
Epigenetic Regulation of Breast Cancer Stem Cells Contributing to Carcinogenesis and Therapeutic Implications


Abstract
:1. Introduction
1.1. Breast Cancer Stem Cells (BCSCs)
1.2. Epigenetic Regulation in Normal Function
1.2.1. DNA Methylation and Demethylation
1.2.2. Histone Modifications
2. Epigenetic Regulation in Breast Cancer and BCSCs
2.1. DNMT1
2.2. TET1
2.3. HMTs
2.3.1. Polycomb Group (PcG) Protein
2.3.2. SETDB1
2.4. LSD1
2.5. HATs
2.6. HDACs
2.7. ncRNAs
2.7.1. miRNAs
2.7.2. lncRNAs
3. Therapeutic Implications
3.1. Methylation-Based Therapy
3.2. Demethylation-Based Therapy
3.3. Chromatin Modifier Therapy
3.4. miRNA-Based Therapy
3.5. Combination Therapy
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Veronesi, U.; Boyle, P.; Goldhirsch, A.; Orecchia, R.; Viale, G. Breast cancer. Lancet 2005, 365, 1727–1741. [Google Scholar] [CrossRef]
- Allison, K.H. Molecular pathology of breast cancer: What a pathologist needs to know. Am. J. Clin. Pathol. 2012, 138, 770–780. [Google Scholar] [CrossRef]
- Taurin, S.; Alkhalifa, H. Breast cancers, mammary stem cells, and cancer stem cells, characteristics, and hypotheses. Neoplasia 2020, 22, 663–678. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Neo, S.Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [Green Version]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheang, M.C.; Martin, M.; Nielsen, T.O.; Prat, A.; Voduc, D.; Rodriguez-Lescure, A.; Ruiz, A.; Chia, S.; Shepherd, L.; Ruiz-Borrego, M.; et al. Defining breast cancer intrinsic subtypes by quantitative receptor expression. Oncologist 2015, 20, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol. 2010, 10, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Sampieri, K.; Fodde, R. Cancer stem cells and metastasis. Semin. Cancer Biol. 2012, 22, 187–193. [Google Scholar] [CrossRef]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The role of microRNAs in the regulation of cancer stem cells. Front. Genet. 2014, 4, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Saeg, F.; Anbalagan, M. Breast cancer stem cells and the challenges of eradication: A review of novel therapies. Stem Cell Investig. 2018, 5, 39. [Google Scholar] [CrossRef]
- Collina, F.; Di Bonito, M.; Li Bergolis, V.; De Laurentiis, M.; Vitagliano, C.; Cerrone, M.; Nuzzo, F.; Cantile, M.; Botti, G. Prognostic Value of Cancer Stem Cells Markers in Triple-Negative Breast Cancer. Biomed. Res. Int. 2015, 2015, 158682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef]
- Crabtree, J.S.; Miele, L. Breast Cancer Stem Cells. Biomedicines 2018, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Keller, P.J.; Arendt, L.M.; Skibinski, A.; Logvinenko, T.; Klebba, I.; Dong, S.; Smith, A.E.; Prat, A.; Perou, C.M.; Gilmore, H.; et al. Defining the cellular precursors to human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2772–2777. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridrichova, I.; Zmetakova, I. MicroRNAs Contribute to Breast Cancer Invasiveness. Cells 2019, 8, 1361. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Jiang, J.; Liang, X.H.; Tang, Y.L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets 2015, 8, 2973–2980. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, C.; Kuperwasser, C. Human breast cancer stem cell markers CD44 and CD24: Enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007, 9, 303. [Google Scholar] [CrossRef] [Green Version]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gu, L.N.; Shan, B.E.; Geng, C.Z.; Sang, M.X. Biomarkers for EMT and MET in breast cancer: An update. Oncol. Lett. 2016, 12, 4869–4876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador, M.A.; Birnbaum, D.; Charafe-Jauffret, E.; Ginestier, C. Breast cancer stem cells programs: Enter the (non)-code. Brief. Funct. Genom. 2016, 15, 186–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Choudhury, S.; Kowalczyk, A.; Bessarabova, M.; Beresford-Smith, B.; Conway, T.; Kaspi, A.; Wu, Z.; Nikolskaya, T.; Merino, V.F.; et al. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet. 2011, 7, e1001369. [Google Scholar] [CrossRef] [PubMed]
- Lustberg, M.B.; Ramaswamy, B. Epigenetic targeting in breast cancer: Therapeutic impact and future direction. Drug News Perspect 2009, 22, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [Green Version]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol. 2007, 211, 261–268. [Google Scholar] [CrossRef]
- Zare, M.; Bastami, M.; Solali, S.; Alivand, M.R. Aberrant miRNA promoter methylation and EMT-involving miRNAs in breast cancer metastasis: Diagnosis and therapeutic implications. J. Cell Physiol. 2018, 233, 3729–3744. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [Green Version]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.F.; Chan, W.Y. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics 2014, 9, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Hua, S.; He, X.; Zhang, Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim Biophys Sin. 2010, 42, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012, 4, 26. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, X.; Clark, E.; Mulcahey, M.; Huang, S.; Shi, Y.G. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010, 20, 1390–1393. [Google Scholar] [CrossRef] [Green Version]
- Alivand, M.R.; Soheili, Z.S.; Pornour, M.; Solali, S.; Sabouni, F. Novel Epigenetic Controlling of Hypoxia Pathway Related to Overexpression and Promoter Hypomethylation of TET1 and TET2 in RPE Cells. J. Cell Biochem 2017, 118, 3193–3204. [Google Scholar] [CrossRef]
