
PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases

 , , ,
, , ,
Abstract
:1. Introduction
2. Overviews of PPARs α, β/δ and γ
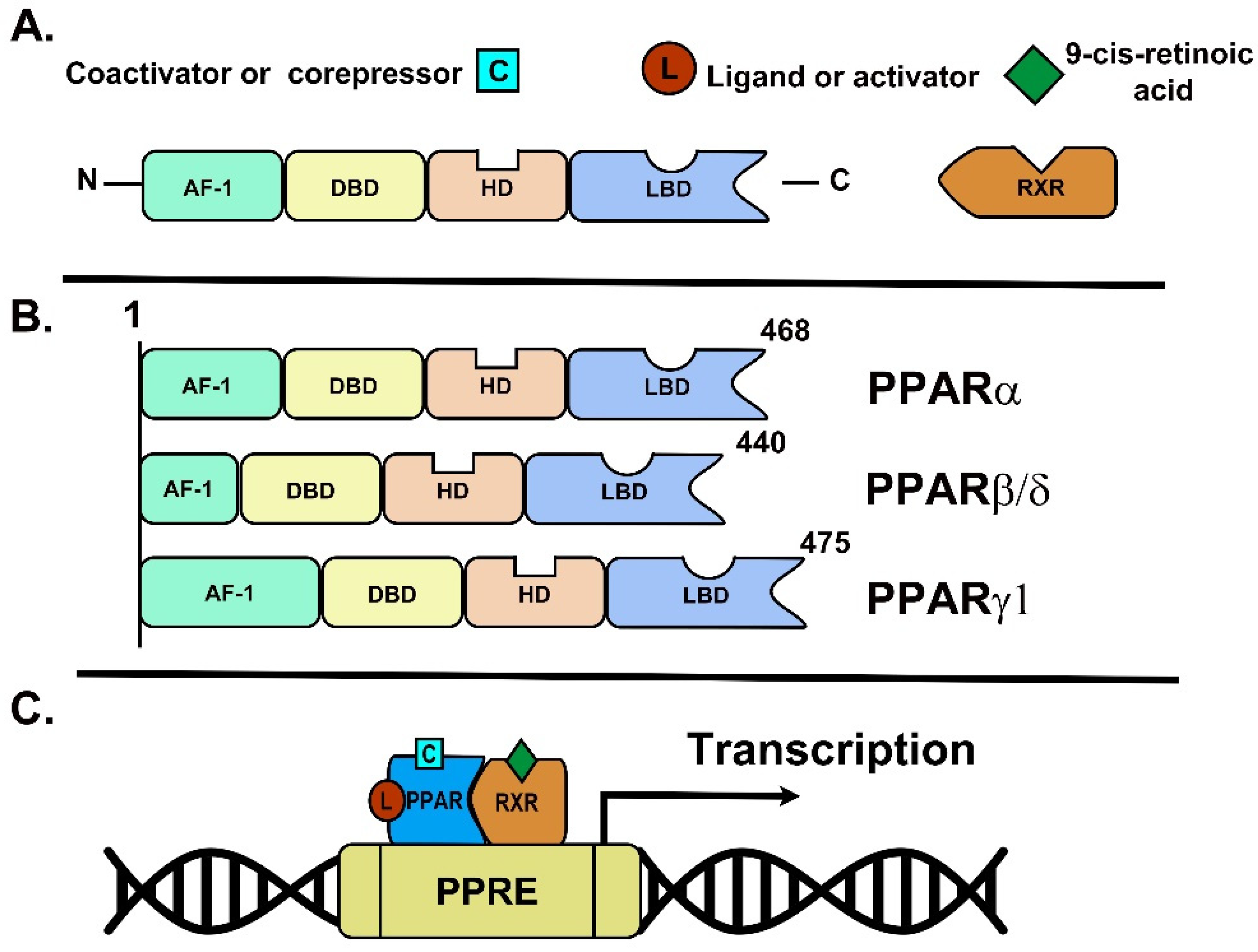
2.1. Structure and Molecular Characteristics
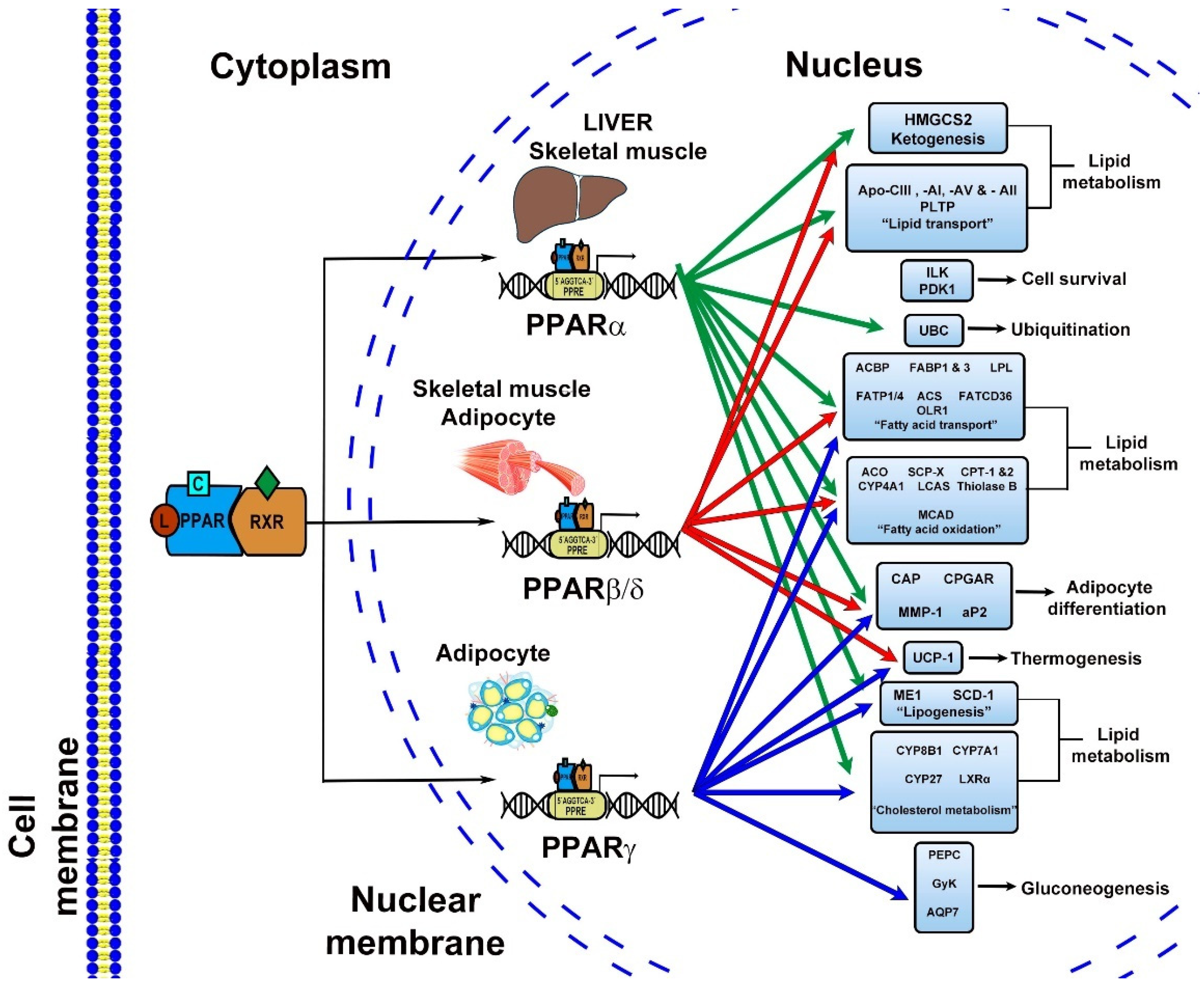
2.2. Mechanisms of Action
Signal Pathways
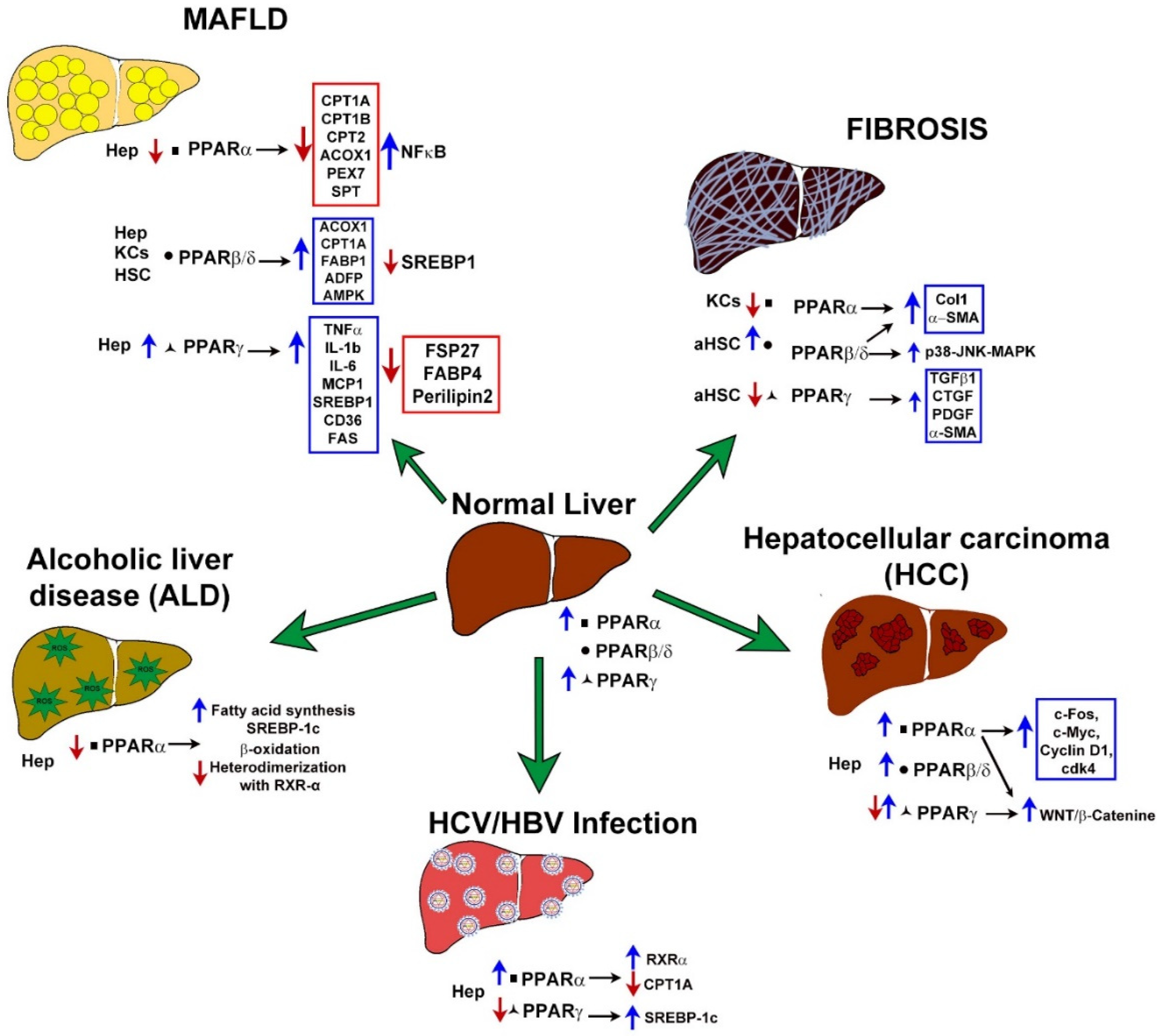
3. Role of PPARs in Liver Diseases
3.1. Gene Expression Alterationof PPARs in MAFLD
3.1.1. PPARα
3.1.2. PPARβ/δ
3.1.3. PPARγ
3.1.4. Clinical Trials of PPAR-Related Drugs in NASH
3.2. PPARs Expression in Liver Fibrosis
3.2.1. PPARα
3.2.2. PPARβ/δ
3.2.3. PPARγ
3.2.4. Clinical Trials of PPAR-Related Drugs in Liver Fibrosis
3.3. PPARs in Hepatocellular Carcinoma
3.3.1. PPARα
3.3.2. PPARβ/δ
3.3.3. PPARγ
3.3.4. Clinical Trials of PPAR-Related Drugs in HCC
3.4. PPARs in HBV and HCV Infections
3.4.1. PPARα
3.4.2. Clinical Trials of PPAR-Related Drugs in Infection HBV/HCV
3.5. PPARs and Their Role in the Development of ALD
3.5.1. PPARα
3.5.2. Clinical Information about PPARs Activity in ALD
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Jones, J.G. Hepatic glucose and lipid metabolism. Diabetology 2016, 59, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.-R.; Wang, J.-L.; Ren, H.-Z.; Shi, X.-L. Lipometabolism and Glycometabolism in Liver Diseases. BioMed Res. Int. 2018, 2018, 1287127. [Google Scholar] [CrossRef]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef]
- Tailleux, A.; Wouters, K.; Staels, B. Roles of PPARs in NAFLD: Potential therapeutic targets. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.-C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef]
- Darbre, P.D. Disruption of Other Receptor Systems: Progesterone and Glucocorticoid Receptors, Peroxisome Proliferator-Activated Receptors, Pregnane X Receptor, and Aryl Hydrocarbon Receptor. In Endocrine Disruption and Human Health, 1st ed.; Philippa, D., Ed.; Academic Press: London, UK, 2015; pp. 111–122. [Google Scholar] [CrossRef]
- Lee, W.-S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y. Peroxisome Proliferator-Activated Receptor Family and Its Relationship to Renal Complications of the Metabolic Syndrome. J. Am. Soc. Nephrol. 2004, 15, 2801–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [Green Version]
- Usuda, D.; Kanda, T. Peroxisome proliferator-activated receptors for hypertension. World J. Cardiol. 2014, 6, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bobiński, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, S.; Lee, C.-H. PPARδ as a therapeutic target in metabolic disease. FEBS Lett. 2008, 582, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, N.; Wagner, K.D. PPAR Beta/Delta and the Hallmarks of Cancer. Cells 2020, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Heming, M.; Gran, S.; Jauch, S.-L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schäfers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-γ Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front. Immunol. 2018, 9, 893–909. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Kim, T.-H.; Kim, M.-Y.; Jo, S.-H.; Park, J.-M.; Ahn, Y.-H. Modulation of the Transcriptional Activity of Peroxisome Proliferator-Activated Receptor Gamma by Protein-Protein Interactions and Post-Translational Modifications. Yonsei Med. J. 2013, 54, 545–559. [Google Scholar] [CrossRef]
- Sayiner, M.; Koenig, A.; Henry, L.; Younossi, Z.M. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in the United States and the Rest of the World. Clin. Liver Dis. 2016, 20, 205–214. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Terlecky, S.R.; Terlecky, L.J.; Giordano, C.R. Peroxisomes, oxidative stress, and inflammation. World J. Biol. Chem. 2012, 3, 93–97. [Google Scholar] [CrossRef]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, C.; Zhu, Y.; Reddy, J.K. Peroxisome Proliferator-Activated Receptors, Coactivators, and Downstream Targets. Cell Biochem. Biophys. 2000, 32, 187–204. [Google Scholar] [CrossRef]
- Pawlak, M.; Baugé, E.; Bourguet, W.; De Bosscher, K.; Lalloyer, F.; Tailleux, A.; Lebherz, C.; Lefebvre, P.; Staels, B. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology 2014, 60, 1593–1606. [Google Scholar] [CrossRef]
- Evans, R.M.; Barrish, G.D.; Wang, Y.-X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Berghe, W.V.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.-C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome Proliferator-activated Receptor α Negatively Regulates the Vascular Inflammatory Gene Response by Negative Cross-talk with Transcription Factors NF-κB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.-R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Park, H.-S.; Jang, J.E.; Ko, M.S.; Woo, S.H.; Kim, B.J.; Kim, H.S.; Park, H.S.; Park, I.-S.; Koh, E.H.; Lee, K.-U. Statins Increase Mitochondrial and Peroxisomal Fatty Acid Oxidation in the Liver and Prevent Non-Alcoholic Steatohepatitis in Mice. Diabetes Metab. J. 2016, 40, 376–385. [Google Scholar] [CrossRef]
- Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Meza-Rios, A.; Garcia-Bañuelos, J.; Vera-Cruz, J.; Gutiérrez-Cuevas, J.; Silva-Gomez, J.; Staels, B.; Dominguez-Rosales, J.; Galicia-Moreno, M.; et al. Pirfenidone Is an Agonistic Ligand for PPARα and Improves NASH by Activation of SIRT1/LKB1/pAMPK. Hepatol. Commun. 2020, 4, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Singh, I. Modulation of Rho-Rock signaling pathway protects oligodendrocytes against cytokine toxicity via PPAR-α-dependent mechanism. Glia 2013, 61, 1500–1517. [Google Scholar] [CrossRef] [Green Version]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free. Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef]
- Brandt, J.M.; Djouadi, F.; Kelly, D.P. Fatty Acids Activate Transcription of the Muscle Carnitine Palmitoyltransferase I Gene in Cardiac Myocytes via the Peroxisome Proliferator-activated Receptor α. J. Biol. Chem. 1998, 273, 23786–23792. [Google Scholar] [CrossRef] [Green Version]
- Patsouris, D.; Reddy, J.K.; Muller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor α Mediates the Effects of High-Fat Diet on Hepatic Gene Expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Sohn, I.; Ahn, J.-I.; Lee, K.-H.; Lee, Y.S.; Lee, Y.S. Hepatic gene expression profiles in a long-term high-fat diet-induced obesity mouse model. Gene 2004, 340, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Redonnet, A.; Groubet, R.; L-Suberville, C.N.; Bonilla, S.; Martínez, A.; Higueret, P. Exposure to an obesity-inducing diet early affects the pattern of expression of peroxisome proliferator, retinoic acid, and triiodothyronine nuclear receptors in the rat. Metabolism 2001, 50, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.-H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.-J. PPAR Expression Protects Male Mice from High Fat–Induced Nonalcoholic Fatty Liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Lalloyer, F.; Wouters, K.; Baron, M.; Caron, S.; Vallez, E.; Vanhoutte, J.; Baugé, E.; Shiri-Sverdlov, R.; Hofker, M.; Staels, B.; et al. Peroxisome Proliferator–Activated Receptor-α Gene Level Differently Affects Lipid Metabolism and Inflammation in Apolipoprotein E2 Knock-In Mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1573–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Muller, M. Peroxisome Proliferator-Activated Receptor α Protects against Obesity-Induced Hepatic Inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the potent PPAR? agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Shiri-Sverdlov, R.; Wouters, K.; van Gorp, P.; Gijbels, M.J.; Noel, B.; Buffat, L.; Staels, B.; Maeda, N.; van Bilsen, M.; Hofker, M.H. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol. 2006, 44, 732–741. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. General molecular biology and architecture of nuclear receptors. Curr. Top. Med. Chem. 2012, 12, 486–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasawa, T.; Inada, Y.; Nakano, S.; Tamura, T.; Takahashi, T.; Maruyama, K.; Yamazaki, Y.; Kuroda, J.; Shibata, N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARδ agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur. J. Pharmacol. 2006, 536, 182–191. [Google Scholar] [CrossRef]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor induces fatty acid -oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [Green Version]
- Bojic, L.A.; Telford, D.E.; Fullerton, M.D.; Ford, R.J.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Gros, R.; Kemp, B.; Steinberg, G.; et al. PPARδ activation attenuates hepatic steatosis in Ldlr mice by enhanced fat oxidation, reduced lipogenesis, and improved insulin sensitivity. J. Lipid Res. 2014, 55, 1254–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risérus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of Peroxisome Proliferator-Activated Receptor (PPAR) Promotes Reversal of Multiple Metabolic Abnormalities, Reduces Oxidative Stress, and Increases Fatty Acid Oxidation in Moderately Obese Men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.-J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, A Novel Peroxisome Proliferator Receptor-δ Agonist: Lipid and Other Metabolic Effects in Dyslipidemic Overweight Patients Treated with and without Atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Xie, X.; Fan, Y.; Tian, J.; Guan, Y.; Wang, X.; Zhu, Y.; Wang, N. Peroxisome proliferator-activated receptor-δ induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology 2008, 48, 432–441. [Google Scholar] [CrossRef]
