
Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment


Abstract
1. Introduction
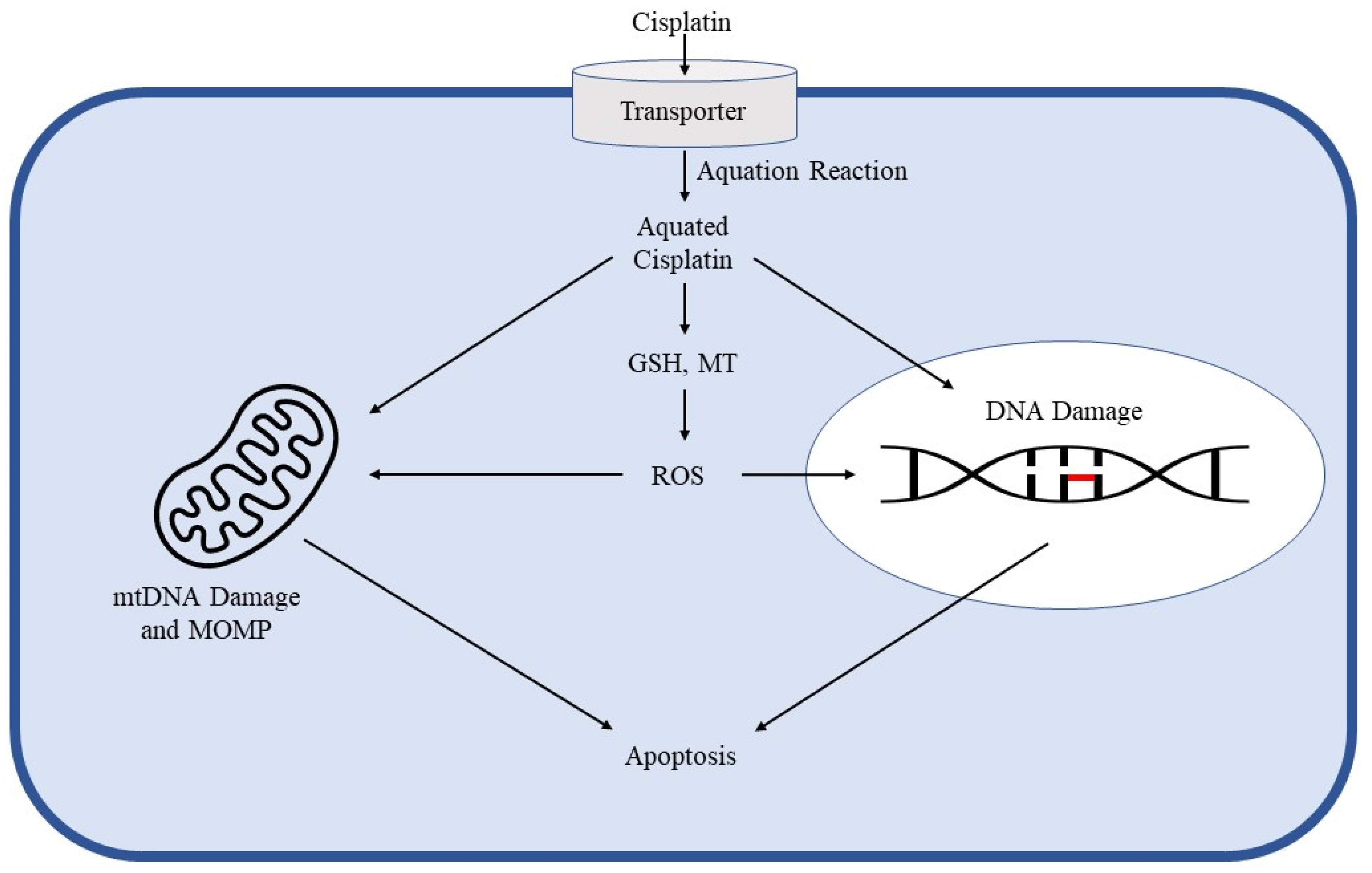
2. Cisplatin Mechanism of Action in Cancer Control and Toxicity
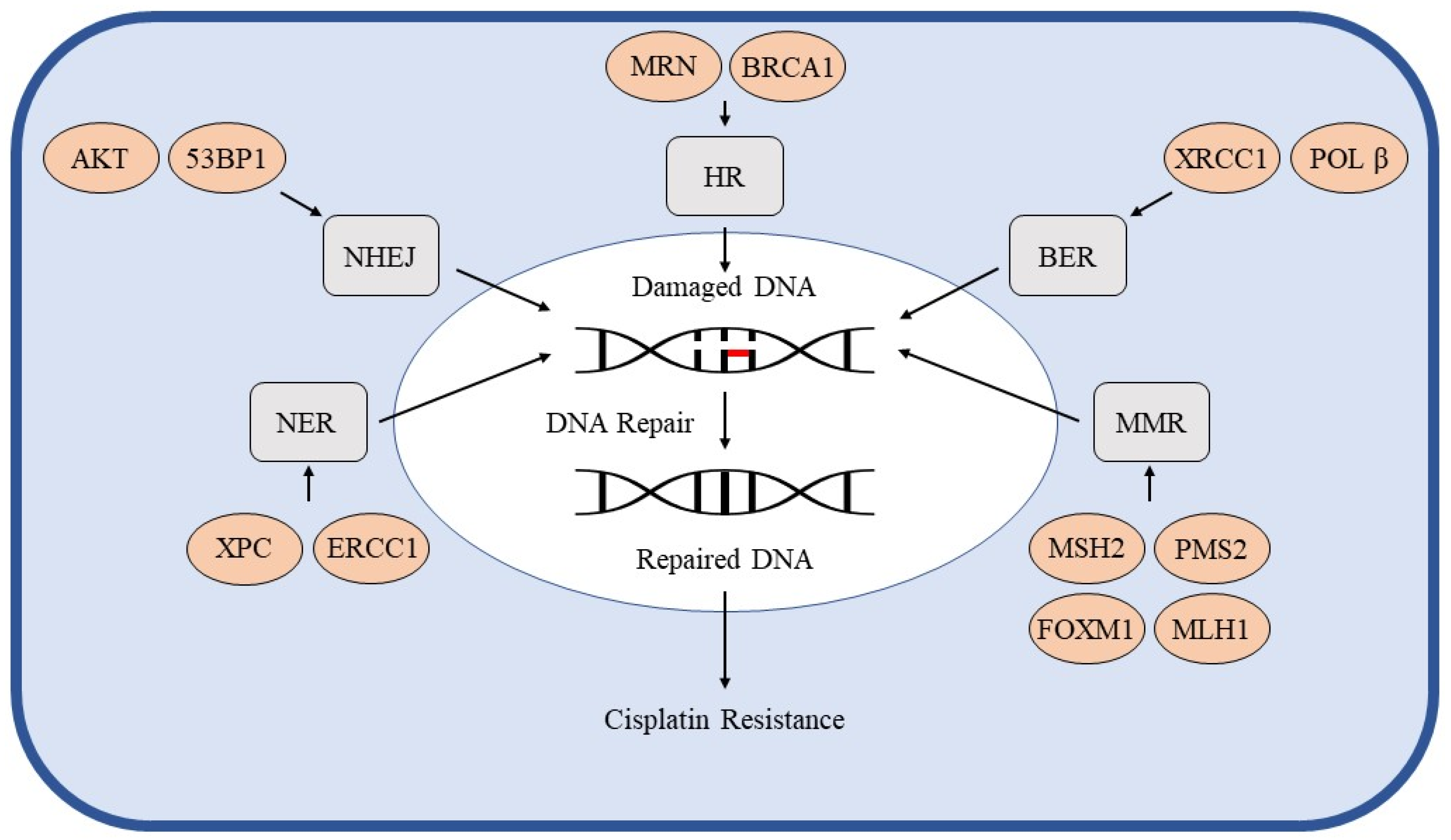
3. DNA Repair Pathways
3.1. Homologous Recombination
3.2. Non-Homologous End Joining
3.3. Nucleotide Excision Repair
3.4. Base Excision Repair
3.5. Mismatch Repair
4. Predictive Markers of Cisplatin Resistance
4.1. ERCC1
4.2. MDR1, MRP1, and β-Catenin
4.3. c-IAP1, XIAP, Apollon, and Livin
4.4. EGFR/FAK/NF-kB Activation
4.5. BRCA1
4.6. Genomic Analysis
5. Current Treatment Strategies and Future Perspectives
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Balayssac, D.; Ferrier, J.; Descoeur, J.; Ling, B.; Pezet, D.; Eschalier, A.; Authier, N. Chemotherapy-induced peripheral neuropathies: From clinical relevance to preclinical evidence. Expert Opin. Drug Saf. 2011, 10, 407–417. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Sharma, V.M.; Wilson, W.R. Radiosensitization of advanced squamous cell carcinoma of the head and neck with cisplatin during concomitant radiation therapy. Eur. Arch. Otorhinolaryngol. 1999, 256, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Gutiérrez, G.; Sereno, M.; Miralles, A.; Casado-Sáenz, E.; Gutiérrez-Rivas, E. Chemotherapy-induced peripheral neuropathy: Clinical features, diagnosis, prevention and treatment strategies. Clin. Transl. Oncol. 2010, 12, 81–91. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; Imperatori, L.; Stefanini, M.; D’Incalci, M. Sensitivity of CHO mutant cell lines with specific defects in nucleotide excision repair to different anti-cancer agents. Int. J. Cancer 1996, 66, 779–783. [Google Scholar] [CrossRef]
- Damia, G.; D’Incalci, M. Targeting DNA repair as a promising approach in cancer therapy. Eur. J. Cancer 2007, 43, 1791–1801. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Traganos, F.; Wlodkowic, D. Impaired DNA damage response—An Achilles’ heel sensitizing cancer to chemotherapy and radiotherapy. Eur. J. Pharmacol. 2009, 625, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.H.; Li, J.; Chen, P.; Jiang, H.G.; Wu, M.; Chen, Y.C. RNA interferences targeting the Fanconi anemia/BRCA pathway upstream genes reverse cisplatin resistance in drug-resistant lung cancer cells. J. Biomed. Sci. 2015, 22, 77. [Google Scholar] [CrossRef]
- Liu, J.J.; Kim, Y.; Yan, F.; Ding, Q.; Ip, V.; Jong, N.N.; Mercer, J.F.; McKeage, M.J. Contributions of rat Ctr1 to the uptake and toxicity of copper and platinum anticancer drugs in dorsal root ganglion neurons. Biochem. Pharmacol. 2013, 85, 207–215. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Lutter, D.; Planells-Cases, R.; Ullrich, F.; Voss, F.K. VRAC: Molecular identification as LRRC8 heteromers with differential functions. Pflugers Arch. 2016, 468, 385–393. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef] [PubMed]
- Wilke, B.U.; Kummer, K.K.; Leitner, M.G.; Kress, M. Chloride—The Underrated Ion in Nociceptors. Front. Neurosci. 2020, 14, 287. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Leblanc, A.F.; Gibson, A.A.; Hong, K.W.; Kim, J.Y.; Janke, L.J.; Li, L.; Vasilyeva, A.; Finkelstein, D.B.; Sprowl, J.A.; et al. Identification of OAT1/OAT3 as Contributors to Cisplatin Toxicity. Clin. Transl. Sci. 2017, 10, 412–420. [Google Scholar] [CrossRef] [PubMed]
- EL-Arabey, A.A. Dual function of OCT2 and MATE1 in cisplatin induced nephrotoxicity. Pharmacol. Res. 2017, 119, 493. [Google Scholar] [CrossRef] [PubMed]
- EL-Arabey, A.A.; Abdalla, M. New Insight of OCT2 Regulation as Mediator for Cisplatin- Induced Nephrotoxicity. Asian Pac. J. Cancer Prev. 2017, 18, 1459–1460. [Google Scholar] [PubMed]
