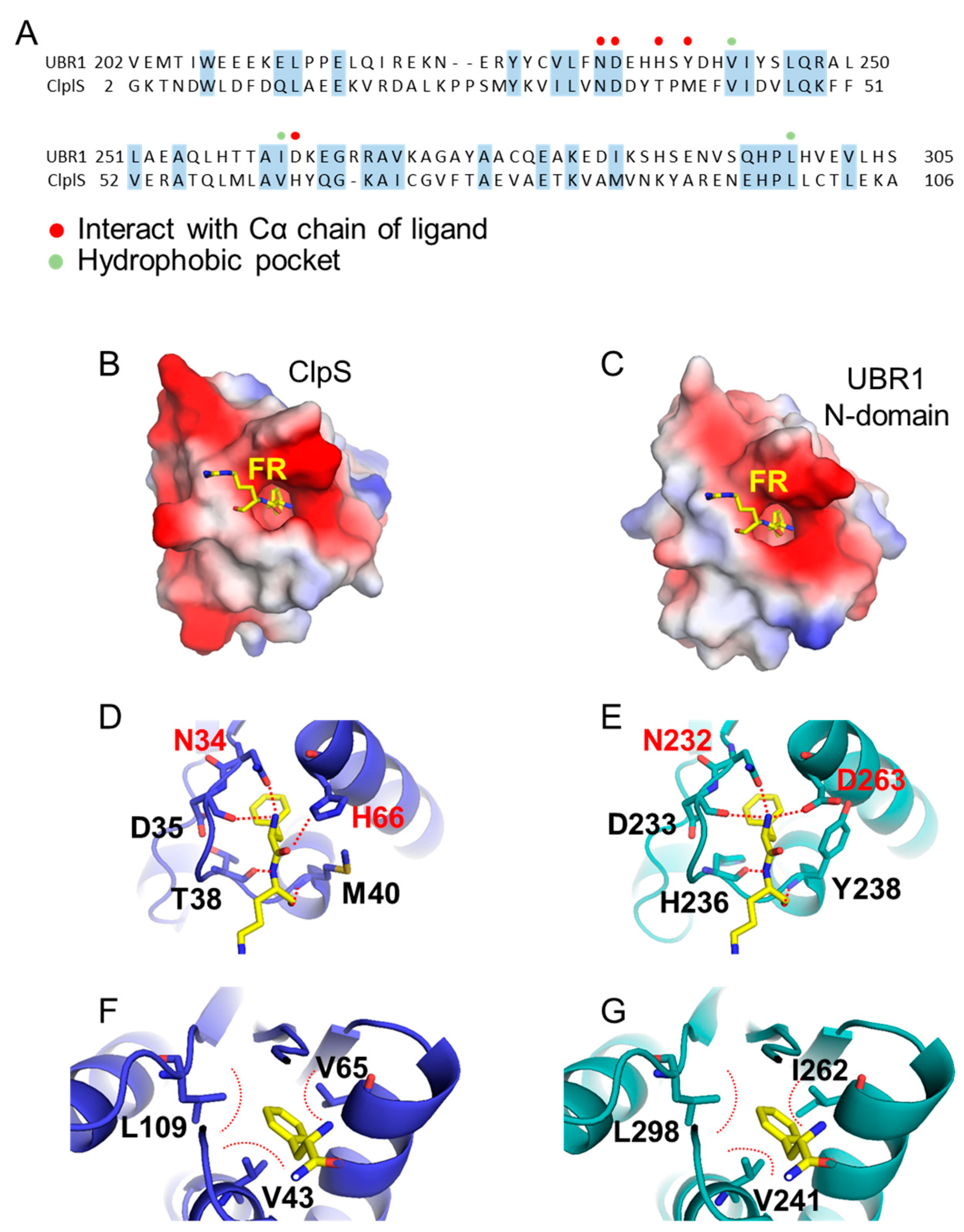
3.2.2. N-Domain
4. Signaling Pathways Controlled by UBR Box N-Recognins in Mammals
4.1. G-Protein Signaling Pathway
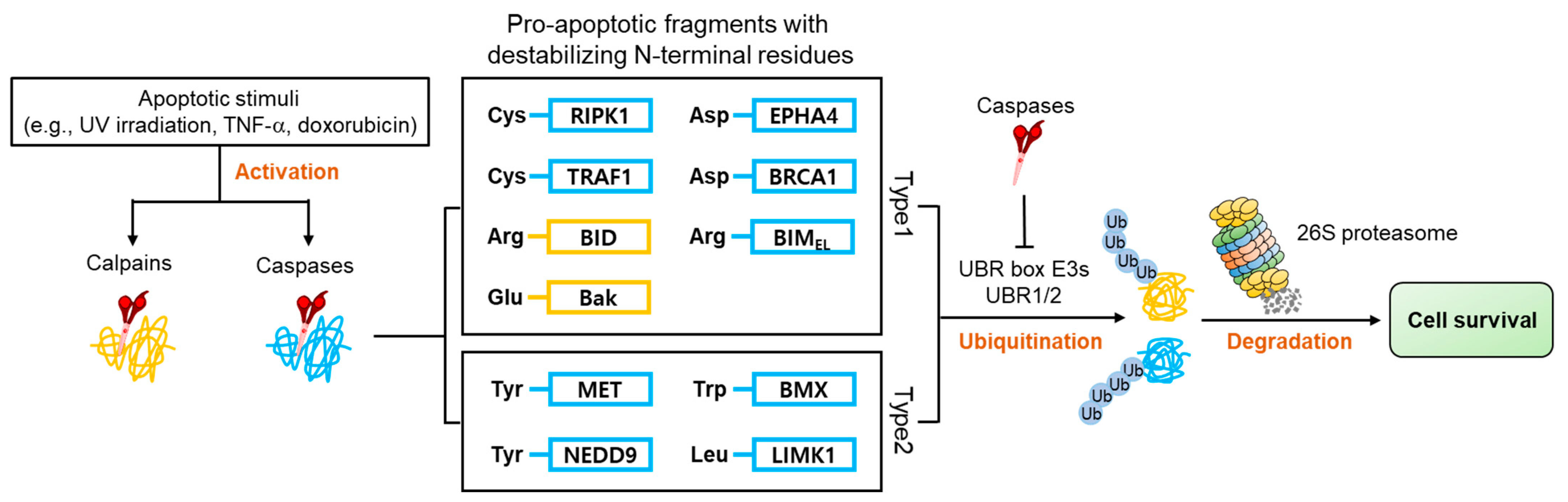
4.2. Apoptosis Signaling Pathway
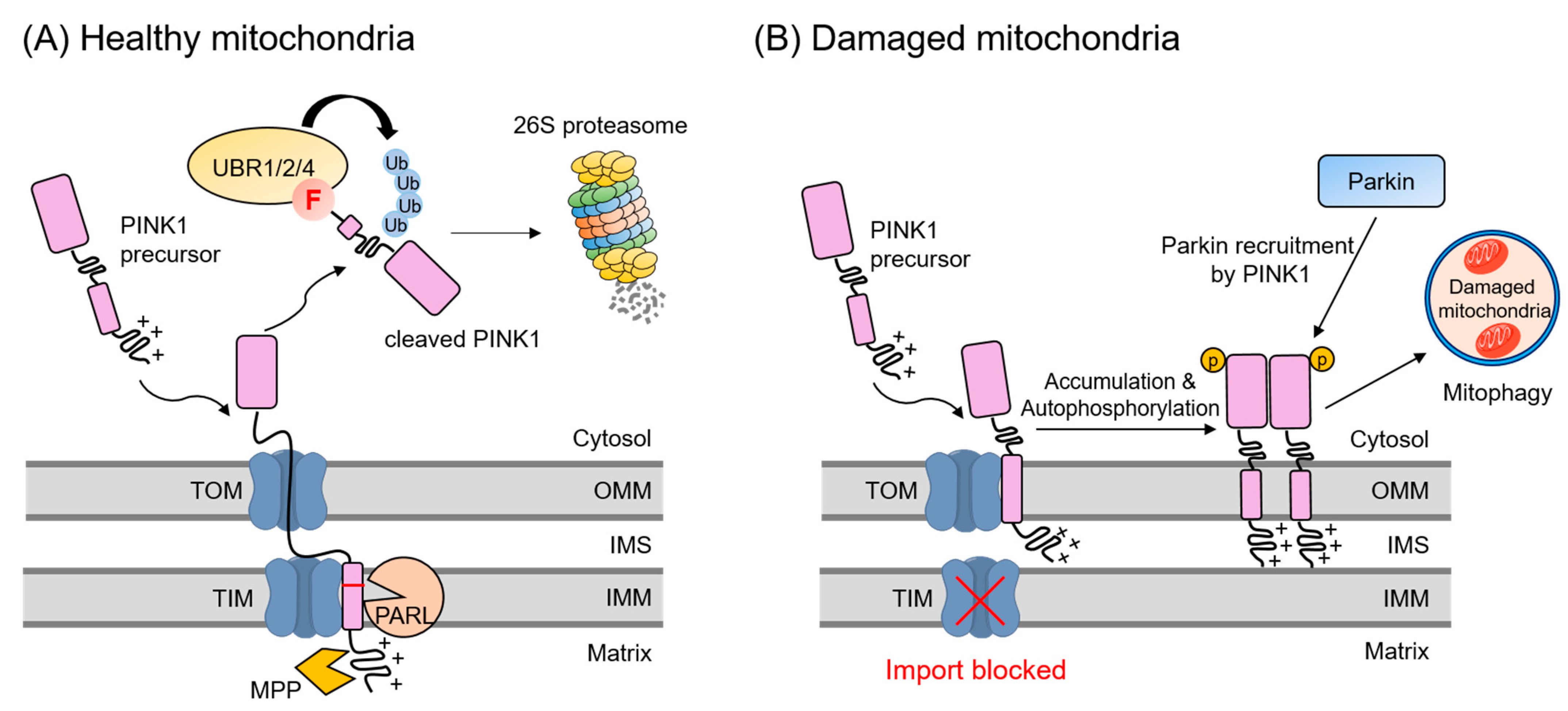
4.3. Mitochondrial Quality Control Pathway
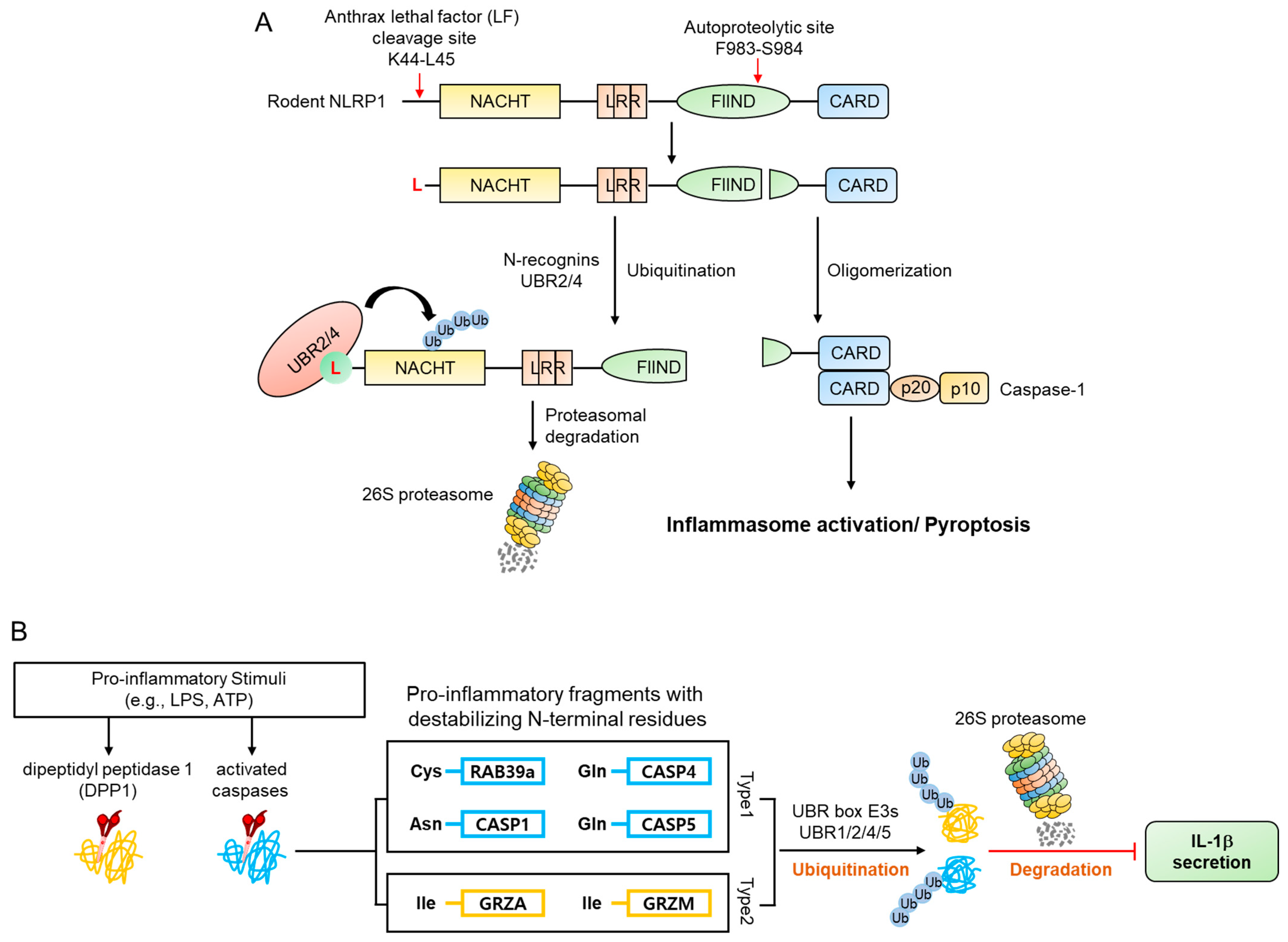
4.4. Inflammatory Signaling Pathways
4.5. DNA Damage Response Pathway
5. Concluding Remarks
6. Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolk, O.; Schenke, C.; Sarikas, A. The ubiquitin-proteasome system: Focus on the heart. Cardiovasc. Res. 2006, 70, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Rahimi, N. The ubiquitin-proteasome system meets angiogenesis. Mol. Cancer Ther. 2012, 11, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, R.Z. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 1999, 6, 303–313. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tokheim, C.; Wang, X.; Timms, R.T.; Zhang, B.; Mena, E.L.; Wang, B.; Chen, C.; Ge, J.; Chu, J.; Zhang, W.; et al. Systematic characterization of mutations altering protein degradation in human cancers. Mol. Cell 2021, 81, 1292–1308. [Google Scholar] [CrossRef] [PubMed]
- Atkin, G.; Paulson, H. Ubiquitin pathways in neurodegenerative disease. Front. Mol. Neurosci. 2014, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Das, C.; Hoang, Q.Q.; Kreinbring, C.A.; Luchansky, S.J.; Meray, R.K.; Ray, S.S.; Lansbury, P.T.; Ringe, D.; Petsko, G.A. Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. USA 2006, 103, 4675–4680. [Google Scholar] [CrossRef] [Green Version]
- Staub, O.; Gautschi, I.; Ishikawa, T.; Breitschopf, K.; Ciechanover, A.; Schild, L.; Rotin, D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovsepian, J.; Becuwe, M.; Kleifeld, O.; Glickman, M.H.; Leon, S. Studying Protein Ubiquitylation in Yeast. Methods Mol. Biol. 2016, 1449, 117–142. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.T.; Walden, H.; Duda, D.; Schulman, B.A. Ubiquitin-like protein activation. Oncogene 2004, 23, 1958–1971. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ella, H.; Reiss, Y.; Ravid, T. The Hunt for Degrons of the 26S Proteasome. Biomolecules 2019, 9, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Barcena, C.; Osinalde, N.; Ramirez, J.; Mayor, U. How to Inactivate Human Ubiquitin E3 Ligases by Mutation. Front. Cell Dev. Biol. 2020, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Medvar, B.; Raghuram, V.; Pisitkun, T.; Sarkar, A.; Knepper, M.A. Comprehensive database of human E3 ubiquitin ligases: Application to aquaporin-2 regulation. Physiol. Genom. 2016, 48, 502–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Kumar, S. Nedd4 and Nedd4-2: Closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010, 17, 68–77. [Google Scholar] [CrossRef]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef]