- Liu, S.; Brown, J.D.; Stanya, K.; Homan, E.A.; Leidl, M.; Inouye, K.; Bhargava, P.; Gangl, M.R.; Dai, L.; Hatano, B.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.-W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.; et al. PPAR regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Pettinelli, P.; Videla, L.A. Up-Regulation of PPAR-γ mRNA Expression in the Liver of Obese Patients: An Additional Reinforcing Lipogenic Mechanism to SREBP-1c Induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef]
- Nakamuta, M.; Kohjima, M.; Morizono, S.; Kotoh, K.; Yoshimoto, T.; Miyagi, I.; Enjoji, M. Evaluation of fatty acid metabo-lism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2005, 16, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.K.; Dietz, K.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M. Liver Peroxisome Proliferator-activated Receptor γ Contributes to Hepatic Steatosis, Triglyceride Clearance, and Regulation of Body Fat Mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, S.; Xiao, X.; Chen, T.; Lou, J. PPAR Ligands Function as Suppressors That Target Biological Actions of HMGB1. PPAR Res. 2016, 2016, 2612743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necela, B.M.; Su, W.; Thompson, E.A. Toll-like receptor 4 mediates cross-talk between peroxisome proliferator-activated receptor γ and nuclear factor-κB in macrophages. Immunology 2008, 125, 344–358. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-inflammatory Properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Li-ver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Matsusue, K.; Kashireddy, P.; Cao, W.-Q.; Yeldandi, V.; Yeldandi, A.V.; Rao, M.S.; Gonzalez, F.J.; Reddy, J.K. Adipocyte-specific Gene Expression and Adipogenic Steatosis in the Mouse Liver Due to Peroxisome Proliferator-activated Receptor γ1 (PPARγ1) Overexpression. J. Biol. Chem. 2003, 278, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Scherer, P.E. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol. Metab. 2013, 2, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator−Activated Receptor−α and −δ, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [Green Version]
- Gastaldelli, A.; Miyazaki, Y.; Mahankali, A.; Berria, R.; Pettiti, M.; Buzzigoli, E.; Ferrannini, E.; DeFronzo, R.A. The Effect of Pioglitazone on the Liver: Role of adiponectin. Diabetes Care 2006, 29, 2275–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, S.H.; Argo, C.K.; Al-Osaimi, A.M. Therapy of NAFLD: Insulin sensitizing agents. J. Clin. Gastroenterol. 2006, 40 (Suppl. 1), S61–S66. [Google Scholar] [PubMed]
- Aithal, G.P.; Thomas, J.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, Placebo-Controlled Trial of Pioglitazone in Nondiabetic Subjects with Nonalcoholic Steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [Green Version]
- Gastaldelli, A.; Harrison, S.; Belfort-Aguiar, R.; Hardies, J.; Balas, B.; Schenker, S.; Cusi, K. Pioglitazone in the treatment of NASH: The role of adiponectin. Aliment. Pharmacol. Ther. 2010, 32, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the mana-gement of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Massagué, J.; Chen, Y.G. Controlling TGF-beta signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar] [CrossRef]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor a target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.M.; Van Rooijen, N.; Staels, B.; Kersten, S.; Muller, M. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 2009, 51, 511–522. [Google Scholar] [CrossRef]
- Chen, L.; Li, L.; Chen, J.; Li, L.; Zheng, Z.; Ren, J.; Qiu, Y. Oleoylethanolamide, an endogenous PPAR-α ligand, atte-nuates liver fibrosis targeting hepatic stellate cells. Oncotarget 2015, 6, 42530–42540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostadinova, R.; Montagner, A.; Gouranton, E.; Fleury, S.; Guillou, H.; Dombrowicz, D.; Desreumaux, P.; Wahli, W. GW501516-activated PPARβ/δ promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci. 2012, 2, 34. [Google Scholar] [CrossRef] [Green Version]
- Hellemans, K.; Michalik, L.; Dittie, A.; Knorr, A.; Rombouts, K.; de Jong, J.; Heirman, C.; Quartier, E.; Schuit, F.; Wahli, W.; et al. Peroxisome proliferator-activated receptor-β signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology 2003, 124, 184–201. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Haimerl, M.; Paik, Y.-H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor agonist. Proc. Natl. Acad. Sci. USA 2012, 109, E1369–E1376. [Google Scholar] [CrossRef] [Green Version]