- Rausch, C.; Hastert, F.D.; Cardoso, M.C. DNA Modification Readers and Writers and Their Interplay. J. Mol. Biol. 2020, 432, 1731–1746. [Google Scholar] [CrossRef]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. Adv. Genet. 2010, 70, 57–85. [Google Scholar]
- Li, Q.; Chen, H. Silencing of Wnt5a during colon cancer metastasis involves histone modifications. Epigenetics 2012, 7, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef]
- Mohn, F.; Schubeler, D. Genetics and epigenetics: Stability and plasticity during cellular differentiation. Trends Genet. 2009, 25, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Lin, C.; Zhong, L.L.; Zhao, L.; Zhang, G.; Lu, A.; Wu, J.; Bian, Z. Targeting histone methylation for colorectal cancer. Ther. Adv. Gastroenterol. 2017, 10, 114–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Xu, A.M. SET and MYND domain containing protein 3 in cancer. Am. J. Transl. Res. 2017, 9, 1–14. [Google Scholar]
- Hino, S.; Kohrogi, K.; Nakao, M. Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Sci. 2016, 107, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Hillringhaus, L.; Yue, W.W.; Rose, N.R.; Ng, S.S.; Gileadi, C.; Loenarz, C.; Bello, S.H.; Bray, J.E.; Schofield, C.J.; Oppermann, U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J. Biol. Chem. 2011, 286, 41616–41625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.Y.; Lei, P.J.; Zhang, X.; Zheng, J.Y.; Wang, H.Y.; Zhao, J.; Li, Y.M.; Ye, M.; Li, L.; Wei, G.; et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin. Epigenetics 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O.; et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 2016, 37, 656–668. [Google Scholar] [CrossRef]
- Khan, S.I.; Aumsuwan, P.; Khan, I.A.; Walker, L.A.; Dasmahapatra, A.K. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem. Res. Toxicol. 2012, 25, 61–73. [Google Scholar] [CrossRef]
- Bansal, N.; David, G.; Farias, E.; Waxman, S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Adv. Cancer Res. 2016, 130, 113–135. [Google Scholar]
- Loginov, V.I.; Burdennyy, A.M.; Pronina, I.V.; Khokonova, V.V.; Kurevljov, S.V.; Kazubskaya, T.P.; Kushlinskii, N.E.; Braga, E.A. Novel miRNA genes hypermethylated in breast cancer. Mol. Biol. Mosk 2016, 50, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Baer, C.; Claus, R.; Plass, C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013, 73, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Allegrucci, C. Keeping an open mind: Highlights and controversies of the breast cancer stem cell theory. Breast Cancer Dove Med. Press 2012, 4, 155–166. [Google Scholar]
- Abdel-Hafiz, H.A. Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer. Diseases 2017, 5, 16. [Google Scholar] [CrossRef]
- Li, L.; Bi, Z.; Wadgaonkar, P.; Lu, Y.; Zhang, Q.; Fu, Y.; Thakur, C.; Wang, L.; Chen, F. Metabolic and epigenetic reprogramming in the arsenic-induced cancer stem cells. Semin. Cancer Biol. 2019, 57, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [Green Version]
- El Helou, R.; Wicinski, J.; Guille, A.; Adelaide, J.; Finetti, P.; Bertucci, F.; Chaffanet, M.; Birnbaum, D.; Charafe-Jauffret, E.; Ginestier, C. Brief reports: A distinct DNA methylation signature defines breast cancer stem cells and predicts cancer outcome. Stem Cells 2014, 32, 3031–3036. [Google Scholar] [CrossRef]
- Li, Y.; Tollefsbol, T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem 2010, 17, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Sengupta, D.; Deb, M.; Shilpi, A.; Parbin, S.; Rath, S.K.; Pradhan, N.; Rakshit, M.; Patra, S.K. Expression profiling of DNA methylation-mediated epigenetic gene-silencing factors in breast cancer. Clin. Epigenetics 2014, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 6910. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Blanco, E.; Kim, Y.S.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.; Chen, C.L.; Jin, G.; Landis, M.D.; Burey, L.A.; et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valk-Lingbeek, M.E.; Bruggeman, S.W.; van Lohuizen, M. Stem cells and cancer; the polycomb connection. Cell 2004, 118, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Paranjape, A.N.; Balaji, S.A.; Mandal, T.; Krushik, E.V.; Nagaraj, P.; Mukherjee, G.; Rangarajan, A. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC Cancer 2014, 14, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Bharali, D.J.; Sudha, T.; Khedr, M.; Guest, I.; Sell, S.; Glinsky, G.V.; Mousa, S.A. Downregulation of Bmi1 in breast cancer stem cells suppresses tumor growth and proliferation. Oncotarget 2017, 8, 38731–38742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Poreba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, M.E.; Moore, H.M.; Li, X.; Toy, K.A.; Huang, W.; Sabel, M.S.; Kidwell, K.M.; Kleer, C.G. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 3098–3103. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Xia, L.; Wu, D.Y.; Wang, H.; Chansky, H.A.; Schubach, W.H.; Hickstein, D.D.; Zhang, Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 2002, 21, 148–152. [Google Scholar] [CrossRef] [Green Version]
- Karanth, A.V.; Maniswami, R.R.; Prashanth, S.; Govindaraj, H.; Padmavathy, R.; Jegatheesan, S.K.; Mullangi, R.; Rajagopal, S. Emerging role of SETDB1 as a therapeutic target. Expert Opin. Targets 2017, 21, 319–331. [Google Scholar] [CrossRef]
- Ryu, T.Y.; Kim, K.; Kim, S.K.; Oh, J.H.; Min, J.K.; Jung, C.R.; Son, M.Y.; Kim, D.S.; Cho, H.S. SETDB1 regulates SMAD7 expression for breast cancer metastasis. BMB Rep. 2019, 52, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schule, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.K.; Bert, A.G.; et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef]