- Tasaki, T.; Mulder, L.C.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 2005, 25, 7120–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, S.M.; Kim, B.Y.; Kwon, Y.T. The N-end rule pathway: Emerging functions and molecular principles of substrate recognition. Nat. Rev. Mol. Cell Biol. 2011, 12, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Frottin, F.; Martinez, A.; Peynot, P.; Mitra, S.; Holz, R.C.; Giglione, C.; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteom. 2006, 5, 2336–2349. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.L.; Bradshaw, R.A. Isolation and characterization of the methionine aminopeptidase from porcine liver responsible for the co-translational processing of proteins. J. Biol. Chem. 1992, 267, 20667–20673. [Google Scholar] [CrossRef]
- Rao, H.; Uhlmann, F.; Nasmyth, K.; Varshavsky, A. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature 2001, 410, 955–959. [Google Scholar] [CrossRef]
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storr, S.J.; Carragher, N.O.; Frame, M.C.; Parr, T.; Martin, S.G. The calpain system and cancer. Nat. Rev. Cancer 2011, 11, 364–374. [Google Scholar] [CrossRef]
- Piatkov, K.I.; Brower, C.S.; Varshavsky, A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. USA 2012, 109, E1839–E1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatkov, K.I.; Oh, J.H.; Liu, Y.; Varshavsky, A. Calpain-generated natural protein fragments as short-lived substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2014, 111, E817–E826. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Payoe, R.; Fahlman, R.P. The C-terminal proteolytic fragment of the breast cancer susceptibility type 1 protein (BRCA1) is degraded by the N-end rule pathway. J. Biol. Chem. 2012, 287, 7495–7502. [Google Scholar] [CrossRef] [Green Version]
- Grigoryev, S.; Stewart, A.E.; Kwon, Y.T.; Arfin, S.M.; Bradshaw, R.A.; Jenkins, N.A.; Copeland, N.G.; Varshavsky, A. A mouse amidase specific for N-terminal asparagine. The gene, the enzyme, and their function in the N-end rule pathway. J. Biol. Chem. 1996, 271, 28521–28532. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Balogh, S.A.; Davydov, I.V.; Kashina, A.S.; Yoon, J.K.; Xie, Y.; Gaur, A.; Hyde, L.; Denenberg, V.H.; Varshavsky, A. Altered activity, social behavior, and spatial memory in mice lacking the NTAN1p amidase and the asparagine branch of the N-end rule pathway. Mol. Cell. Biol. 2000, 20, 4135–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Piatkov, K.I.; Brower, C.S.; Varshavsky, A. Glutamine-specific N-terminal amidase, a component of the N-end rule pathway. Mol. Cell 2009, 34, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.G.; Sheng, J.; Qi, X.; Xu, Z.; Takahashi, T.T.; Varshavsky, A. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 2005, 437, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Kashina, A.S.; Davydov, I.V.; Hu, R.G.; An, J.Y.; Seo, J.W.; Du, F.; Varshavsky, A. An essential role of N-terminal arginylation in cardiovascular development. Science 2002, 297, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Tasaki, T.; Moroi, K.; An, J.Y.; Kimura, S.; Davydov, I.V.; Kwon, Y.T. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 15030–15035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, N.; Keeley, T.P.; Giuntoli, B.; White, M.D.; Puerta, M.L.; Perata, P.; Hopkinson, R.J.; Flashman, E.; Licausi, F.; Ratcliffe, P.J. Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 2019, 365, 65–69. [Google Scholar] [CrossRef]
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasaki, T.; Zakrzewska, A.; Dudgeon, D.D.; Jiang, Y.; Lazo, J.S.; Kwon, Y.T. The substrate recognition domains of the N-end rule pathway. J. Biol. Chem. 2009, 284, 1884–1895. [Google Scholar] [CrossRef] [Green Version]