- Marsillach, J.; Camps, J.; Ferré, N.; Beltran, R.; Rull, A.; Mackness, B.; Mackness, M.; Joven, J. Paraoxonase-1 is related to inflammation, fibrosis and PPAR delta in experimental liver disease. BMC Gastroenterol. 2009, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, C.; Lu, J.; Huang, K.; Han, Y.; Chen, J.; Yang, Y.; Liu, B. PPAR δ inhibition protects against palmitic acid-LPS induced lipidosis and injury in cultured hepatocyte L02 cell. Int. J. Med. Sci. 2019, 16, 1593–1603. [Google Scholar] [CrossRef]
- Kang, K.; Reilly, S.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.-H. Adipocyte-Derived Th2 Cytokines and Myeloid PPARδ Regulate Macrophage Polarization and Insulin Sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.-C.; Kang, K.; Reilly, S.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of Peroxisome Proliferator-activated Receptor δ/β in Hepatic Metabolic Regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, W.; Palkar, P.S.; Murray, I.A.; McDevitt, E.I.; Kennett, M.J.; Kang, B.H.; Isom, H.C.; Perdew, G.H.; Gonzalez, F.J.; Peters, J.M. Ligand Activation of Peroxisome Proliferator–Activated Receptor β/δ (PPARβ/δ) Attenuates Carbon Tetrachloride Hepatotoxicity by Downregulating Proinflammatory Gene Expression. Toxicol. Sci. 2008, 105, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Fu, M.; Zhang, J.; Zhu, X.; Myles, D.E.; Willson, T.M.; Liu, X.; Chen, Y.E. Peroxisome Proliferator-activated Receptor γ Inhibits Transforming Growth Factor β-induced Connective Tissue Growth Factor Expression in Human Aortic Smooth Muscle Cells by Interfering with Smad3. J. Biol. Chem. 2001, 276, 45888–45894. [Google Scholar] [CrossRef] [Green Version]
- Saidi, A.; Kasabova, M.; Vanderlynden, L.; Wartenberg, M.; Kara-Ali, G.H.; Marc, D.; Lecaille, F.; Lalmanach, G. Curcumin inhibits the TGF-β1-dependent differentiation of lung fibroblasts via PPARγ-driven upregulation of cathepsins B and L. Sci. Rep. 2019, 9, 491. [Google Scholar] [CrossRef]
- Xia, Y.; Li, J.; Chen, K.; Feng, J.; Guo, C. Bergenin Attenuates Hepatic Fibrosis by Regulating Autophagy Mediated by the PPAR-γ/TGF-β Pathway. PPAR Res. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Zhang, F.; Kong, D.; Lu, Y.; Zheng, S. Peroxisome proliferator-activated receptor-γ as a therapeutic target for hepatic fibrosis: From bench to bedside. Cell. Mol. Life Sci. 2013, 70, 259–276. [Google Scholar] [CrossRef]
- Hazra, S.; Xiong, S.; Wang, J.; Rippe, R.A.; Krishna, V.; Chatterjee, K.; Tsukamoto, H. Peroxisome Proliferator-activated Receptor γ Induces a Phenotypic Switch from Activated to Quiescent Hepatic Stellate Cells. J. Biol. Chem. 2004, 279, 11392–11401. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Hong, B.; Bian, M.; Jin, H.; Chen, J.; Shao, J.; Zhang, F.; Zheng, S. Docosahexaenoic acid inhibits hepatic stellate cell activation to attenuate liver fibrosis in a PPARγ-dependent manner. Int. Immunopharmacol. 2019, 75, 105816. [Google Scholar] [CrossRef]
- Panebianco, C.; Oben, J.A.; Vinciguerra, M.; Pazienza, V. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: A putative synergy between retinoic acid and PPAR-gamma signalings. Clin. Exp. Med. 2017, 17, 269–280. [Google Scholar] [CrossRef]
- She, H.; Xiong, S.; Hazra, S.; Tsukamoto, H. Adipogenic Transcriptional Regulation of Hepatic Stellate Cells. J. Biol. Chem. 2005, 280, 4959–4967. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, S.; Chu, E.S.; Go, M.Y.; Lau, R.H.; Zhao, J.; Wu, C.-W.; Tong, L.; Zhao, J.; Poon, T.C.; et al. Peroxisome proliferator-activated receptors gamma reverses hepatic nutritional fibrosis in mice and suppresses activation of hepatic stellate cells in vitro. Int. J. Biochem. Cell Biol. 2010, 42, 948–957. [Google Scholar] [CrossRef]
- Deleve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.Z.; Usher, M.G.; Mortensen, R.M. Peroxisome Proliferator-Activated Receptor-γ–Mediated Effects in the Vasculature. Circ. Res. 2008, 102, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Naidenow, J.; Hrgovic, I.; Doll, M.; Hailemariam-Jahn, T.; Lang, V.; Kleemann, J.; Kippenberger, S.; Kaufmann, R.; Zöller, N.; Meissner, M. Peroxisome proliferator-activated receptor (PPAR) α and δ activators induce ICAM-1 expression in quiescent non stimulated endothelial cells. J. Inflamm. 2016, 13, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Morán-Salvador, E.; López-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Clària, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef]