- Sprowl, J.A.; Ciarimboli, G.; Lancaster, C.S.; Giovinazzo, H.; Gibson, A.A.; Du, G.; Janke, L.J.; Cavaletti, G.; Shields, A.F.; Sparreboom, A. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. USA 2013, 110, 11199–11204. [Google Scholar] [CrossRef]
- Kanno, Y.; Chen, C.Y.; Lee, H.L.; Chiou, J.F.; Chen, Y.J. Molecular Mechanisms of Chemotherapy Resistance in Head and Neck Cancers. Front. Oncol. 2021, 11, 640392. [Google Scholar] [CrossRef] [PubMed]
- Boulikas, T.; Vougiouka, M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol. Rep. 2003, 10, 1663–1682. [Google Scholar] [CrossRef]
- Englander, E.W. DNA damage response in peripheral nervous system: Coping with cancer therapy-induced DNA lesions. DNA Repair 2013, 12, 685–690. [Google Scholar] [CrossRef]
- McDonald, E.S.; Randon, K.R.; Knight, A.; Windebank, A.J. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: A potential mechanism for neurotoxicity. Neurobiol. Dis. 2005, 18, 305–313. [Google Scholar] [CrossRef]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Lempiäinen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef]
- Ryan, C.J.; Bajrami, I.; Lord, C.J. Synthetic Lethality and Cancer—Penetrance as the Major Barrier. Trends Cancer 2018, 4, 671–683. [Google Scholar] [CrossRef]
- Kanaar, R.; Hoeijmakers, J.H.; van Gent, D.C. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998, 8, 483–489. [Google Scholar] [CrossRef]
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal 2014, 21, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Yarden, R.I.; Metsuyanim, S.; Pickholtz, I.; Shabbeer, S.; Tellio, H.; Papa, M.Z. BRCA1-dependent Chk1 phosphorylation triggers partial chromatin disassociation of phosphorylated Chk1 and facilitates S-phase cell cycle arrest. Int. J. Biochem. Cell Biol. 2012, 44, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011, 102, 663–669. [Google Scholar] [CrossRef]
- Tran, H.M.; Shi, G.; Li, G.; Carney, J.P.; O’Malley, B.; Li, D. Mutant Nbs1 enhances cisplatin-induced DNA damage and cytotoxicity in head and neck cancer. Otolaryngol. Head Neck Surg. 2004, 131, 477–484. [Google Scholar] [CrossRef]
- Altan, B.; Yokobori, T.; Ide, M.; Bai, T.; Yanoma, T.; Kimura, A.; Kogure, N.; Suzuki, M.; Bao, P.; Mochiki, E.; et al. High Expression of MRE11-RAD50-NBS1 Is Associated with Poor Prognosis and Chemoresistance in Gastric Cancer. Anticancer Res. 2016, 36, 5237–5247. [Google Scholar] [PubMed]
- Abuzeid, W.M.; Jiang, X.; Shi, G.; Wang, H.; Paulson, D.; Araki, K.; Jungreis, D.; Carney, J.; O’Malley, B.W., Jr.; Li, D. Molecular disruption of RAD50 sensitizes human tumor cells to cisplatin-based chemotherapy. J. Clin. Investig. 2009, 119, 1974–1985. [Google Scholar] [CrossRef]
- Araki, K.; Yamashita, T.; Reddy, N.; Wang, H.; Abuzeid, W.M.; Khan, K.; O’Malley, B.W., Jr.; Li, D. Molecular disruption of NBS1 with targeted gene delivery enhances chemosensitisation in head and neck cancer. Br. J. Cancer 2010, 103, 1822–1830. [Google Scholar] [CrossRef]
- O’Malley, B.W., Jr.; Li, D.; Carney, J.; Rhee, J.; Suntharalingam, M. Molecular disruption of the MRN(95) complex induces radiation sensitivity in head and neck cancer. Laryngoscope 2003, 113, 1588–1594. [Google Scholar] [CrossRef]
- Rhee, J.G.; Li, D.; Suntharalingam, M.; Guo, C.; O’Malley, B.W., Jr.; Carney, J.P. Radiosensitization of head/neck squamous cell carcinoma by adenovirus-mediated expression of the Nbs1 protein. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 273–278. [Google Scholar] [CrossRef]