- Wu, M.Y.; Fu, J.; Xiao, X.; Wu, J.; Wu, R.C. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett. 2014, 354, 311–319. [Google Scholar] [CrossRef]
- Han, X.; Yan, S.; Weijie, Z.; Feng, W.; Liuxing, W.; Mengquan, L.; Qingxia, F. Critical role of miR-10b in transforming growth factor-beta1-induced epithelial-mesenchymal transition in breast cancer. Cancer Gene 2014, 21, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Bahena-Ocampo, I.; Espinosa, M.; Ceballos-Cancino, G.; Lizarraga, F.; Campos-Arroyo, D.; Schwarz, A.; Garcia-Lopez, P.; Maldonado, V.; Melendez-Zajgla, J. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016, 17, 1081. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, W.; Liu, C.; Li, W.; Yu, H.; Lei, B.; Ren, Y.; Li, Z.; Pang, D.; Qian, C. MiR-23a promotes TGF-beta1-induced EMT and tumor metastasis in breast cancer cells by directly targeting CDH1 and activating Wnt/beta-catenin signaling. Oncotarget 2017, 8, 69538–69550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Li, J.; Zhang, Y.; Wang, N.; Liang, H.; Liu, Y.; Zhang, C.Y.; Zen, K.; Gu, H. Slug-upregulated miR-221 promotes breast cancer progression through suppressing E-cadherin expression. Sci. Rep. 2016, 6, 25798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscigno, G.; Quintavalle, C.; Donnarumma, E.; Puoti, I.; Diaz-Lagares, A.; Iaboni, M.; Fiore, D.; Russo, V.; Todaro, M.; Romano, G.; et al. MiR-221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget 2016, 7, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Lu, Y.; Wang, H.; Han, X.; Mao, J.; Li, J.; Yu, L.; Wang, B.; Fan, S.; Yu, X.; et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed. Pharm. 2016, 79, 93–101. [Google Scholar] [CrossRef]
- Li, B.; Lu, Y.; Yu, L.; Han, X.; Wang, H.; Mao, J.; Shen, J.; Wang, B.; Tang, J.; Li, C.; et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-kappaB/COX-2 activation. Chem. Biol. Interact. 2017, 277, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Li, F.; Li, X.; Tian, Y.; Zhang, Y.; Sheng, X.; Song, Y.; Meng, Q.; Yuan, S.; Luan, L.; et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 2017, 8, 1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Lang, T.Y.; Zou, D.L.; Zhou, L.; Lou, M.; Liu, J.S.; Li, Y.Z.; Ding, D.Y.; Li, Y.C.; Zhang, N.; et al. miR-520b Promotes Breast Cancer Stemness Through Hippo/YAP Signaling Pathway. Onco Targets 2019, 12, 11691–11700. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padua Alves, C.; Fonseca, A.S.; Muys, B.R.; de Barros, E.L.B.R.; Burger, M.C.; de Souza, J.E.; Valente, V.; Zago, M.A.; Silva, W.A., Jr. Brief report: The lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells 2013, 31, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, M.; Jiang, R.; An, N.; Wang, X.; Liu, B. Long Non-Coding RNA HOTAIR Regulates the Proliferation, Self-Renewal Capacity, Tumor Formation and Migration of the Cancer Stem-Like Cell (CSC) Subpopulation Enriched from Breast Cancer Cells. PLoS ONE 2017, 12, e0170860. [Google Scholar] [CrossRef] [Green Version]
- Askarian-Amiri, M.E.; Seyfoddin, V.; Smart, C.E.; Wang, J.; Kim, J.E.; Hansji, H.; Baguley, B.C.; Finlay, G.J.; Leung, E.Y. Emerging role of long non-coding RNA SOX2OT in SOX2 regulation in breast cancer. PLoS ONE 2014, 9, e102140. [Google Scholar] [CrossRef]
- Li, H.; Zhu, L.; Xu, L.; Qin, K.; Liu, C.; Yu, Y.; Su, D.; Wu, K.; Sheng, Y. Long noncoding RNA linc00617 exhibits oncogenic activity in breast cancer. Mol. Carcinog 2017, 56, 3–17. [Google Scholar] [CrossRef]
- Zhou, M.; Hou, Y.; Yang, G.; Zhang, H.; Tu, G.; Du, Y.E.; Wen, S.; Xu, L.; Tang, X.; Tang, S.; et al. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells 2016, 34, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Gabory, A.; Jammes, H.; Dandolo, L. The H19 locus: Role of an imprinted non-coding RNA in growth and development. Bioessays 2010, 32, 473–480. [Google Scholar] [CrossRef]
- Matouk, I.J.; Raveh, E.; Abu-lail, R.; Mezan, S.; Gilon, M.; Gershtain, E.; Birman, T.; Gallula, J.; Schneider, T.; Barkali, M.; et al. Oncofetal H19 RNA promotes tumor metastasis. Biochim Biophys Acta 2014, 1843, 1414–1426. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.Y.; Liu, G.F.; Qian, X.L.; Tang, L.B.; Huang, Q.Y.; Xiong, L.X. Long Non-Coding RNA: Dual Effects on Breast Cancer Metastasis and Clinical Applications. Cancers 2019, 11, 1802. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Li, T.T.; Wang, K.L.; Xiao, G.Q.; Wang, J.H.; Zhao, H.D.; Kang, Z.J.; Fan, W.J.; Zhu, L.L.; Li, M.; et al. H19/let-7/LIN28 reciprocal negative regulatory circuit promotes breast cancer stem cell maintenance. Cell Death Dis 2017, 8, e2569. [Google Scholar] [CrossRef] [Green Version]
- Shima, H.; Kida, K.; Adachi, S.; Yamada, A.; Sugae, S.; Narui, K.; Miyagi, Y.; Nishi, M.; Ryo, A.; Murata, S.; et al. Lnc RNA H19 is associated with poor prognosis in breast cancer patients and promotes cancer stemness. Breast Cancer Res. Treat. 2018, 170, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, Y.; Xiao, G.D.; Zheng, X.Q.; Wang, J.C.; Xu, C.W.; Qin, S.; Ren, H.; Tang, S.C.; Sun, X. H19 regulation of oestrogen induction of symmetric division is achieved by antagonizing Let-7c in breast cancer stem-like cells. Cell Prolif 2019, 52, e12534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Venzor, A.; Mandujano-Tinoco, E.A.; Lizarraga, F.; Zampedri, C.; Krotzsch, E.; Salgado, R.M.; Davila-Borja, V.M.; Encarnacion-Guevara, S.; Melendez-Zajgla, J.; Maldonado, V. Microenvironment-regulated lncRNA-HAL is able to promote stemness in breast cancer cells. Biochim Biophys Acta Mol. Cell Res. 2019, 1866, 118523. [Google Scholar] [CrossRef]