- Nillegoda, N.B.; Theodoraki, M.A.; Mandal, A.K.; Mayo, K.J.; Ren, H.Y.; Sultana, R.; Wu, K.; Johnson, J.; Cyr, D.M.; Caplan, A.J. Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol. Biol. Cell 2010, 21, 2102–2116. [Google Scholar] [CrossRef] [Green Version]
- Tasaki, T.; Kim, S.T.; Zakrzewska, A.; Lee, B.E.; Kang, M.J.; Yoo, Y.D.; Cha-Molstad, H.J.; Hwang, J.; Soung, N.K.; Sung, K.S.; et al. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 3800–3805. [Google Scholar] [CrossRef] [Green Version]
- Hochstrasser, M.; Varshavsky, A. In vivo degradation of a transcriptional regulator: The yeast alpha 2 repressor. Cell 1990, 61, 697–708. [Google Scholar] [CrossRef]
- Barlow, P.N.; Luisi, B.; Milner, A.; Elliott, M.; Everett, R. Structure of the C3HC4 domain by 1H-nuclear magnetic resonance spectroscopy. A new structural class of zinc-finger. J. Mol. Biol. 1994, 237, 201–211. [Google Scholar] [CrossRef]
- Borden, K.L.; Boddy, M.N.; Lally, J.; O’Reilly, N.J.; Martin, S.; Howe, K.; Solomon, E.; Freemont, P.S. The solution structure of the RING finger domain from the acute promyelocytic leukaemia proto-oncoprotein PML. EMBO. J. 1995, 14, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Varshavsky, A. The E2-E3 interaction in the N-end rule pathway: The RING-H2 finger of E3 is required for the synthesis of multiubiquitin chain. EMBO. J. 1999, 18, 6832–6844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, H.; Zhang, Y.; Wu, B.; Wu, S.; You, S.; Zhang, N.; Sun, Y. Structure and Function of HECT E3 Ubiquitin Ligases and their Role in Oxidative Stress. J. Transl. Int. Med. 2020, 8, 71–79. [Google Scholar] [CrossRef]
- Shah, S.S.; Kumar, S. Adaptors as the regulators of HECT ubiquitin ligases. Cell Death Differ. 2021, 28, 455–472. [Google Scholar] [CrossRef]
- Abbas, T.; Mueller, A.C.; Shibata, E.; Keaton, M.; Rossi, M.; Dutta, A. CRL1-FBXO11 promotes Cdt2 ubiquitylation and degradation and regulates Pr-Set7/Set8-mediated cellular migration. Mol. Cell 2013, 49, 1147–1158. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J. Biol. Chem. 2007, 282, 1797–1804. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Schulman, B.A.; Song, L.; Miller, J.J.; Jeffrey, P.D.; Wang, P.; Chu, C.; Koepp, D.M.; Elledge, S.J.; Pagano, M.; et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002, 416, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Sen, P.; Hofmann, K.; Ma, L.; Goebl, M.; Harper, J.W.; Elledge, S.J. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 1996, 86, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Schindler, U.; Beckmann, H.; Cashmore, A.R. HAT3.1, a novel Arabidopsis homeodomain protein containing a conserved cysteine-rich region. Plant J. 1993, 4, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Aasland, R.; Gibson, T.J.; Stewart, A.F. The PHD finger: Implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci. 1995, 20, 56–59. [Google Scholar] [CrossRef]
- Pascual, J.; Martinez-Yamout, M.; Dyson, H.J.; Wright, P.E. Structure of the PHD zinc finger from human Williams-Beuren syndrome transcription factor. J. Mol. Biol. 2000, 304, 723–729. [Google Scholar] [CrossRef]