- Shen, B.; Huang, D.; Ramsey, A.J.; Ig-Izevbekhai, K.; Zhang, K.; Lajud, S.A.; O’Malley, B.W.; Li, D. PD-L1 and MRN synergy in platinum-based chemoresistance of head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 640–647. [Google Scholar] [CrossRef]
- Shen, M.; Xu, Z.; Xu, W.; Jiang, K.; Zhang, F.; Ding, Q.; Xu, Z.; Chen, Y. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 149. [Google Scholar] [CrossRef]
- Lee, J.O.; Kang, M.J.; Byun, W.S.; Kim, S.A.; Seo, I.H.; Han, J.A.; Moon, J.W.; Kim, J.H.; Kim, S.J.; Lee, E.J.; et al. Metformin overcomes resistance to cisplatin in triple-negative breast cancer (TNBC) cells by targeting RAD51. Breast Cancer Res. 2019, 21, 115. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Pantelias, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20. [Google Scholar] [CrossRef]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Chan, D.W.; Chen, B.P.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef]
- Katsube, T.; Mori, M.; Tsuji, H.; Shiomi, T.; Shiomi, N.; Onoda, M. Differences in sensitivity to DNA-damaging Agents between XRCC4- and Artemis-deficient human cells. J. Radiat. Res. 2011, 52, 415–424. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef]
- Stronach, E.A.; Chen, M.; Maginn, E.N.; Agarwal, R.; Mills, G.B.; Wasan, H.; Gabra, H. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011, 13, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Li, X.; Zhao, Y.; Yang, Q.; Kong, B. 53BP1 inhibits the migration and regulates the chemotherapy resistance of ovarian cancer cells. Oncol. Lett. 2018, 15, 9917–9922. [Google Scholar] [CrossRef]
- Pavón, M.A.; Parreño, M.; León, X.; Sancho, F.J.; Céspedes, M.V.; Casanova, I.; Lopez-Pousa, A.; Mangues, M.A.; Quer, M.; Barnadas, A.; et al. Ku70 predicts response and primary tumor recurrence after therapy in locally advanced head and neck cancer. Int. J. Cancer 2008, 123, 1068–1079. [Google Scholar] [CrossRef]
- Acklin, S.; Xia, F. The Role of Nucleotide Excision Repair in Cisplatin-Induced Peripheral Neuropathy: Mechanism, Prevention, and Treatment. Int. J. Mol. Sci. 2021, 22, 1975. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Kanat, O.; Ertas, H.; Caner, B. Platinum-induced neurotoxicity: A review of possible mechanisms. World J. Clin. Oncol. 2017, 8, 329–335. [Google Scholar] [CrossRef]
- Duan, M.; Ulibarri, J.; Liu, K.J.; Mao, P. Role of Nucleotide Excision Repair in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 9248. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, J.; Meng, Y.; Qu, C.; Shen, F.; Xu, L. Overexpression of xeroderma pigmentosum group C decreases the chemotherapeutic sensitivity of colorectal carcinoma cells to cisplatin. Oncol. Lett. 2018, 15, 6336–6344. [Google Scholar]
- Pajuelo-Lozano, N.; Bargiela-Iparraguirre, J.; Dominguez, G.; Quiroga, A.G.; Perona, R.; Sanchez-Perez, I. XPA, XPC, and XPD Modulate Sensitivity in Gastric Cisplatin Resistance Cancer Cells. Front. Pharmacol. 2018, 9, 1197. [Google Scholar] [CrossRef]
- Teng, X.; Fan, X.F.; Li, Q.; Liu, S.; Wu, D.Y.; Wang, S.Y.; Shi, Y.; Dong, M. XPC inhibition rescues cisplatin resistance via the Akt/mTOR signaling pathway in A549/DDP lung adenocarcinoma cells. Oncol. Rep. 2019, 41, 1875–1882. [Google Scholar] [CrossRef]
- Köberle, B.; Masters, J.R.; Hartley, J.A.; Wood, R.D. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol. 1999, 9, 273–276. [Google Scholar] [CrossRef]
- Chen, H.Y.; Shao, C.J.; Chen, F.R.; Kwan, A.L.; Chen, Z.P. Role of ERCC1 promoter hypermethylation in drug resistance to cisplatin in human gliomas. Int. J. Cancer 2010, 126, 1944–1954. [Google Scholar] [CrossRef]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair 2010, 9, 745–753. [Google Scholar] [CrossRef]