- Tu, Z.; Schmollerl, J.; Cuiffo, B.G.; Karnoub, A.E. Microenvironmental Regulation of Long Noncoding RNA LINC01133 Promotes Cancer Stem Cell-Like Phenotypic Traits in Triple-Negative Breast Cancers. Stem Cells 2019, 37, 1281–1292. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, B.; Zhang, M.; Guo, W.; Wu, Z.; Wang, Y.; Jia, L.; Li, S.; Cancer Genome Atlas Research, N.; Xie, W.; et al. lncRNA Epigenetic Landscape Analysis Identifies EPIC1 as an Oncogenic lncRNA that Interacts with MYC and Promotes Cell-Cycle Progression in Cancer. Cancer Cell 2018, 33, 706–720 e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.D.; Jiao, D.C.; Qiao, J.H.; Yang, S.; Liu, Z.Z. Long non-coding RNA SOX21-AS1 modulates breast cancer stem cells properties and carcinogenesis via targeting SOX2. Oncotarget 2017, 5, 1–11. [Google Scholar] [CrossRef]
- Li, L.; Meng, D.; Wang, R. Long non-coding RNA SOX21-AS1 enhances the stemness of breast cancer cells via the Hippo pathway. FEBS Open Biol. 2021, 11, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ye, Q.; Zou, C. Long Non-Coding RNA THOR Enhances the Stem Cell-Like Traits of Triple-Negative Breast Cancer Cells Through Activating beta-Catenin Signaling. Med. Sci. Monit. 2020, 26, e923507. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Guo, C.; Xia, T.; Zhang, R.; Zen, K.; Pan, Y.; Jin, L. LncCCAT1 Promotes Breast Cancer Stem Cell Function through Activating WNT/beta-catenin Signaling. Theranostics 2019, 9, 7384–7402. [Google Scholar] [CrossRef]
- Zheng, A.; Song, X.; Zhang, L.; Zhao, L.; Mao, X.; Wei, M.; Jin, F. Long non-coding RNA LUCAT1/miR-5582-3p/TCF7L2 axis regulates breast cancer stemness via Wnt/beta-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Cen, Y.; Chen, J. Long non-coding RNA MALAT-1 contributes to maintenance of stem cell-like phenotypes in breast cancer cells. Oncol. Lett. 2018, 15, 2117–2122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Sun, L.; Zhang, Y.; Lu, G.; Li, Y.; Wei, Z. Long non-coding RNA FEZF1-AS1 promotes breast cancer stemness and tumorigenesis via targeting miR-30a/Nanog axis. J. Cell Physiol. 2018, 233, 8630–8638. [Google Scholar] [CrossRef]
- Lu, G.; Li, Y.; Ma, Y.; Lu, J.; Chen, Y.; Jiang, Q.; Qin, Q.; Zhao, L.; Huang, Q.; Luo, Z.; et al. Long noncoding RNA LINC00511 contributes to breast cancer tumourigenesis and stemness by inducing the miR-185-3p/E2F1/Nanog axis. J. Exp. Clin. Cancer Res. 2018, 37, 289. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.L.; Gong, C.; Chen, C.H.; Lee, D.Y.; Hu, H.; Huang, P.; Yao, Y.; Guo, W.; Reinhardt, F.; Wulf, G.; et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res. 2014, 74, 3603–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Garikapati, K.R.; Makani, V.K.K.; Nair, A.D.; Vangara, N.; Bhadra, U.; Pal Bhadra, M. Regulating BMI1 expression via miRNAs promote Mesenchymal to Epithelial Transition (MET) and sensitizes breast cancer cell to chemotherapeutic drug. PLoS ONE 2018, 13, e0190245. [Google Scholar] [CrossRef] [Green Version]
- Sossey-Alaoui, K.; Pluskota, E.; Szpak, D.; Schiemann, W.P.; Plow, E.F. The Kindlin-2 regulation of epithelial-to-mesenchymal transition in breast cancer metastasis is mediated through miR-200b. Sci. Rep. 2018, 8, 7360. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Qin, S.; Fan, C.; Xu, C.; Du, N.; Ren, H. Let-7: A regulator of the ERalpha signaling pathway in human breast tumors and breast cancer stem cells. Oncol. Rep. 2013, 29, 2079–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Xu, C.; Tang, S.C.; Wang, J.; Wang, H.; Wang, P.; Du, N.; Qin, S.; Li, G.; Xu, S.; et al. Let-7c blocks estrogen-activated Wnt signaling in induction of self-renewal of breast cancer stem cells. Cancer Gene 2016, 23, 83–89. [Google Scholar] [CrossRef]
- di Gennaro, A.; Damiano, V.; Brisotto, G.; Armellin, M.; Perin, T.; Zucchetto, A.; Guardascione, M.; Spaink, H.P.; Doglioni, C.; Snaar-Jagalska, B.E.; et al. A p53/miR-30a/ZEB2 axis controls triple negative breast cancer aggressiveness. Cell Death Differ. 2018, 25, 2165–2180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, L.C.; Jiang, N.; Wang, X.L.; Zhou, X.N.; Luo, X.L.; Ren, J. MicroRNA miR-590-5p inhibits breast cancer cell stemness and metastasis by targeting SOX2. Eur. Rev. Med. Pharm. Sci. 2017, 21, 87–94. [Google Scholar]
- Zhang, Y.; Eades, G.; Yao, Y.; Li, Q.; Zhou, Q. Estrogen receptor alpha signaling regulates breast tumor-initiating cells by down-regulating miR-140 which targets the transcription factor SOX2. J. Biol. Chem. 2012, 287, 41514–41522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, Y.; Kong, D.; Ahmad, A.; Banerjee, S.; Sarkar, F.H. Cross-talk between miRNA and Notch signaling pathways in tumor development and progression. Cancer Lett. 2010, 292, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, H.; Li, T.; Yu, L.; Liu, G.; Peng, X.; Zhao, J. Kruppel-like factor 8 promotes tumorigenic mammary stem cell induction by targeting miR-146a. Am. J. Cancer Res. 2013, 3, 356–373. [Google Scholar]