- Adhikary, S.; Chakravarti, D.; Terranova, C.; Sengupta, I.; Maitituoheti, M.; Dasgupta, A.; Srivastava, D.K.; Ma, J.; Raman, A.T.; Tarco, E.; et al. Atypical plant homeodomain of UBR7 functions as an H2BK120Ub ligase and breast tumor suppressor. Nat. Commun. 2019, 10, 1398. [Google Scholar] [CrossRef] [PubMed]
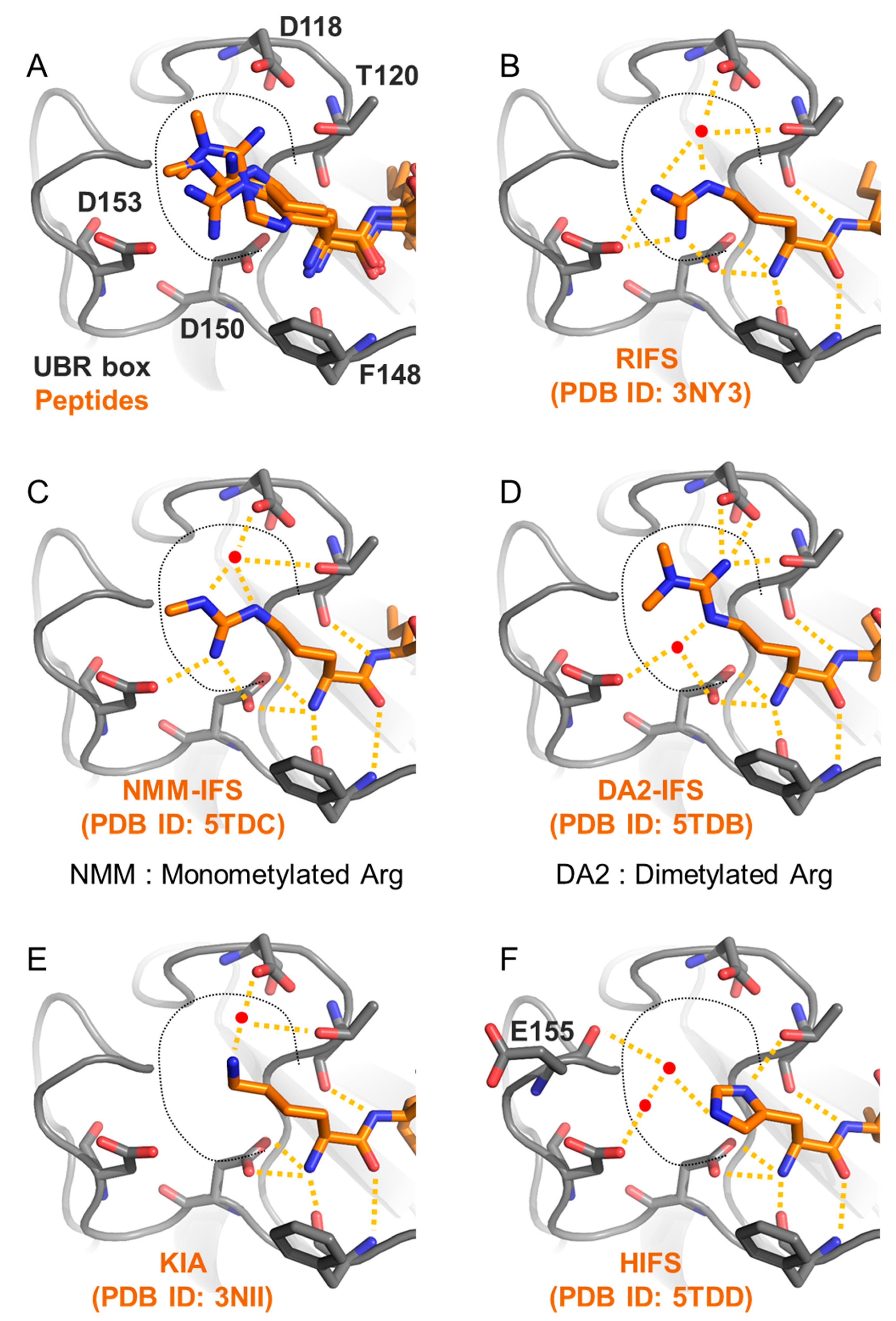
- Choi, W.S.; Jeong, B.C.; Joo, Y.J.; Lee, M.R.; Kim, J.; Eck, M.J.; Song, H.K. Structural basis for the recognition of N-end rule substrates by the UBR box of ubiquitin ligases. Nat. Struct. Mol. Biol. 2010, 17, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Escobar, J.; Matta-Camacho, E.; Cho, C.; Kozlov, G.; Gehring, K. Bound Waters Mediate Binding of Diverse Substrates to a Ubiquitin Ligase. Structure 2017, 25, 719–729.e713. [Google Scholar] [CrossRef] [Green Version]
- Matta-Camacho, E.; Kozlov, G.; Li, F.F.; Gehring, K. Structural basis of substrate recognition and specificity in the N-end rule pathway. Nat. Struct. Mol. Biol. 2010, 17, 1182–1187. [Google Scholar] [CrossRef]
- Kim, L.; Kwon, D.H.; Heo, J.; Park, M.R.; Song, H.K. Use of the LC3B-fusion technique for biochemical and structural studies of proteins involved in the N-degron pathway. J. Biol. Chem. 2020, 295, 2590–2600. [Google Scholar] [CrossRef] [PubMed]
- Erbse, A.; Schmidt, R.; Bornemann, T.; Schneider-Mergener, J.; Mogk, A.; Zahn, R.; Dougan, D.A.; Bukau, B. ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 2006, 439, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Roman-Hernandez, G.; Hou, J.Y.; Grant, R.A.; Sauer, R.T.; Baker, T.A. The ClpS adaptor mediates staged delivery of N-end rule substrates to the AAA+ ClpAP protease. Mol. Cell 2011, 43, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Roman-Hernandez, G.; Grant, R.A.; Sauer, R.T.; Baker, T.A. Molecular basis of substrate selection by the N-end rule adaptor protein ClpS. Proc. Natl. Acad. Sci. USA 2009, 106, 8888–8893. [Google Scholar] [CrossRef] [Green Version]
- Schuenemann, V.J.; Kralik, S.M.; Albrecht, R.; Spall, S.K.; Truscott, K.N.; Dougan, D.A.; Zeth, K. Structural basis of N-end rule substrate recognition in Escherichia coli by the ClpAP adaptor protein ClpS. EMBO. Rep. 2009, 10, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Hamm, H.E. The many faces of G protein signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprang, S.R. G protein mechanisms: Insights from structural analysis. Annu. Rev. Biochem. 1997, 66, 639–678. [Google Scholar] [CrossRef] [Green Version]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Park, J.; Clavio, N.A.B.; Kang, S.; Choi, S. Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef]
- Bebelman, M.P.; Crudden, C.; Pegtel, D.M.; Smit, M.J. The Convergence of Extracellular Vesicle and GPCR Biology. Trends Pharmacol. Sci. 2020, 41, 627–640. [Google Scholar] [CrossRef]
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO. J. 1999, 18, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Goricanec, D.; Stehle, R.; Egloff, P.; Grigoriu, S.; Pluckthun, A.; Wagner, G.; Hagn, F. Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding. Proc. Natl. Acad. Sci. USA 2016, 113, E3629–E3638. [Google Scholar] [CrossRef] [Green Version]