- Sabatino, M.A.; Marabese, M.; Ganzinelli, M.; Caiola, E.; Geroni, C.; Broggini, M. Down-regulation of the nucleotide excision repair gene XPG as a new mechanism of drug resistance in human and murine cancer cells. Mol. Cancer 2010, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Im, J.; Nho, R.S. Fibroblasts from patients with idiopathic pulmonary fibrosis are resistant to cisplatin-induced cell death via enhanced CK2-dependent XRCC1 activity. Apoptosis 2019, 24, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Zhang, L.; Zhong, Z. Cytoplasmic APE1 promotes resistance response in osteosarcoma patients with cisplatin treatment. Cell Biochem. Funct. 2020, 38, 195–203. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Wu, L.; Dong, Y.; Zhang, J.; Chen, F.; Xie, W.; Huang, J.; Lu, N. Apurinic endonuclease 1 promotes the cisplatin resistance of lung cancer cells by inducing Parkin-mediated mitophagy. Oncol. Rep. 2019, 42, 2245–2254. [Google Scholar] [CrossRef]
- Nemec, A.A.; Abriola, L.; Merkel, J.S.; de Stanchina, E.; DeVeaux, M.; Zelterman, D.; Glazer, P.M.; Sweasy, J.B. DNA Polymerase Beta Germline Variant Confers Cellular Response to Cisplatin Therapy. Mol. Cancer Res. 2017, 15, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef]
- Li, G.M. New insights and challenges in mismatch repair: Getting over the chromatin hurdle. DNA Repair 2014, 19, 48–54. [Google Scholar] [CrossRef]
- Fink, D.; Nebel, S.; Aebi, S.; Nehme, A.; Howell, S. Loss of DNA mismatch repair due to knockout of MSH2 or PMS2 results in resistance to cisplatin and carboplatin. Int. J. Oncol. 1997, 11, 539–542. [Google Scholar] [CrossRef]
- Takahashi, M.; Koi, M.; Balaguer, F.; Boland, C.R.; Goel, A. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J. Biol. Chem. 2011, 286, 12157–12165. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Wang, Y.; Peng, J.; Fang, F.; Yang, X. MLH1 enhances the sensitivity of human endometrial carcinoma cells to cisplatin by activating the MLH1/c-Abl apoptosis signaling pathway. BMC Cancer 2018, 18, 1294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, Y.; Wang, Y.; Yin, X.; He, Y.; Chen, L.; Wang, W.; Liu, T.; Di, W. FOXM1 modulates cisplatin sensitivity by regulating EXO1 in ovarian cancer. PLoS ONE 2014, 9, e96989. [Google Scholar]
- Asada, K.; Kaneko, S.; Takasawa, K.; Machino, H.; Takahashi, S.; Shinkai, N.; Shimoyama, R.; Komatsu, M.; Hamamoto, R. Integrated Analysis of Whole Genome and Epigenome Data Using Machine Learning Technology: Toward the Establishment of Precision Oncology. Front. Oncol. 2021, 11, 666937. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.P.; Harper, A.; Malcolm, J.; McAndrews, M.S.; Mockus, S.M.; Patterson, S.E.; Reynolds, T.; Baker, E.J.; Bult, C.J.; Chesler, E.J.; et al. Cisplatin-resistant triple-negative breast cancer subtypes: Multiple mechanisms of resistance. BMC Cancer 2019, 19, 1039. [Google Scholar] [CrossRef]
- Koutsoukos, K.; Andrikopoulou, A.; Dedes, N.; Zagouri, F.; Bamias, A.; Dimopoulos, M.A. Clinical Perspectives of ERCC1 in Bladder Cancer. Int. J. Mol. Sci. 2020, 21, 8829. [Google Scholar] [CrossRef]
- Pan, C.H.; Chen, S.Y.; Wang, J.Y.; Tsao, S.P.; Huang, H.Y.; Wei-Chen Chiu, P.; Wu, C.H. Sclareol ameliorated ERCC1-mediated cisplatin resistance in A549 human lung adenocarcinoma cells and a murine xenograft tumor model by suppressing AKT-GSK3β-AP1/Snail and JNK-AP1 pathways. Chem. Biol. Interact. 2020, 332, 109304. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hua, R.X.; Jiang, J.; Zhao, L.Q.; Sun, X.; Luan, J.; Lang, Y.; Sun, Y.; Shang, K.; Peng, S.; et al. Association studies of ERCC1 polymorphisms with lung cancer susceptibility: A systematic review and meta-analysis. PLoS ONE 2014, 9, e97616. [Google Scholar] [CrossRef]