- Liang, R.; Li, Y.; Wang, M.; Tang, S.C.; Xiao, G.; Sun, X.; Li, G.; Du, N.; Liu, D.; Ren, H. MiR-146a promotes the asymmetric division and inhibits the self-renewal ability of breast cancer stem-like cells via indirect upregulation of Let-7. Cell Cycle 2018, 17, 1445–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Helou, R.; Pinna, G.; Cabaud, O.; Wicinski, J.; Bhajun, R.; Guyon, L.; Rioualen, C.; Finetti, P.; Gros, A.; Mari, B.; et al. miR-600 Acts as a Bimodal Switch that Regulates Breast Cancer Stem Cell Fate through WNT Signaling. Cell Rep. 2017, 18, 2256–2268. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, N.; Liu, L.; Dong, H.; Liu, X. microRNA-128-3p overexpression inhibits breast cancer stem cell characteristics through suppression of Wnt signalling pathway by down-regulating NEK2. J. Cell Mol. Med. 2020, 24, 7353–7369. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Huang, Y.; Lou, C.; He, Y.; Zhang, Y.; Zhang, Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin beta3/Wnt signaling under miR-137 regulation. Cancer Biol. 2019, 20, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Guo, Q.; Li, X.; Yang, X.; Ni, H.; Wang, T.; Zhao, Q.; Liu, H.; Xing, Y.; Xi, T.; et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine 2019, 41, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Liu, X.; Zhou, S.; Li, W.; Liu, C.; Chadwick, M.; Qian, C. Long non-coding RNA FGF13-AS1 inhibits glycolysis and stemness properties of breast cancer cells through FGF13-AS1/IGF2BPs/Myc feedback loop. Cancer Lett. 2019, 450, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Zhang, R.; Xiao, W.; Niu, X.; Ye, X.; Li, Z.; Guo, Y.; Tan, J.; Li, Y. The EZH2- H3K27me3-DNMT1 complex orchestrates epigenetic silencing of the wwc1 gene, a Hippo/YAP pathway upstream effector, in breast cancer epithelial cells. Cell Signal. 2018, 51, 243–256. [Google Scholar] [CrossRef]
- Fukagawa, A.; Ishii, H.; Miyazawa, K.; Saitoh, M. deltaEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer Med. 2015, 4, 125–135. [Google Scholar] [CrossRef]
- Al-Kharashi, L.A.; Al-Mohanna, F.H.; Tulbah, A.; Aboussekhra, A. The DNA methyl-transferase protein DNMT1 enhances tumor-promoting properties of breast stromal fibroblasts. Oncotarget 2018, 9, 2329–2343. [Google Scholar] [CrossRef]
- Sengupta, D.; Deb, M.; Rath, S.K.; Kar, S.; Parbin, S.; Pradhan, N.; Patra, S.K. DNA methylation and not H3K4 trimethylation dictates the expression status of miR-152 gene which inhibits migration of breast cancer cells via DNMT1/CDH1 loop. Exp. Cell Res. 2016, 346, 176–187. [Google Scholar] [CrossRef]
- Shi, Z.; Li, Y.; Qian, X.; Hu, Y.; Liu, J.; Zhang, S.; Zhang, J. MiR-340 Inhibits Triple-Negative Breast Cancer Progression by Reversing EZH2 Mediated miRNAs Dysregulated Expressions. J. Cancer 2017, 8, 3037–3048. [Google Scholar] [CrossRef]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [Green Version]
- Good, C.R.; Panjarian, S.; Kelly, A.D.; Madzo, J.; Patel, B.; Jelinek, J.; Issa, J.J. TET1-Mediated Hypomethylation Activates Oncogenic Signaling in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 4126–4137. [Google Scholar] [CrossRef] [Green Version]
- Wahl, G.M.; Spike, B.T. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 2017, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Joo, H.S.; Won, H.Y.; Min, K.W.; Kim, H.Y.; Son, T.; Oh, Y.H.; Lee, J.Y.; Kong, G. Role of RBP2-Induced ER and IGF1R-ErbB Signaling in Tamoxifen Resistance in Breast Cancer. J. Natl. Cancer Inst. 2018, 110, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Mendaza, S.; Ulazia-Garmendia, A.; Monreal-Santesteban, I.; Cordoba, A.; Azua, Y.R.; Aguiar, B.; Beloqui, R.; Armendariz, P.; Arriola, M.; Martin-Sanchez, E.; et al. ADAM12 is A Potential Therapeutic Target Regulated by Hypomethylation in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 903. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Fan, X.; Hao, M.; Wang, J.; Zhou, X.; Sun, X. Higher levels of TIMP-1 expression are associated with a poor prognosis in triple-negative breast cancer. Mol. Cancer 2016, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Yang, L.; Zou, Y.; Liang, J.Y.; Liu, P.; Gao, G.; Yang, A.; Tang, H.; Xie, X. Long non-coding RNA HUMT hypomethylation promotes lymphangiogenesis and metastasis via activating FOXK1 transcription in triple-negative breast cancer. J. Hematol. Oncol. 2020, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Teslow, E.A.; Mitrea, C.; Boerner, J.L.; Dyson, G.; Bollig-Fischer, A. Role of TET1 and 5hmC in an Obesity-Linked Pathway Driving Cancer Stem Cells in Triple-Negative Breast Cancer. Mol. Cancer Res. 2020, 18, 1803–1814. [Google Scholar] [CrossRef]
- Huang, J.; Dorsey, J.; Chuikov, S.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [Green Version]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhao, Y.; Zhang, J.; Lu, Y.; Liu, X.; Geng, P.; Huang, B.; Zhang, Y.; Lu, J. The dual function of PRMT1 in modulating epithelial-mesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB1. Sci. Rep. 2016, 6, 19874. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Xia, W.; Liao, H.W.; Saito, M.; Hung, M.C.; Yamaguchi, H. The role of PRMT1 in EGFR methylation and signaling in MDA-MB-468 triple-negative breast cancer cells. Breast Cancer 2018, 25, 74–80. [Google Scholar] [CrossRef]
- Liu, L.M.; Sun, W.Z.; Fan, X.Z.; Xu, Y.L.; Cheng, M.B.; Zhang, Y. Methylation of C/EBPalpha by PRMT1 Inhibits Its Tumor-Suppressive Function in Breast Cancer. Cancer Res. 2019, 79, 2865–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.H.; Feng, Y.; Zhang, R.; Xu, L.H.; Li, M.Z.; Kung, H.F.; Song, L.B.; Zeng, M.S. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol. Cancer 2011, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Siddique, H.R.; Parray, A.; Tarapore, R.S.; Wang, L.; Mukhtar, H.; Karnes, R.J.; Deng, Y.; Konety, B.R.; Saleem, M. BMI1 polycomb group protein acts as a master switch for growth and death of tumor cells: Regulates TCF4-transcriptional factor-induced BCL2 signaling. PLoS ONE 2013, 8, e60664. [Google Scholar] [CrossRef]