- Wall, M.A.; Posner, B.A.; Sprang, S.R. Structural basis of activity and subunit recognition in G protein heterotrimers. Structure 1998, 6, 1169–1183. [Google Scholar] [CrossRef] [Green Version]
- Ford, C.E.; Skiba, N.P.; Bae, H.; Daaka, Y.; Reuveny, E.; Shekter, L.R.; Rosal, R.; Weng, G.; Yang, C.S.; Iyengar, R.; et al. Molecular basis for interactions of G protein betagamma subunits with effectors. Science 1998, 280, 1271–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sternweis, P.M.; Charnecki, S.; Smith, T.F.; Gilman, A.G.; Neer, E.J.; Kozasa, T. Sites for Galpha binding on the G protein beta subunit overlap with sites for regulation of phospholipase Cbeta and adenylyl cyclase. J. Biol. Chem. 1998, 273, 16265–16272. [Google Scholar] [CrossRef] [Green Version]
- Sprang, S.R. Invited review: Activation of G proteins by GTP and the mechanism of Galpha-catalyzed GTP hydrolysis. Biopolymers 2016, 105, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Hepler, J.R.; Berman, D.M.; Gilman, A.G.; Kozasa, T. RGS4 and GAIP are GTPase-activating proteins for Gq alpha and block activation of phospholipase C beta by gamma-thio-GTP-Gq alpha. Proc. Natl. Acad. Sci. USA 1997, 94, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Berman, D.M.; Wilkie, T.M.; Gilman, A.G. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell 1996, 86, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Sjogren, B.; Neubig, R.R. Thinking outside of the “RGS box”: New approaches to therapeutic targeting of regulators of G protein signaling. Mol. Pharmacol. 2010, 78, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydov, I.V.; Varshavsky, A. RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J. Biol. Chem. 2000, 275, 22931–22941. [Google Scholar] [CrossRef] [Green Version]
- An, J.Y.; Seo, J.W.; Tasaki, T.; Lee, M.J.; Varshavsky, A.; Kwon, Y.T. Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 6212–6217. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Kim, S.; Lee, J.H.; Kwon, Y.T.; Lee, M.J. Ablation of Arg-tRNA-protein transferases results in defective neural tube development. BMB. Rep. 2016, 49, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Choi, W.H.; Lee, J.H.; Han, D.H.; Kim, J.H.; Chung, Y.S.; Kim, S.H.; Lee, M.J. A neurostimulant para-chloroamphetamine inhibits the arginylation branch of the N-end rule pathway. Sci. Rep. 2014, 4, 6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Hay, B.A. Cell proliferation and apoptosis. Curr. Opin. Cell Biol. 1999, 11, 745–752. [Google Scholar] [CrossRef]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef]
- Mak, T.W.; Yeh, W.C. Signaling for survival and apoptosis in the immune system. Arthritis Res. 2002, 4 (Suppl. S3), S243–S252. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Fahlman, R.P. Phosphorylation Impacts N-end Rule Degradation of the Proteolytically Activated Form of BMX Kinase. J. Biol. Chem. 2016, 291, 22757–22768. [Google Scholar] [CrossRef] [Green Version]
- Agarwalla, P.; Banerjee, R. N-end rule pathway inhibition assists colon tumor regression via necroptosis. Mol. Ther. Oncolytics 2016, 3, 16020. [Google Scholar] [CrossRef]