- Chiu, T.J.; Chen, C.H.; Chien, C.Y.; Li, S.H.; Tsai, H.T.; Chen, Y.J. High ERCC1 expression predicts cisplatin-based chemotherapy resistance and poor outcome in unresectable squamous cell carcinoma of head and neck in a betel-chewing area. J. Transl. Med. 2011, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Piljić Burazer, M.; Mladinov, S.; Matana, A.; Kuret, S.; Bezić, J.; Glavina Durdov, M. Low ERCC1 expression is a good predictive marker in lung adenocarcinoma patients receiving chemotherapy based on platinum in all TNM stages—A single-center study. Diagn. Pathol. 2019, 14, 105. [Google Scholar] [CrossRef]
- Warta, R.; Theile, D.; Mogler, C.; Herpel, E.; Grabe, N.; Lahrmann, B.; Plinkert, P.K.; Herold-Mende, C.; Weiss, J.; Dyckhoff, G. Association of drug transporter expression with mortality and progression-free survival in stage IV head and neck squamous cell carcinoma. PLoS ONE 2014, 9, e108908. [Google Scholar] [CrossRef]
- Li, L.; Liu, H.C.; Wang, C.; Liu, X.; Hu, F.C.; Xie, N.; Lü, L.; Chen, X.; Huang, H.Z. Overexpression of β-Catenin Induces Cisplatin Resistance in Oral Squamous Cell Carcinoma. BioMed Res. Int. 2016, 2016, 5378567. [Google Scholar] [CrossRef]
- Tanimoto, T.; Tsuda, H.; Imazeki, N.; Ohno, Y.; Imoto, I.; Inazawa, J.; Matsubara, O. Nuclear expression of cIAP-1, an apoptosis inhibiting protein, predicts lymph node metastasis and poor patient prognosis in head and neck squamous cell carcinomas. Cancer Lett. 2005, 224, 141–151. [Google Scholar] [CrossRef]
- Yang, X.H.; Feng, Z.E.; Yan, M.; Hanada, S.; Zuo, H.; Yang, C.Z.; Han, Z.G.; Guo, W.; Chen, W.T.; Zhang, P. XIAP is a predictor of cisplatin-based chemotherapy response and prognosis for patients with advanced head and neck cancer. PLoS ONE 2012, 7, e31601. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, B.L.; Zhou, Y.W.; Guo, R.W.; Shuai, M.T.; Zeng, J.X.; Leng, A.M. Expression and clinical significance of Apollon in esophageal squamous cell carcinoma. Mol. Med. Rep. 2016, 14, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.M.; Kim, S.A.; Lee, D.H.; Lee, J.K.; Park, Y.L.; Lee, K.H.; Chung, I.J.; Joo, Y.E.; Lim, S.C. Livin enhances chemoresistance in head and neck squamous cell carcinoma. Oncol. Rep. 2017, 37, 3667–3673. [Google Scholar] [CrossRef] [PubMed]
- Kuang, C.M.; Fu, X.; Hua, Y.J.; Shuai, W.D.; Ye, Z.H.; Li, Y.; Peng, Q.H.; Li, Y.Z.; Chen, S.; Qian, C.N.; et al. BST2 confers cisplatin resistance via NF-κB signaling in nasopharyngeal cancer. Cell Death Dis. 2017, 8, e2874. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Lee, J.W.; Slebos, R.J.; Howard, J.D.; Perez, J.; Kang, H.; Fertig, E.J.; Considine, M.; Gilbert, J.; Murphy, B.A.; et al. A 3′-UTR KRAS-variant is associated with cisplatin resistance in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2014, 25, 2230–2236. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.S.; Xia, F. BRCA1 16 years later: DNA damage-induced BRCA1 shuttling. FEBS J. 2010, 277, 3079–3085. [Google Scholar] [CrossRef]
- Paul, A.; Paul, S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front. Biosci. 2014, 19, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Vatish, J.; Wilkinson, B.; Al-Ishaq, Z.; Pujji, O.; Isgar, B.; Vidya, R.; Matey, P.; Sircar, T.; Mylvaganam, S. The use of genomic assays reduces rates of chemotherapy: A single-institution experience. Ir. J. Med. Sci. 2021. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Tang, G.; Shak, S.; Kim, C.; Baker, J.; Kim, W.; Cronin, M.; Baehner, F.L.; Watson, D.; Bryant, J.; et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J. Clin. Oncol. 2006, 24, 3726–3734. [Google Scholar] [CrossRef]