- Wei, Z.; Zhao, D.; Zhang, Y.; Chen, Y.; Zhang, S.; Li, Q.; Zeng, P.; Li, X.; Zhang, W.; Duan, Y.; et al. Rosiglitazone ameliorates bile duct ligation-induced liver fibrosis by down-regulating NF-κB-TNF-α signaling pathway in a PPARγ-dependent manner. Biochem. Biophys. Res. Commun. 2019, 519, 854–860. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.; Riccardi, L.; Morelli, A.; Pruzanski, M.; Pellicciari, R. Cross-Talk between Farnesoid-X-Receptor (FXR) and Peroxisome Proliferator-Activated Receptor γ Contributes to the Antifibrotic Activity of FXR Ligands in Rodent Models of Liver Cirrhosis. J. Pharmacol. Exp. Ther. 2005, 315, 58–68. [Google Scholar] [CrossRef]
- Ko, K.-L.; Mak, L.-Y.; Cheung, K.-S.; Yuen, M.-F. Hepatocellular carcinoma: Recent advances and emerging medical therapies. F1000Research 2020, 9, 620. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Chu, E.S.H.; Zhao, G.; Man, K.; Wu, C.-W.; Cheng, J.T.Y.; Li, G.; Nie, Y.; Lo, C.M.; Teoh, N.; et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. Br. J. Cancer 2012, 106, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Bo, Q.-F.; Sun, X.-M.; Liu, J.; Sui, X.-M.; Li, G.-X. Antitumor action of the peroxisome proliferator-activated receptor-γ agonist rosiglitazone in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 1979–1984. [Google Scholar] [CrossRef] [Green Version]
- Misra, P.; Viswakarma, N.; Reddy, J.K. Peroxisome Proliferator-Activated Receptor-α Signaling in Hepatocarcinogenesis. Subcell. Biochem. 2013, 69, 77–99. [Google Scholar] [CrossRef]
- Xiao, Y.-B.; Cai, S.-H.; Liu, L.-L.; Yang, X.; Yun, J.-P. Decreased expression of peroxisome proliferator-activated receptor alpha indicates unfavorable outcomes in hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Bayha, C.; Klein, K.; Müller, S.; Weiss, T.S.; Schwab, M.; Zanger, U.M. The truncated splice variant of peroxisome proliferator-activated receptor alpha, PPARα-tr, autonomously regulates proliferative and pro-inflammatory genes. BMC Cancer 2015, 15, 488–503. [Google Scholar] [CrossRef] [Green Version]
- Senni, N.; Savall, M.; Granados, D.C.; Alves-Guerra, M.-C.; Sartor, C.; Lagoutte, I.; Gougelet, A.; Terris, B.; Gilgenkrantz, H.; Perret, C.; et al. β-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut 2019, 68, 322–334. [Google Scholar] [CrossRef]
- Vacca, M.; D’Amore, S.; Graziano, G.; D’Orazio, A.; Cariello, M.; Massafra, V.; Salvatore, L.; Martelli, N.; Murzilli, S.; Sasso, G.L.; et al. Clustering Nuclear Receptors in Liver Regeneration Identifies Candidate Modulators of Hepatocyte Proliferation and Hepatocarcinoma. PLoS ONE 2014, 9, e104449. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-J.; Choi, Y.-K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.-G.; Jeon, H.-J.; Kim, J.-H.; Kim, J.-G.; et al. PPARδ Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol. Cancer Res. 2017, 15, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Shen, B.; Chu, E.S.H.; Teoh, N.; Cheung, K.-F.; Wu, C.-W.; Wang, S.; Lam, C.N.Y.; Feng, H.; Zhao, J.; et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology 2010, 51, 2008–2019. [Google Scholar] [CrossRef]
- Zuo, Q.; He, J.; Zhang, S.; Wang, H.; Jin, G.; Jin, H.; Cheng, Z.; Tao, X.; Yu, C.; Li, B.; et al. PPARγ Coactivator-1α Suppresses Metastasis of Hepatocellular Carcinoma by Inhibiting Warburg Effect by PPARγ–Dependent WNT/β-Catenin/Pyruvate Dehydrogenase Kinase Isozyme 1 Axis. Hepatology 2021, 73, 644–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, F.; Quan, Y.; Shen, J. Avicularin ameliorates human hepatocellular carcinoma via the regulation of NF-κB/COX-2/PPAR-γ activities. Mol. Med. Rep. 2019, 19, 5417–5423. [Google Scholar] [CrossRef] [Green Version]
- Saber, S.; Khodir, A.E.; Soliman, W.E.; Salama, M.M.; Abdo, W.S.; Elsaeed, B.; Nader, K.; Abdelnasser, A.; Megahed, N.; Basuony, M.; et al. Telmisartan attenuates N-nitrosodiethylamine-induced hepatocellular carcinoma in mice by modulating the NF-κB-TAK1-ERK1/2 axis in the context of PPARγ agonistic activity. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elbaset, M.; Mansour, A.M.; Ahmed, O.M.; Abo-Youssef, A.M. The potential chemotherapeutic effect of β-ionone and/or sorafenib against hepatocellular carcinoma via its antioxidant effect, PPAR-γ, FOXO-1, Ki-67, Bax, and Bcl-2 signaling pathways. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Dai, W.; Mao, Y.; Wu, L.; Li, J.; Chen, K.; Yu, Q.; Kong, R.; Li, S.; Zhang, J.; et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1α/PPAR-γ/PKM2-mediated glycolysis. J. Exp. Clin. Cancer Res. 2020, 39, 24. [Google Scholar] [CrossRef] [Green Version]