- Shimbo, K.; Hsu, G.W.; Nguyen, H.; Mahrus, S.; Trinidad, J.C.; Burlingame, A.L.; Wells, J.A. Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc. Natl. Acad. Sci. USA 2012, 109, 12432–12437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, E.D.; Seaman, J.E.; Agard, N.; Hsu, G.W.; Julien, O.; Mahrus, S.; Nguyen, H.; Shimbo, K.; Yoshihara, H.A.; Zhuang, M.; et al. The DegraBase: A database of proteolysis in healthy and apoptotic human cells. Mol. Cell Proteom. 2013, 12, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, Y.; Sorimachi, H. Calpains: An elaborate proteolytic system. Biochim. Biophys. Acta 2012, 1824, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Leboeuf, D.; Abakumova, T.; Prikazchikova, T.; Rhym, L.; Anderson, D.G.; Zatsepin, T.S.; Piatkov, K.I. Downregulation of the Arg/N-degron Pathway Sensitizes Cancer Cells to Chemotherapy In Vivo. Mol. Ther. 2020, 28, 1092–1104. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Xu, C.; Li, X.; Li, M.; Wu, X.; Pu, W.; Zhou, B.; Wang, H.; Li, D.; et al. Ubiquitination of RIPK1 suppresses programmed cell death by regulating RIPK1 kinase activation during embryogenesis. Nat. Commun. 2019, 10, 4158. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Pal, K.; Tasaki, T.; Roy, S.; Jiang, Y.; An, J.Y.; Banerjee, R.; Kwon, Y.T. Synthetic heterovalent inhibitors targeting recognition E3 components of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.Y.; Qiu, Y.; He, J.; Kung, H.J.; Oleinick, N.L. Etk/Bmx, a PH-domain containing tyrosine kinase, protects prostate cancer cells from apoptosis induced by photodynamic therapy or thapsigargin. Oncogene 1999, 18, 3391–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.L.; Storey, A. BMX Negatively Regulates BAK Function, Thereby Increasing Apoptotic Resistance to Chemotherapeutic Drugs. Cancer Res. 2015, 75, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.M.; Huang, C.L.; Kung, H.J.; Huang, C.Y. Proteolytic activation of ETK/Bmx tyrosine kinase by caspases. J. Biol. Chem. 2001, 276, 17672–17678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekman, N.; Arighi, E.; Rajantie, I.; Saharinen, P.; Ristimaki, A.; Silvennoinen, O.; Alitalo, K. The Bmx tyrosine kinase is activated by IL-3 and G-CSF in a PI-3K dependent manner. Oncogene 2000, 19, 4151–4158. [Google Scholar] [CrossRef] [Green Version]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC. Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO. Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Ragheb, M.A. N-degron-mediated degradation and regulation of mitochondrial PINK1 kinase. Curr. Genet. 2020, 66, 693–701. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
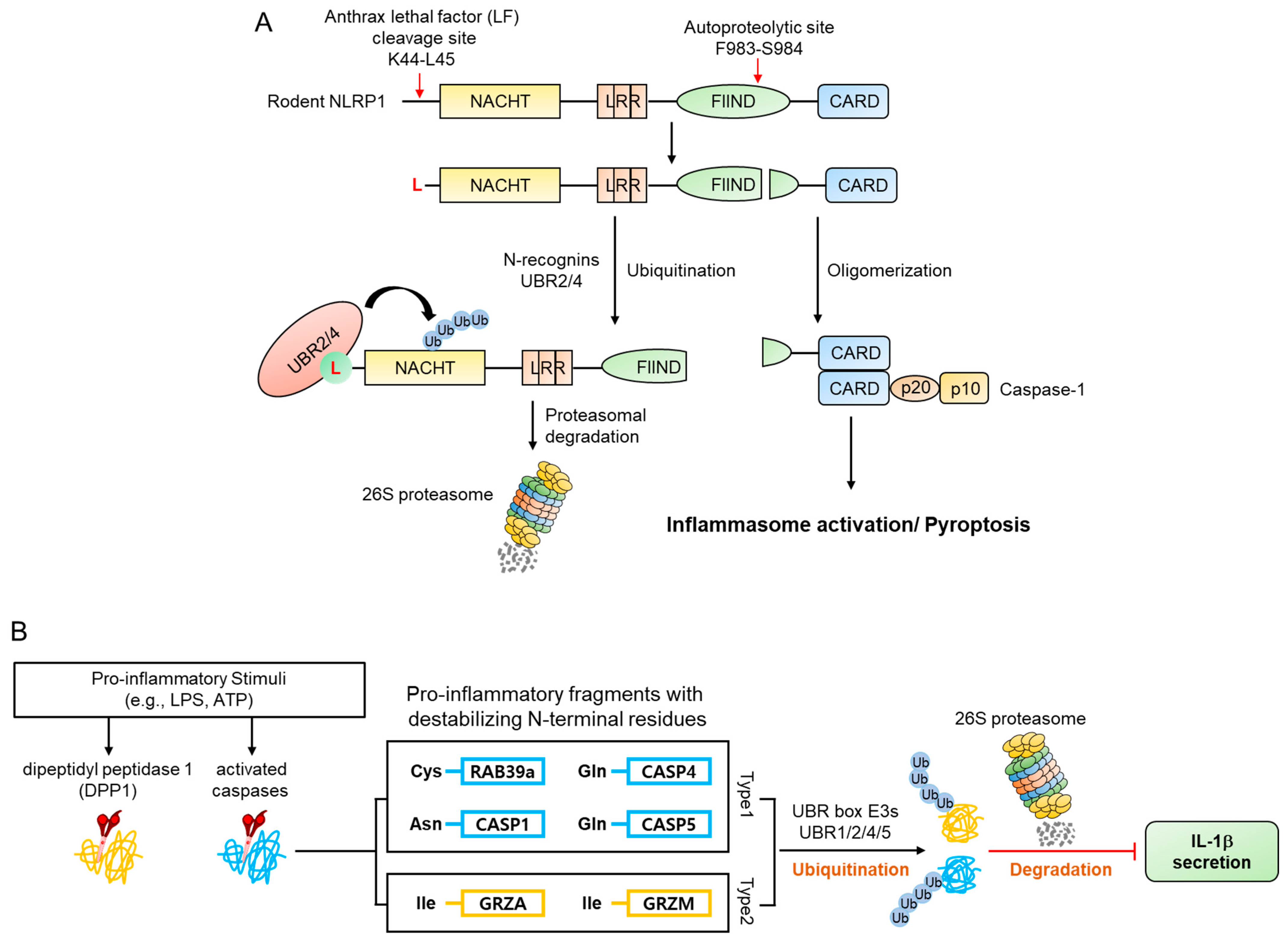
- Chui, A.J.; Okondo, M.C.; Rao, S.D.; Gai, K.; Griswold, A.R.; Johnson, D.C.; Ball, D.P.; Taabazuing, C.Y.; Orth, E.L.; Vittimberga, B.A.; et al. N-terminal degradation activates the NLRP1B inflammasome. Science 2019, 364, 82–85. [Google Scholar] [CrossRef]
- Xu, H.; Shi, J.; Gao, H.; Liu, Y.; Yang, Z.; Shao, F.; Dong, N. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO. J. 2019, 38, e101996. [Google Scholar] [CrossRef] [PubMed]
- Taabazuing, C.Y.; Griswold, A.R.; Bachovchin, D.A. The NLRP1 and CARD8 inflammasomes. Immunol. Rev. 2020, 297, 13–25. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Frew, B.C.; Joag, V.R.; Mogridge, J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS. Pathog. 2012, 8, e1002659. [Google Scholar] [CrossRef] [Green Version]