- Albain, K.S.; Barlow, W.E.; Shak, S.; Hortobagyi, G.N.; Livingston, R.B.; Yeh, I.T.; Ravdin, P.; Bugarini, R.; Baehner, F.L.; Davidson, N.E.; et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: A retrospective analysis of a randomised trial. Lancet Oncol. 2010, 11, 55–65. [Google Scholar] [CrossRef]
- Dowsett, M.; Cuzick, J.; Wale, C.; Forbes, J.; Mallon, E.A.; Salter, J.; Quinn, E.; Dunbier, A.; Baum, M.; Buzdar, A.; et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: A TransATAC study. J. Clin. Oncol. 2010, 28, 1829–1834. [Google Scholar] [CrossRef]
- Habel, L.A.; Shak, S.; Jacobs, M.K.; Capra, A.; Alexander, C.; Pho, M.; Baker, J.; Walker, M.; Watson, D.; Hackett, J.; et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res. 2006, 8, R25. [Google Scholar] [CrossRef] [PubMed]
- Toi, M.; Iwata, H.; Yamanaka, T.; Masuda, N.; Ohno, S.; Nakamura, S.; Nakayama, T.; Kashiwaba, M.; Kamigaki, S.; Kuroi, K. Clinical significance of the 21-gene signature (Oncotype DX) in hormone receptor-positive early stage primary breast cancer in the Japanese population. Cancer 2010, 116, 3112–3118. [Google Scholar] [CrossRef]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E., Jr.; Dees, E.C.; Goetz, M.P.; Olson, J.A., Jr.; et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N. Engl. J. Med. 2018, 379, 111–121. [Google Scholar] [CrossRef]
- Sjöström, M.; Chang, S.L.; Hartman, L.; Holmberg, E.; Feng, F.Y.; Speers, C.W.; Pierce, L.J.; Malmström, P.; Fernö, M.; Karlsson, P. Discovery and validation of a genomic signature to identify women with early-stage invasive breast cancer who may safely omit adjuvant radiotherapy after breast-conserving surgery. J. Clin. Oncol. 2021, 39, 512-512. [Google Scholar] [CrossRef]
- Cimmino, F.; Lasorsa, V.A.; Vetrella, S.; Iolascon, A.; Capasso, M. A Targeted Gene Panel for Circulating Tumor DNA Sequencing in Neuroblastoma. Front. Oncol. 2020, 10, 596191. [Google Scholar] [CrossRef] [PubMed]
- Surrey, L.F.; MacFarland, S.P.; Chang, F.; Cao, K.; Rathi, K.S.; Akgumus, G.T.; Gallo, D.; Lin, F.; Gleason, A.; Raman, P.; et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019, 11, 32. [Google Scholar] [CrossRef]
- Aristei, C.; Perrucci, E.; Alì, E.; Marazzi, F.; Masiello, V.; Saldi, S.; Ingrosso, G. Personalization in Modern Radiation Oncology: Methods, Results and Pitfalls. Personalized Interventions and Breast Cancer. Front. Oncol. 2021, 11, 616042. [Google Scholar] [CrossRef] [PubMed]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Yang, B.; Ke, W.; Wan, Y.; Li, T. Targeting RNF8 effectively reverses cisplatin and doxorubicin resistance in endometrial cancer. Biochem. Biophys. Res. Commun. 2021, 545, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Min, L.; Liu, C.; Chen, S.; Gao, C.; Ma, J.; Wu, X.; Li, W.; Wu, L.; Zhu, L. RNF8 Promotes Epithelial-Mesenchymal Transition in Lung Cancer Cells via Stabilization of Slug. Mol. Cancer Res. 2020, 18, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Z.; Meyer, M.B.; Saha, S.; Yu, M.; Guo, A.; Wisinski, K.B.; Huang, W.; Cai, W.; Pike, J.W.; et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 2014, 25, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, W. Histone H3R17me2a mark recruits human RNA polymerase-associated factor 1 complex to activate transcription. Proc. Natl. Acad. Sci. USA 2012, 109, 5675–5680. [Google Scholar] [CrossRef]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e6. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef]