- Maiti, R. Metronomic chemotherapy. J. Pharmacol. Pharmacother. 2014, 5, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, I.; Schulz, U.; Vogelhuber, M.; Wiedmann, K.; Endlicher, E.; Klebl, F.; Andreesen, R.; Herr, W.; Ghibelli, L.; Hackl, C.; et al. Communicative reprogramming non-curative hepatocellular carcinoma with low-dose metronomic chemotherapy, COX-2 inhibitor and PPAR-gamma agonist: A phase II trial. Med. Oncol. 2017, 34, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Hu, J. Formation of Hepatitis B Virus Covalently Closed Circular DNA: Removal of Genome-Linked Protein. J. Virol. 2007, 81, 6164–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turton, K.L.; Meier-Stephenson, V.; Badmalia, M.D.; Coffin, C.S.; Patel, T.R. Host Transcription Factors in Hepatitis B Virus RNA Synthesis. Viruses 2020, 12, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Cheng, X.; Li, Y.; Valdez, K.; Chen, W.; Liang, T.J. Hepatitis B Virus Deregulates the Cell Cycle To Promote Viral Replication and a Premalignant Phenotype. J. Virol. 2018, 92, e00722-18. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Ma, Y.; Liu, M.; Yan, L.; Tang, H. Peroxisome Proliferators Activated Receptor (PPAR) agonists activate hepatitis B virus replication in vivo. Virol. J. 2017, 14, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Li, Y.; Huang, C.; Ying, L.; Xue, J.; Wu, H.; Chen, Z.; Yang, Z. Resveratrol enhances HBV replication through activating Sirt1-PGC-1α-PPARα pathway. Sci. Rep. 2016, 6, 24744–25756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.-L. Metabolic alterations and hepatitis C: From bench to bedside. World J. Gastroenterol. 2016, 22, 1461–1476. [Google Scholar] [CrossRef]
- Shirasaki, T.; Honda, M.; Shimakami, T.; Horii, R.; Yamashita, T.; Sakai, Y.; Sakai, A.; Okada, H.; Watanabe, R.; Murakami, S.; et al. MicroRNA-27a Regulates Lipid Metabolism and Inhibits Hepatitis C Virus Replication in Human Hepatoma Cells. J. Virol. 2013, 87, 5270–5286. [Google Scholar] [CrossRef] [Green Version]
- Portius, D.; Sobolewski, C.; Foti, M. MicroRNAs-Dependent Regulation of PPARs in Metabolic Diseases and Cancers. PPAR Res. 2017, 2017, 7058424. [Google Scholar] [CrossRef] [Green Version]
- Moss, H.B. The Impact of Alcohol on Society: A Brief Overview. Soc. Work. Public Health 2013, 28, 175–177. [Google Scholar] [CrossRef]
- World Health Organization. Global Status Report on Alcohol and Health 2018; Poznyak, V., Rekve, D., Eds.; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9. [Google Scholar]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Liu, J. Ethanol and liver: Recent insights into the mechanisms of ethanol-induced fatty liver. World J. Gastroenterol. 2014, 20, 14672–14685. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.-Z.; Chandimali, N.; Han, Y.-H.; Lee, D.S.; Kim, J.-S.; Kim, S.-U.; Kim, T.-D.; Jeong, D.K.; Sun, H.-N.; Kwon, T. Pathogenesis, Early Diagnosis, and Therapeutic Management of Alcoholic Liver Disease. Int. J. Mol. Sci. 2019, 20, 2712. [Google Scholar] [CrossRef] [Green Version]
- Stickel, F.; Datz, C.; Hampe, J.; Bataller, R. Pathophysiology and Management of Alcoholic Liver Disease: Update 2016. Gut Liver 2017, 11, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Gitto, S.; Micco, L.; Conti, F.; Andreone, P.; Bernardi, M. Alcohol and viral hepatitis: A mini-review. Dig. Liver Dis. 2009, 41, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Salameh, H.; Raff, E.; Erwin, A.; Seth, D.; Nischalke, H.D.; Falleti, E.; Burza, M.A.; Leathert, J.; Romeo, S.; Molinaro, A.; et al. PNPLA3 Gene Polymorphism Is Associated with Predisposition to and Severity of Alcoholic Liver Disease. Am. J. Gastroenterol. 2015, 110, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucher, S.S.; Stickel, F.; Trépo, E.; Way, M.M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.M.; Rosendahl, J.J.; Berg, T.; Ridinger, M.M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic Liver Disease: Pathogenesis and New Therapeutic Targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol. Res. 2017, 38, 147–161. [Google Scholar]
- Donohue, T.D., Jr. Alcohol-induced steatosis in liver cells. World J. Gastroenterol. 2007, 13, 4974–4978. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Gao, B.; Song, B.-J. Molecular Mechanisms of Alcoholic Fatty Liver. Alcohol. Clin. Exp. Res. 2009, 33, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Galli, A.; Pinaire, J.; Fischer, M.; Dorris, R.; Crabb, D.W. The Transcriptional and DNA Binding Activity of Peroxisome Proliferator-activated Receptor α Is Inhibited by Ethanol Metabolism. A novel mechanism for the de-velopment of ethanol-induced fatty liver. J. Biol. Chem. 2001, 276, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.-G.; Zhang, X.-N.; Liu, S.-X.; Wang, Y.-R.; Zeng, T. Roles of peroxisome proliferator-activated receptor α in the pathogenesis of ethanol-induced liver disease. Chem. Biol. Interact. 2020, 327, 109176. [Google Scholar] [CrossRef]