- D’Osualdo, A.; Weichenberger, C.X.; Wagner, R.N.; Godzik, A.; Wooley, J.; Reed, J.C. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS ONE 2011, 6, e27396. [Google Scholar] [CrossRef] [Green Version]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8, e1002638. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, K.A.; Levinsohn, J.L.; Fattah, R.; Newman, Z.L.; Maier, N.; Sastalla, I.; Liu, S.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS ONE 2012, 7, e49741. [Google Scholar] [CrossRef]
- Chavarria-Smith, J.; Vance, R.E. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 2013, 9, e1003452. [Google Scholar] [CrossRef] [Green Version]
- Leboeuf, D.; Pyatkov, M.; Zatsepin, T.S.; Piatkov, K. The Arg/N-Degron Pathway-A Potential Running Back in Fine-Tuning the Inflammatory Response? Biomolecules 2020, 10, 903. [Google Scholar] [CrossRef]
- Wilhelm, T.; Said, M.; Naim, V. DNA Replication Stress and Chromosomal Instability: Dangerous Liaisons. Genes 2020, 11, 642. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broers, J.L.; Hutchison, C.J.; Ramaekers, F.C. Laminopathies. J. Pathol. 2004, 204, 478–488. [Google Scholar] [CrossRef]
- Friedberg, E.C. A brief history of the DNA repair field. Cell Res. 2008, 18, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Essers, J.; Theil, A.F.; Baldeyron, C.; van Cappellen, W.A.; Houtsmuller, A.B.; Kanaar, R.; Vermeulen, W. Nuclear dynamics of PCNA in DNA replication and repair. Mol. Cell Biol. 2005, 25, 9350–9359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair. 2007, 6, 891–899. [Google Scholar] [CrossRef]
- Li, X.Q.; Ren, J.; Chen, P.; Chen, Y.J.; Wu, M.; Wu, Y.; Chen, K.; Li, J. Co-inhibition of Pol eta and ATR sensitizes cisplatin-resistant non-small cell lung cancer cells to cisplatin by impeding DNA damage repair. Acta Pharmacol. Sin. 2018, 39, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Kashiwaba, S.; Kanao, R.; Masuda, Y.; Kusumoto-Matsuo, R.; Hanaoka, F.; Masutani, C. USP7 Is a Suppressor of PCNA Ubiquitination and Oxidative-Stress-Induced Mutagenesis in Human Cells. Cell Rep. 2015, 13, 2072–2080. [Google Scholar] [CrossRef] [Green Version]
- Niimi, A.; Brown, S.; Sabbioneda, S.; Kannouche, P.L.; Scott, A.; Yasui, A.; Green, C.M.; Lehmann, A.R. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Hibbert, R.G.; Huang, A.; Boelens, R.; Sixma, T.K. E3 ligase Rad18 promotes monoubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6. Proc. Natl. Acad. Sci. USA 2011, 108, 5590–5595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Qin, Z.; Zhang, X.; Xiao, W. Roles of sequential ubiquitination of PCNA in DNA-damage tolerance. FEBS. Lett. 2011, 585, 2786–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
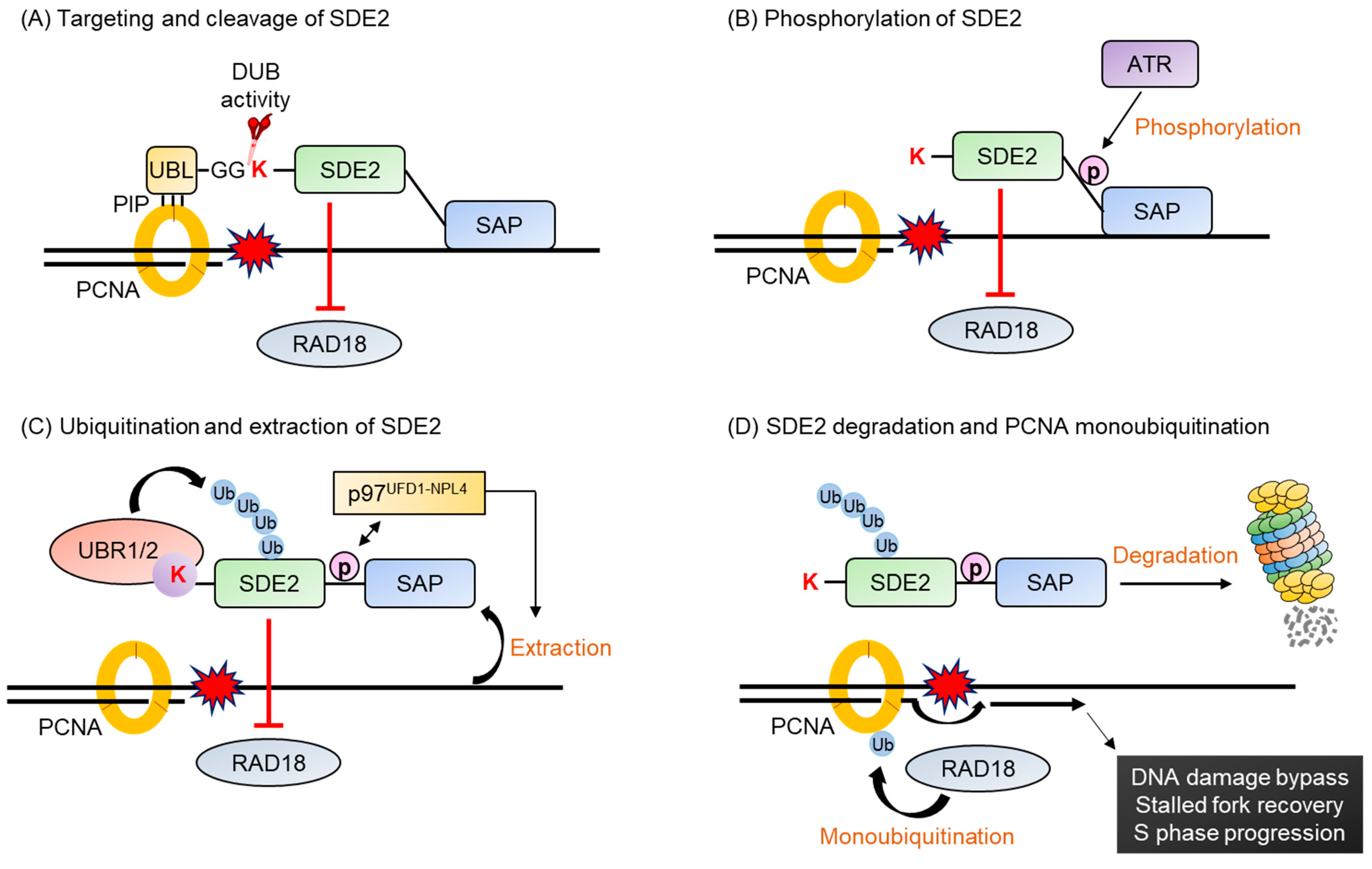
- Jo, U.; Cai, W.; Wang, J.; Kwon, Y.; D’Andrea, A.D.; Kim, H. PCNA-Dependent Cleavage and Degradation of SDE2 Regulates Response to Replication Stress. PLoS Genet. 2016, 12, e1006465. [Google Scholar] [CrossRef]