- Oei, A.L.; Vriend, L.E.; van Leeuwen, C.M.; Rodermond, H.M.; Ten Cate, R.; Westermann, A.M.; Stalpers, L.J.; Crezee, J.; Kanaar, R.; Kok, H.P.; et al. Sensitizing thermochemotherapy with a PARP1-inhibitor. Oncotarget 2017, 8, 16303–16312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Lee, J.M.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2014, 25, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Moutafi, M.; Economopoulou, P.; Rimm, D.; Psyrri, A. PARP inhibitors in head and neck cancer: Molecular mechanisms, preclinical and clinical data. Oral. Oncol. 2021, 117, 105292. [Google Scholar] [CrossRef]
- Jin, M.H.; Oh, D.Y. ATM in DNA repair in cancer. Pharmacol. Ther. 2019, 203, 107391. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A.; Jang, H.; Kim, S.; Lee, M.; Kim, D.K.; Yang, Y.; Kim, H.J.; Lee, K.H.; Kim, J.W.; et al. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol. Cancer Ther. 2017, 16, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.P.; George, J.; Nowak, M.; Leeser, U.; Büttner, R.; Perner, S.; Peifer, M.; et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef] [PubMed]
- Weston, V.J.; Oldreive, C.E.; Skowronska, A.; Oscier, D.G.; Pratt, G.; Dyer, M.J.; Smith, G.; Powell, J.E.; Rudzki, Z.; Kearns, P.; et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, S.; Ayadi, M.; Brino, L.; Egly, J.M.; Larsen, A.K.; Coin, F. A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem. Biol. 2014, 21, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Matsuura, K.; Kawai, H.; Wakasugi, M.; Matsunaga, T. Spironolactone-induced XPB degradation depends on CDK7 kinase and SCF(FBXL18) E3 ligase. Genes Cells 2019, 24, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Krishnamurthy, S.; Kent, M.N.; Schumacher, D.L.; Sharma, P.; Excoffon, K.; Travers, J.B. Spironolactone Depletes the XPB Protein and Inhibits DNA Damage Responses in UVB-Irradiated Human Skin. J. Investig. Dermatol. 2019, 139, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Neher, T.M.; Shuck, S.C.; Liu, J.Y.; Zhang, J.T.; Turchi, J.J. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem. Biol. 2010, 5, 953–965. [Google Scholar] [CrossRef]
- Jiang, H.; Yang, L.Y. Cell cycle checkpoint abrogator UCN-01 inhibits DNA repair: Association with attenuation of the interaction of XPA and ERCC1 nucleotide excision repair proteins. Cancer Res. 1999, 59, 4529–4534. [Google Scholar] [PubMed]
- Arora, S.; Heyza, J.; Zhang, H.; Kalman-Maltese, V.; Tillison, K.; Floyd, A.M.; Chalfin, E.M.; Bepler, G.; Patrick, S.M. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget 2016, 7, 75104–75117. [Google Scholar] [CrossRef]
- Long, K.; Gu, L.; Li, L.; Zhang, Z.; Li, E.; Zhang, Y.; He, L.; Pan, F.; Guo, Z.; Hu, Z. Small-molecule inhibition of APE1 induces apoptosis, pyroptosis, and necroptosis in non-small cell lung cancer. Cell Death Dis. 2021, 12, 503. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Li, M.; Sui, J.; Ren, T.; Li, Z.; Zhang, L.; Zhou, L.; Cheng, Y.; Wang, D. Identification of a novel potential antitumor activity of gossypol as an APE1/Ref-1 inhibitor. Drug Des. Dev. Ther. 2014, 8, 485–496. [Google Scholar]
- Wang, Y.; Li, X.; Zhang, L.; Li, M.; Dai, N.; Luo, H.; Shan, J.; Yang, X.; Xu, M.; Feng, Y.; et al. A randomized, double-blind, placebo-controlled study of B-cell lymphoma 2 homology 3 mimetic gossypol combined with docetaxel and cisplatin for advanced non-small cell lung cancer with high expression of apurinic/apyrimidinic endonuclease 1. Investig. New Drugs 2020, 38, 1862–1871. [Google Scholar] [CrossRef]