- Nakajima, T.; Kamijo, Y.; Tanaka, N.; Sugiyama, E.; Tanaka, E.; Kiyosawa, K.; Fukushima, Y.; Peters, J.M.; Gonzalez, F.J.; Aoyama, T. Peroxisome proliferator-activated receptor? protects against alcohol-induced liver damage. Hepatology 2004, 40, 972–980. [Google Scholar] [CrossRef]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome Proliferator-activated Receptor α (PPARα) Agonist Treatment Reverses PPARα Dysfunction and Abnormalities in Hepatic Lipid Metabolism in Ethanol-fed Mice. J. Biol. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Ren, W.; Li, W.; Zhao, S.; Mi, H.; Wang, R.; Zhang, Y.; Wu, W.; Nan, Y.; Yu, J. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol induced steatohepatitis in mice. Lipids Health Dis. 2011, 10, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Wo, L.; Du, Z.; Tang, L.; Song, Z.; Dou, X. Danshen protects against early-stage alcoholic liver disease in mice via inducing PPARα activation and subsequent 4-HNE degradation. PLoS ONE 2017, 12, e0186357. [Google Scholar] [CrossRef]
- Hsu, J.-Y.; Lin, H.-H.; Hsu, C.-C.; Chen, B.-C.; Chen, J.-H. Aqueous Extract of Pepino (Solanum muriactum Ait) Leaves Ameliorate Lipid Accumulation and Oxidative Stress in Alcoholic Fatty Liver Disease. Nutrients 2018, 10, 931. [Google Scholar] [CrossRef] [Green Version]
- Blednov, Y.A.; Black, M.; Benavidez, J.M.; Stamatakis, E.E.; Harris, R.A. PPAR Agonists: II. Fenofibrate and Tesaglitazar Alter Behaviors Related to Voluntary Alcohol Consumption. Alcohol. Clin. Exp. Res. 2016, 40, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, L.B.; Most, D.; Blednov, Y.A.; Harris, R.A. PPAR agonists regulate brain gene expression: Relationship to their effects on ethanol consumption. Neuropharmacology 2014, 86, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, D.; Rivera-Meza, M.; Flores-Bastías, O.; Quintanilla, M.E.; Karahanian, E. Fenofibrate-a PPARα agonist-increases alcohol dehydrogenase levels in the liver: Implications for its possible use as an ethanol-aversive drug. Adicciones 2020, 32, 208–215. [Google Scholar] [CrossRef]
- Le Foll, B.; Di Ciano, P.; Panlilio, L.V.; Goldberg, S.R.; Ciccocioppo, R. Peroxisome proliferator-activated receptor (PPAR) agonists as promising new medications for drug addiction: Preclinical evidence. Curr. Drug Targets 2013, 14, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, D.M.; Jang, J.Y.; Kim, H.-J.; Han, B.W. Differential Effects of Cancer-Associated Mutations Enriched in Helix H3 of PPARγ. Cancers 2020, 12, 3580. [Google Scholar] [CrossRef]
- Issemann, I.; Prince, R.A.; Tugwood, J.D.; Green, S. The peroxisome proliferator-activated receptor:retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J. Mol. Endocrinol. 1993, 11, 37–47. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- Kosgei, V.J.; Coelho, D.; Guéant-Rodriguez, R.-M.; Guéant, J.-L. Sirt1-PPARS Cross-Talk in Complex Metabolic Diseases and Inherited Disorders of the One Carbon Metabolism. Cells 2020, 9, 1882. [Google Scholar] [CrossRef]
- Hajri, T.; Zaiou, M.; Fungwe, T.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Peyrou, M.; Ramadori, P.; Bourgoin, L.; Foti, M. PPARs in Liver Diseases and Cancer: Epigenetic Regulation by MicroRNAs. PPAR Res. 2012, 2012, 757803. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| PPAR Subtype | PPARα | PPAR β/δ | PPARγ |
|---|---|---|---|
| Gene location | Human chromosome 22q12.2–13.1 | Human chromosome 6p21.1–21.2 | Human chromosome 3p25 |
| Isoforms | None | None | PPARγ1, PPARγ2, PPARγ3 |
| Tissue distribution | Liver, heart, skeletal muscle (tissues with high fatty acid oxidation rates); brown adipose tissue, kidney, adrenal gland. | Liver, kidney, skeletal and cardiac muscle, adipose tissue, brain, colon, vasculature, esophagus, gut. Ubiquitous. | Mainly in adipose tissue (white and brown). Other tissues such as liver, gut, kidney, retina, immunologic system, muscles, spleen, urinary bladder, heart, lung, brain, vasculature. |
| Endogenous Ligands | Unsaturated and saturated fatty acids and their derivatives (8-S-hydroxyeicosatetraenoic acid, arachidonic acid lipoxygenase metabolite LTB4, arachidonate monooxygenase metabolite epoxyeicosatrienoic acids), leukotriene derivatives, VLDL hydrolysis products. | Unsaturated fatty acids, arachidonic acid cyclooxygenase metabolite prostacyclin, the linoleic acid 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid, carbaprostacyclin, components of VLDL. | Polyunsaturated fatty acids, prostanoids (15-deoxy-Δ12, 14-prostaglandin J2 (15-dPGJ2)), 13-hydroxyeicosatetraenoic acid, components of oxidized LDL, eicosanoids, oxidized alkyl phospholipids. |