- Rageul, J.; Park, J.J.; Jo, U.; Weinheimer, A.S.; Vu, T.T.M.; Kim, H. Conditional degradation of SDE2 by the Arg/N-End rule pathway regulates stress response at replication forks. Nucleic Acids Res. 2019, 47, 3996–4010. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. SAP—A putative DNA-binding motif involved in chromosomal organization. Trends Biochem. Sci. 2000, 25, 112–114. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.4.1; Schrödinger, LLC: New York, NY, USA, 2021.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Function | Species | Substrate | Pro-N-degron | N-degron | Modifications | Ref. |
|---|---|---|---|---|---|---|
| G-protein signaling | Mus musculus | RGS4 | Cys2 | Arg-Cys* | MetAPs cleavage, oxidation, arginylation | [34,36,84] |
| Mus musculus | RGS5 | Cys2 | Arg-Cys* | |||
| Mus musculus | RGS16 | Cys2 | Arg-Cys* | |||
| Apoptosis | Mus musculus | RIPK1 | Cys325 | Arg-Cys* | Endoproteolytic cleavage by caspase, oxidation, arginylation | [28,30,93] |
| Mus musculus | TRAF1 | Cys157 | Arg-Cys* | |||
| Mus musculus | BRCA1 | Asp1119 | Arg-Asp | Endoproteolytic cleavage by caspase, arginylation | ||
| Mus musculus | EPHA4 | Asp774 | Arg-Asp | |||
| Mus musculus | BIMEL | - | Arg12 | Endoproteolytic cleavage by caspase | ||
| Mus musculus | MET | - | Tyr1001 | |||
| Mus musculus | NEDD9 | - | Tyr631 | |||
| Homo sapiens | LIMK1 | - | Leu241 | |||
| Homo sapiens | BMX | - | Trp243 | |||
| Homo sapiens | BID | - | Arg71 | Endoproteolytic cleavage by calpain, arginylation | [29] | |
| Homo sapiens | Bak | Glu16 | Arg-Glu | |||
| Mitochondrial quality control | Homo sapiens | PINK1 | - | Phe104 | Endoproteolytic cleavage by PARL | [112] |
| mNLRP1B Inflammasome | Mus musculus | NLRP1B | - | Leu45 | Endoproteolytic cleavage by anthrax lethal factor | [124,125] |
| Inflammatory response | Homo sapiens | Caspase-1 | Asn120 | Arg-Asp | Auto-cleavage, deamidation, arginylation | [134] |
| Homo sapiens | Caspase-4 | Gln81 | Arg-Glu | |||
| Homo sapiens | Caspase-5 | Gln138 | Arg-Glu | |||
| Homo sapiens | RAB39a | Cys149 | Arg-Cys* | Endoproteolytic cleavage by caspase-1, oxidation, arginylation | ||
| Mus musculus | Granzyme A | - | Ile29 | Endoproteolytic cleavage by DPP1 | ||
| Mus musculus | Granzyme M | - | Ile27 | |||
| Genome stability | Homo sapiens | SDE2 | - | Lys78 | Endoproteolytic cleavage by DUB | [152,153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.G.; Shin, H.-C.; Seo, T.; Nawale, L.; Han, G.; Kim, B.Y.; Kim, S.J.; Cha-Molstad, H. Signaling Pathways Regulated by UBR Box-Containing E3 Ligases. Int. J. Mol. Sci. 2021, 22, 8323. https://doi.org/10.3390/ijms22158323
Kim JG, Shin H-C, Seo T, Nawale L, Han G, Kim BY, Kim SJ, Cha-Molstad H. Signaling Pathways Regulated by UBR Box-Containing E3 Ligases. International Journal of Molecular Sciences. 2021; 22(15):8323. https://doi.org/10.3390/ijms22158323
Chicago/Turabian StyleKim, Jung Gi, Ho-Chul Shin, Taewook Seo, Laxman Nawale, Goeun Han, Bo Yeon Kim, Seung Jun Kim, and Hyunjoo Cha-Molstad. 2021. "Signaling Pathways Regulated by UBR Box-Containing E3 Ligases" International Journal of Molecular Sciences 22, no. 15: 8323. https://doi.org/10.3390/ijms22158323
APA StyleKim, J. G., Shin, H.-C., Seo, T., Nawale, L., Han, G., Kim, B. Y., Kim, S. J., & Cha-Molstad, H. (2021). Signaling Pathways Regulated by UBR Box-Containing E3 Ligases. International Journal of Molecular Sciences, 22(15), 8323. https://doi.org/10.3390/ijms22158323