- Yin, J.; Zheng, G.; Jia, X.; Zhang, Z.; Zhang, W.; Song, Y.; Xiong, Y.; He, Z. A Bmi1-miRNAs cross-talk modulates chemotherapy response to 5-fluorouracil in breast cancer cells. PLoS ONE 2013, 8, e73268. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.; Farnie, G.; Brennan, K.R.; Clarke, R.B. Breast cancer stem cells: Something out of notching? Cancer Res. 2010, 70, 8973–8976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoey, T.; Yen, W.C.; Axelrod, F.; Basi, J.; Donigian, L.; Dylla, S.; Fitch-Bruhns, M.; Lazetic, S.; Park, I.K.; Sato, A.; et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 2009, 5, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Li, S.; Song, E.; Liu, S. The roles of ncRNAs and histone-modifiers in regulating breast cancer stem cells. Protein Cell 2016, 7, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Batham, J.; Lim, P.S.; Rao, S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers 2019, 11, 1143. [Google Scholar] [CrossRef] [Green Version]
- Suriyamurthy, S.; Baker, D.; Ten Dijke, P.; Iyengar, P.V. Epigenetic Reprogramming of TGF-beta Signaling in Breast Cancer. Cancers 2019, 11, 726. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Katsuno, Y.; Meyer, D.; Budi, E.H.; Chen, S.H.; Koeppen, H.; Wang, H.; Akhurst, R.J.; Derynck, R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018, 19, 135–155. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, S.; Sedukhina, A.S.; Nakagawa, Y.; Maeda, I.; Kubota, M.; Ohnuma, S.; Tsugawa, K.; Ohta, T.; Roche-Molina, M.; Bernal, J.A.; et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS ONE 2015, 10, e0118002. [Google Scholar] [CrossRef]
- Ma, L.; Ma, S.; Zhao, G.; Yang, L.; Zhang, P.; Yi, Q.; Cheng, S. miR-708/LSD1 axis regulates the proliferation and invasion of breast cancer cells. Cancer Med. 2016, 5, 684–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniyan, B.; Sridharan, S.; Howard, C.M.; Tilley, A.M.; Basuroy, T.; De La Serna, I.; Butt, E.; Raman, D. Role of the CXCR4-LASP1 Axis in the Stabilization of Snail1 in Triple-Negative Breast Cancer. Cancers 2020, 12, 2372. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liang, Y.; Cao, X.; Wang, X.; Gao, H.; Lin, S.Y.; Schiff, R.; Wang, X.S.; Li, K. Identification of MYST3 as a novel epigenetic activator of ERalpha frequently amplified in breast cancer. Oncogene 2017, 36, 2910–2918. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci 2017, 18, 1414. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, J.; Huo, M.; Gao, J.; Yang, T.; Yin, X.; Wang, P.; Leng, S.; Feng, D.; Chen, Y.; et al. CUL4B Promotes Breast Carcinogenesis by Coordinating with Transcriptional Repressor Complexes in Response to Hypoxia Signaling Pathway. Adv. Sci Weinh 2021, 8, 2001515. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Lo, P.K.; Wolfson, B.; Zhou, X.; Duru, N.; Gernapudi, R.; Zhou, Q. Noncoding RNAs in breast cancer. Brief. Funct. Genom. 2016, 15, 200–221. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Niinuma, T.; Kai, M. Relationship Between Noncoding RNA Dysregulation and Epigenetic Mechanisms in Cancer. Adv. Exp. Med. Biol. 2016, 927, 109–135. [Google Scholar]
- Das, P.K.; Siddika, M.A.; Asha, S.Y.; Aktar, S.; Rakib, M.A.; Khanam, J.A.; Pillai, S.; Islam, F. MicroRNAs, a Promising Target for Breast Cancer Stem Cells. Mol. Diagn 2020, 24, 69–83. [Google Scholar] [CrossRef]
- Vahidian, F.; Mohammadi, H.; Ali-Hasanzadeh, M.; Derakhshani, A.; Mostaan, M.; Hemmatzadeh, M.; Baradaran, B. MicroRNAs and breast cancer stem cells: Potential role in breast cancer therapy. J. Cell Physiol. 2019, 234, 3294–3306. [Google Scholar] [CrossRef]
- Mulrane, L.; McGee, S.F.; Gallagher, W.M.; O’Connor, D.P. miRNA dysregulation in breast cancer. Cancer Res. 2013, 73, 6554–6562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Huerta, N.; Silva-Cazares, M.B.; Arriaga-Pizano, L.A.; Prieto-Chavez, J.L.; Lopez-Camarillo, C. LncRNAs and microRNAs as Essential Regulators of Stemness in Breast Cancer Stem Cells. Biomolecules 2021, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Sasheva, P.; Grossniklaus, U. Differentially Methylated Region-Representational Difference Analysis (DMR-RDA): A Powerful Method to Identify DMRs in Uncharacterized Genomes. Methods Mol. Biol. 2017, 1456, 113–125. [Google Scholar]
- Hasegawa, T.; Adachi, R.; Iwakata, H.; Takeno, T.; Sato, K.; Sakamaki, T. ErbB2 signaling epigenetically suppresses microRNA-205 transcription via Ras/Raf/MEK/ERK pathway in breast cancer. FEBS Open Biol. 2017, 7, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Wongtrakoongate, P. Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries. World J. Stem Cells 2015, 7, 137–148. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [Green Version]
- Azorin, P.; Bonin, F.; Moukachar, A.; Ponceau, A.; Vacher, S.; Bieche, I.; Marangoni, E.; Fuhrmann, L.; Vincent-Salomon, A.; Lidereau, R.; et al. Distinct expression profiles and functions of Kindlins in breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 281. [Google Scholar] [CrossRef] [PubMed]
- Pronina, I.V.; Loginov, V.I.; Burdennyy, A.M.; Fridman, M.V.; Senchenko, V.N.; Kazubskaya, T.P.; Kushlinskii, N.E.; Dmitriev, A.A.; Braga, E.A. DNA methylation contributes to deregulation of 12 cancer-associated microRNAs and breast cancer progression. Gene 2017, 604, 1–8. [Google Scholar] [CrossRef]