| Functions | Major regulator of the mitochondrial and peroxisomal β-oxidation (fatty acid metabolism), lowers lipid levels, anti-inflammatory activities. | Increase lipid catabolism, improves the plasma HDL-cholesterol levels and insulin resistance, induce cell proliferation and differentiation, anti-inflammatory activities. | Regulate adipocyte differentiation, lipid storage, and glucose metabolism (improves insulin sensitivity), main regulator of metabolic genes, increase fatty acid oxidation, HDL and uncoupling protein, decrease triglycerides, improves vascular integrity, energy balance, anti-inflammatory activities. |
| Target genes | CYP8B1, FATP, FAT/CD36, liver cytosolic FABP, LPL. Lipid/hormone transport genes. (LEPR, SLC27A2, SLC27A4). Acyl-CoA metabolism. (ACOT12, ACSL3, ACSL3, ACSL5, ACSL1, ACSM3, FABP1, FABP3). β-oxidation. (ACAA2, ACADM, ACADS, ACADVL, CPT1A, CPT2, ETFDH, HADHA, HADHB, SLC25A20, SLC22A5, TXNIP). Ketogenesis/ketolysis genes. (FGF21, HMGCS2), Peroxisomal β-oxidation (ABCD2, ABCD3, ACAA1A, ACOX1, ECH1, HSD17B4). Lipogenesis. (ACACB, AGPAT2, ELOVL5, ELOVL6 FADS1, GPAM, MLYCD, MOD1). Lipases and lipid droplet proteins. (ADFP, CIDEC, PNPLA2, S3-12). Lipoprotein metabolism. (ANGPTL4, APOA1, APOA2, APOA5, APOCIII, LIPC, PCTP, VLDLR). Cholesterol and bile metabolism. (ABCA1, ABCB4, CYP7A1, FXR, LXR) | Genes related with lipid uptake, represses genes that participated in lipid metabolism and efflux. LPL, PGAR, IDK, PDK-1, Ubiquitin C, CPT1, AOX, LCAD, UCP1, UCP3, PGC1-alpha. Tumor angiogenesis (Pdgfrβ, Pdgfb, c-kit) | AP2, CAP, IRS2, GLUT4, GLUT2, adiponectin, ACS, PCK2, LPL, FAT/CD36, FABP, GYK fatty acid transport, acyl-CoA synthetase, glucokinase, leptin, perilipin, GK PEPCK, UCP 1, UCP-2, UCP-3, LXR-alpha, TNF-alpha, IL-6. |
| References | [8,10,11,12,13,14,15] | [8,10,11,12,13,14,16,17] | [8,10,11,12,13,14,17,18,19,20] |
| Liver Disease | Expression | Function | Mutation | Reference | |
|---|---|---|---|---|---|
| MAFLD | Hepatocytes Kupffer Cells Hepatic Stellate Cells | ↓ PPARα ↑ PARβ/δ ↑ PPARγ ↑ PARβ/δ ↑ PPARγ | PPARα: Induces lipogenesis PPARβ/δ: Augments liver fat content and decreases insulin sensitivity PPARγ: Promotes steatosis | PPARA: CM003689 association with elevated plasma lipid concentration in diabetes CM025499 CM025500 associated with diabetes PPARG: CM981614, CM981615, CM1617313 associated with Obesity CM066185, CM066187, CM066186, CM066188, CD066392, CX022192 Associated with IR CR032439 association with increased height/lipid metabolism CR057908 association with increased body weight | [25,47,62] The Human Gene Mutation Database, consulted July 2021 |
| Fibrosis | Kupffer Cells Hepatic Stellate Cells (activated) | ↓ PPARα ↑ PPARβ/δ ↓ PPARγ | PPARα: Increases oxidative stress and inflammation PPARβ/δ: Facilitates HSC activation PPARγ: key factor in HSCs activation and regulation of inflammation | No mutations associated with liver fibrosis | [73,74,90] |
| Hepatocellular carcinoma | Hepatocytes | ↑ PPARα ↑ PARβ/δ ↓↑PPARγ | PPARα: Regulates expression of B-catenin, c-Fos, c-Myc, Cyclin D1. PPARγ: Modulates activity of p53, ERK1/2, TAK1 and NF-κB | PPARG: R280C, C285Y, Q286P, F287Y, R288C, R288H S289C mutations are potential loss offunction mutations in various aspects including ligand binding for PPARγ activation | [104,105,106,147] |
| HBV and HCV infections | Hepatocytes | ↑ PPARα ↓ PPARγ | PPARα: Increase fatty acid Synthesis PPARγ: Decrease β-oxidation | No mutations associated with HBV, or HCV infection | [116,117,118,119] |
| Alcoholic liver disease | Hepatocytes | ↑ PPARα | PPARα: Increase fatty acid Synthesis Decrease β-oxidation | No mutations associated with alcoholic liver disease | [134,135,136,137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monroy-Ramirez, H.C.; Galicia-Moreno, M.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Santos, A.; Armendariz-Borunda, J. PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases. Int. J. Mol. Sci. 2021, 22, 8298. https://doi.org/10.3390/ijms22158298
Monroy-Ramirez HC, Galicia-Moreno M, Sandoval-Rodriguez A, Meza-Rios A, Santos A, Armendariz-Borunda J. PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases. International Journal of Molecular Sciences. 2021; 22(15):8298. https://doi.org/10.3390/ijms22158298
Chicago/Turabian StyleMonroy-Ramirez, Hugo Christian, Marina Galicia-Moreno, Ana Sandoval-Rodriguez, Alejandra Meza-Rios, Arturo Santos, and Juan Armendariz-Borunda. 2021. "PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases" International Journal of Molecular Sciences 22, no. 15: 8298. https://doi.org/10.3390/ijms22158298
APA StyleMonroy-Ramirez, H. C., Galicia-Moreno, M., Sandoval-Rodriguez, A., Meza-Rios, A., Santos, A., & Armendariz-Borunda, J. (2021). PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases. International Journal of Molecular Sciences, 22(15), 8298. https://doi.org/10.3390/ijms22158298