- Mesquita, K.A.; Ali, R.; Doherty, R.; Toss, M.S.; Miligy, I.; Alblihy, A.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M., III; et al. FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers. Cancers 2021, 13, 1866. [Google Scholar] [CrossRef] [PubMed]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef]
- Nair, J.; Huang, T.T.; Murai, J.; Haynes, B.; Steeg, P.S.; Pommier, Y.; Lee, J.M. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020, 39, 5520–5535. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; Menéndez, M.D.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353.e6. [Google Scholar] [CrossRef]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M.; Arpino, G.; De Laurentiis, M.; Puglisi, F.; De Placido, S.; et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front. Oncol. 2018, 8, 608. [Google Scholar] [CrossRef]
- Skowron, M.A.; Vermeulen, M.; Winkelhausen, A.; Becker, T.K.; Bremmer, F.; Petzsch, P.; Schönberger, S.; Calaminus, G.; Köhrer, K.; Albers, P.; et al. CDK4/6 inhibition presents as a therapeutic option for paediatric and adult germ cell tumours and induces cell cycle arrest and apoptosis via canonical and non-canonical mechanisms. Br. J. Cancer 2020, 123, 378–391. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Inhibitor | CT Identifier | Other Regimen | Phase | Enrollment | Recruitment Status |
|---|---|---|---|---|---|---|
| CHK1 | Prexasertib | NCT02555644 | Cetuximab, Radiation | I | 70 | Completed |
| NCT02124148 | Cetuximab, G-CSF, Pemetrexed, Fluorouracil, LY3023414, Leucovorin | I | 167 | Completed | ||
| CDK4/6 | Palbociclib | NCT02897375 | Carboplatin | I | 90 | Recruiting |
| NCT03389477 | Cetuximab, radiation | II | 29 | Recruiting | ||
| PARP | Olaparib | NCT02308072 | Radiation | I | 70 | Active, not recruiting |
| NCT02882308 | Durvalumab | II | 41 | Completed | ||
| NCT01562210 | Radiation | I | 28 | Completed | ||
| NCT01296763 | Irinotecan, Mitomycin-C | I | 18 | Completed | ||
| NCT00782574 | - | I | 56 | Active, not recruiting | ||
| NCT00678132 | Gemcitabine | I | 23 | Completed | ||
| NCT02533765 | - | II | 18 | Active, not recruiting | ||
| Niraparib | NCT03983226 | Cisplatin/gemcitabine, Carboplatin/taxane, Carboplatin/gemcitabine, Liposome doxorubicin/carboplatin | II | 96 | Recruiting | |
| Veliparib | NCT01711541 | Carboplatin, Fluorouracil, Hydroxyurea, Paclitaxel, Radiation | I, II | 24 | Active, not recruiting | |
| NCT02723864 | VX-970 | I | 53 | Active, not recruiting | ||
| NCT02595905 | - | II | 333 | Active, not recruiting | ||
| NCT01104259 | Vinorelbine tartrate | I | 50 | Completed | ||
| NCT01585805 | Gemcitabine, Gemcitabine hydrochloride | I | 107 | Active, not recruiting | ||
| NCT01281852 | Paclitaxel | I | 37 | Completed | ||
| NCT01642251 | Etoposide | I, II | 156 | Completed | ||
| NCT01711541 | Carboplatin, Fluorouracil, Hydroxyurea, Paclitaxel, Radiation | I, II | 24 | Active, not recruiting | ||
| APE1 | Gossypol | NCT01977209 | - | III | 204 | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss, R.C.; Xia, F.; Acklin, S. Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 8199. https://doi.org/10.3390/ijms22158199
Kiss RC, Xia F, Acklin S. Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment. International Journal of Molecular Sciences. 2021; 22(15):8199. https://doi.org/10.3390/ijms22158199
Chicago/Turabian StyleKiss, Robert Csaba, Fen Xia, and Scarlett Acklin. 2021. "Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment" International Journal of Molecular Sciences 22, no. 15: 8199. https://doi.org/10.3390/ijms22158199
APA StyleKiss, R. C., Xia, F., & Acklin, S. (2021). Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment. International Journal of Molecular Sciences, 22(15), 8199. https://doi.org/10.3390/ijms22158199