- Doecke, J.D.; Wang, Y.; Baggerly, K. Co-localized genomic regulation of miRNA and mRNA via DNA methylation affects survival in multiple tumor types. Cancer Genet. 2016, 209, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Damiano, V.; Brisotto, G.; Borgna, S.; di Gennaro, A.; Armellin, M.; Perin, T.; Guardascione, M.; Maestro, R.; Santarosa, M. Epigenetic silencing of miR-200c in breast cancer is associated with aggressiveness and is modulated by ZEB1. Genes Chromosomes Cancer 2017, 56, 147–158. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Luo, N.; Hu, Y.; Li, X.; Zhang, K. MiR-137 Suppresses Triple-Negative Breast Cancer Stemness and Tumorigenesis by Perturbing BCL11A-DNMT1 Interaction. Cell Physiol. Biochem. 2018, 47, 2147–2158. [Google Scholar] [CrossRef]
- Janaki Ramaiah, M.; Vaishnave, S. BMI1 and PTEN are key determinants of breast cancer therapy: A plausible therapeutic target in breast cancer. Gene 2018, 678, 302–311. [Google Scholar]
- Soudyab, M.; Iranpour, M.; Ghafouri-Fard, S. The Role of Long Non-Coding RNAs in Breast Cancer. Arch. Iran. Med. 2016, 19, 508–517. [Google Scholar] [PubMed]
- Chen, X.; Fan, S.; Song, E. Noncoding RNAs: New Players in Cancers. Adv. Exp. Med. Biol 2016, 927, 1–47. [Google Scholar] [PubMed]
- Gloss, B.S.; Dinger, M.E. The specificity of long noncoding RNA expression. Biochim Biophys Acta 2016, 1859, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Tracy, K.M.; Tye, C.E.; Ghule, P.N.; Malaby, H.L.H.; Stumpff, J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Mitotically-Associated lncRNA (MANCR) Affects Genomic Stability and Cell Division in Aggressive Breast Cancer. Mol. Cancer Res. 2018, 16, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Wang, J.; Yumoto, K.; Jung, Y.; Cackowski, F.C.; Decker, A.M.; Li, Y.; Franceschi, R.T.; Pienta, K.J.; Taichman, R.S. DNMT1 Regulates Epithelial-Mesenchymal Transition and Cancer Stem Cells, Which Promotes Prostate Cancer Metastasis. Neoplasia 2016, 18, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roboz, G.J.; Kantarjian, H.M.; Yee, K.W.L.; Kropf, P.L.; O’Connell, C.L.; Griffiths, E.A.; Stock, W.; Daver, N.G.; Jabbour, E.; Ritchie, E.K.; et al. Dose, schedule, safety, and efficacy of guadecitabine in relapsed or refractory acute myeloid leukemia. Cancer 2018, 124, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Holowatyj, A.; Wu, J.; Liu, H.; Zhang, L.; Suzuki, T.; Yang, Z.Q. Genetic alterations of KDM4 subfamily and therapeutic effect of novel demethylase inhibitor in breast cancer. Am. J. Cancer Res. 2015, 5, 1519–1530. [Google Scholar]
- Song, X.; Gao, T.; Wang, N.; Feng, Q.; You, X.; Ye, T.; Lei, Q.; Zhu, Y.; Xiong, M.; Xia, Y.; et al. Selective inhibition of EZH2 by ZLD1039 blocks H3K27 methylation and leads to potent anti-tumor activity in breast cancer. Sci. Rep. 2016, 6, 20864. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.C.; Abdullah, L.N.; Pang, Q.Y.; Jha, S.; Chow, E.K.; Yang, H.; Kato, H.; Poellinger, L.; Ueda, J.; Lee, K.L. Inhibition of the H3K9 methyltransferase G9A attenuates oncogenicity and activates the hypoxia signaling pathway. PLoS ONE 2017, 12, e0188051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Jia, L.F.; Gan, Y.H. PTEN activation through K163 acetylation by inhibiting HDAC6 contributes to tumour inhibition. Oncogene 2016, 35, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef]
- Hsieh, H.Y.; Chuang, H.C.; Shen, F.H.; Detroja, K.; Hsin, L.W.; Chen, C.S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef]
- Hayward, D.; Cole, P.A. LSD1 Histone Demethylase Assays and Inhibition. Methods Enzym. 2016, 573, 261–278. [Google Scholar]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Huang, Y.; Marton, L.J.; Woster, P.M.; Davidson, N.E.; Casero, R.A., Jr. Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids 2012, 42, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Metzger, E.; Stepputtis, S.S.; Strietz, J.; Preca, B.T.; Urban, S.; Willmann, D.; Allen, A.; Zenk, F.; Iovino, N.; Bronsert, P.; et al. KDM4 Inhibition Targets Breast Cancer Stem-like Cells. Cancer Res. 2017, 77, 5900–5912. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, R.; Dell’Aversana, C.; De Marchi, T.; Rotili, D.; Liu, N.Q.; Novakovic, B.; Boccella, S.; Di Maro, S.; Cosconati, S.; Baldi, A.; et al. Inhibition of Histone Demethylases LSD1 and UTX Regulates ERalpha Signaling in Breast Cancer. Cancers 2019, 11, 2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuyas, E.; Gumuzio, J.; Verdura, S.; Brunet, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Alarcon, T.; Encinar, J.A.; Martin, A.G.; Menendez, J.A. The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: A potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging Albany NY 2020, 12, 4794–4814. [Google Scholar] [CrossRef]
- Zhou, M.; Venkata, P.P.; Viswanadhapalli, S.; Palacios, B.; Alejo, S.; Chen, Y.; He, Y.; Pratap, U.P.; Liu, J.; Zou, Y.; et al. KDM1A inhibition is effective in reducing stemness and treating triple negative breast cancer. Breast Cancer Res. Treat. 2021, 185, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.; Sachdev, J.; Ejadi, S.; Borad, M.; Kang, Y.K.; Lim, H.; Kim, T.; Bader, A.; Stoudemire, J. Abstract C43: Safety, tolerability, and clinical activity of MRX34, the first-in-class liposomal miR-34 mimic, in patients with advanced solid tumors. AACR 2015. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, F.; Chen, L.; Yang, Y.; Cao, S.; Ye, Y.; Wang, X.; Mu, J.; Li, Z.; Li, L. Blockage of TGFbeta-SMAD2 by demethylation-activated miR-148a is involved in caffeic acid-induced inhibition of cancer stem cell-like properties in vitro and in vivo. FEBS Open Biol. 2015, 5, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Li, Y.; Mu, J.; Hu, C.; Zhou, M.; Wang, X.; Si, L.; Ning, S.; Li, Z. Glabridin inhibits cancer stem cell-like properties of human breast cancer cells: An epigenetic regulation of miR-148a/SMAd2 signaling. Mol. Carcinog 2016, 55, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xu, C.; Xiao, G.; Meng, J.; Wang, J.; Tang, S.C.; Qin, S.; Du, N.; Li, G.; Ren, H.; et al. Breast cancer stem-like cells are sensitized to tamoxifen induction of self-renewal inhibition with enforced Let-7c dependent on Wnt blocking. Int. J. Mol. Med. 2018, 41, 1967–1975. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Shin, D.H.; Kim, J.S. Let-7 miRNA and CDK4 siRNA co-encapsulated in Herceptin-conjugated liposome for breast cancer stem cells. Asian J. Pharm. Sci. 2020, 15, 472–481. [Google Scholar] [CrossRef]
- Lin, X.; Chen, W.; Wei, F.; Xie, X. TV-circRGPD6 Nanoparticle Suppresses Breast Cancer Stem Cell-Mediated Metastasis via the miR-26b/YAF2 Axis. Mol. Ther. 2021, 29, 244–262. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Hopfinger, N.R.; Nguyen, T.D.; Pogash, T.J.; Santucci-Pereira, J.; Russo, J. Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J. Exp. Clin. Cancer Res. 2018, 37, 314. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents-A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, L.J.; Stojanovic, L.; Kogan, A.A.; Rutherford, J.L.; Choi, E.Y.; Yen, R.C.; Xia, L.; Zou, Y.; Lapidus, R.G.; Baylin, S.B.; et al. Pharmacologic induction of innate immune signaling directly drives homologous recombination deficiency. Proc. Natl. Acad. Sci. USA 2020, 117, 17785–17795. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Du, Y.; Nakai, K.; Ding, M.; Chang, S.S.; Hsu, J.L.; Yao, J.; Wei, Y.; Nie, L.; Jiao, S.; et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 2018, 37, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.; Vasilatos, S.N.; Bhargava, R.; Fine, J.L.; Oesterreich, S.; Davidson, N.E.; Huang, Y. Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 2017, 36, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/KappaDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Vasilatos, S.N.; Chen, L.; Wu, H.; Cao, Z.; Fu, Y.; Huang, M.; Vlad, A.M.; Lu, B.; Oesterreich, S.; et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2019, 38, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, T.; Wu, F.; Khanna, K.K.; Yip, D.; Malik, L.; Dahlstrom, J.E.; Rao, S. Optimizing poly (ADP-ribose) polymerase inhibition through combined epigenetic and immunotherapy. Cancer Sci. 2018, 109, 3383–3392. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Qu, J.; Jin, N.; Xu, J.; Lin, C.; Chen, Y.; Yang, X.; He, X.; Tang, S.; Lan, X.; et al. Feedback Activation of Leukemia Inhibitory Factor Receptor Limits Response to Histone Deacetylase Inhibitors in Breast Cancer. Cancer Cell 2016, 30, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Luu, T.; Kim, K.P.; Blanchard, S.; Anyang, B.; Hurria, A.; Yang, L.; Beumer, J.H.; Somlo, G.; Yen, Y. Phase IB trial of ixabepilone and vorinostat in metastatic breast cancer. Breast Cancer Res. Treat. 2018, 167, 469–478. [Google Scholar] [CrossRef]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Ho, S.Y.; Lin, P.; Wang, Y.J. Combination of the novel histone deacetylase inhibitor YCW1 and radiation induces autophagic cell death through the downregulation of BNIP3 in triple-negative breast cancer cells in vitro and in an orthotopic mouse model. Mol. Cancer 2016, 15, 46. [Google Scholar] [CrossRef] [Green Version]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, K.; Cole, J.J.; VanderKraats, N.D.; McBryan, T.; Pchelintsev, N.A.; Clark, W.; Copland, M.; Edwards, J.R.; Adams, P.D. DNMT inhibitors reverse a specific signature of aberrant promoter DNA methylation and associated gene silencing in AML. Genome Biol. 2014, 15, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Liu, T.; Qin, S.; Zhuang, Y.; Liu, D.; Lu, S.W.; Kalari, K.R.; et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Investig. 2018, 128, 2376–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, R.M.; Li, H.; Jankowitz, R.C.; Zhang, Z.; Rudek, M.A.; Jeter, S.C.; Slater, S.A.; Powers, P.; Wolff, A.C.; Fetting, J.H.; et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin. Cancer Res. 2017, 23, 2691–2701. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-Torres, K.; Gluck, C.; Sinha, S.; Gabrilovich, D.; Chakrabarti, R. DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Investig. 2018, 128, 5095–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Zhang, Q.; Shao, N.; Shan, Z.; Cheang, T.; Zhang, Z.; Su, Q.; Wang, S.; Lin, Y. Respiratory hyperoxia reverses immunosuppression by regulating myeloid-derived suppressor cells and PD-L1 expression in a triple-negative breast cancer mouse model. Am. J. Cancer Res. 2019, 9, 529–545. [Google Scholar]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, A.; Sulaiman, B.; Khouri, L.; McGarry, S.; Nessim, C.; Arnaout, A.; Li, X.; Addison, C.; Dimitroulakos, J.; Wang, L. Both bulk and cancer stem cell subpopulations in triple-negative breast cancer are susceptible to Wnt, HDAC, and ERalpha coinhibition. FEBS Lett. 2016, 590, 4606–4616. [Google Scholar] [CrossRef]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, R.; Illert, A.L.; Erbes, T.; Flotho, C.; Lubbert, M.; Duque-Afonso, J. The epigenetics of breast cancer-Opportunities for diagnostics, risk stratification and therapy. Epigenetics 2021, 1–13. [Google Scholar] [CrossRef]
- Arana Yi, C.; Tam, C.S.; Verstovsek, S. Efficacy and safety of ruxolitinib in the treatment of patients with myelofibrosis. Future Oncol. 2015, 11, 719–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
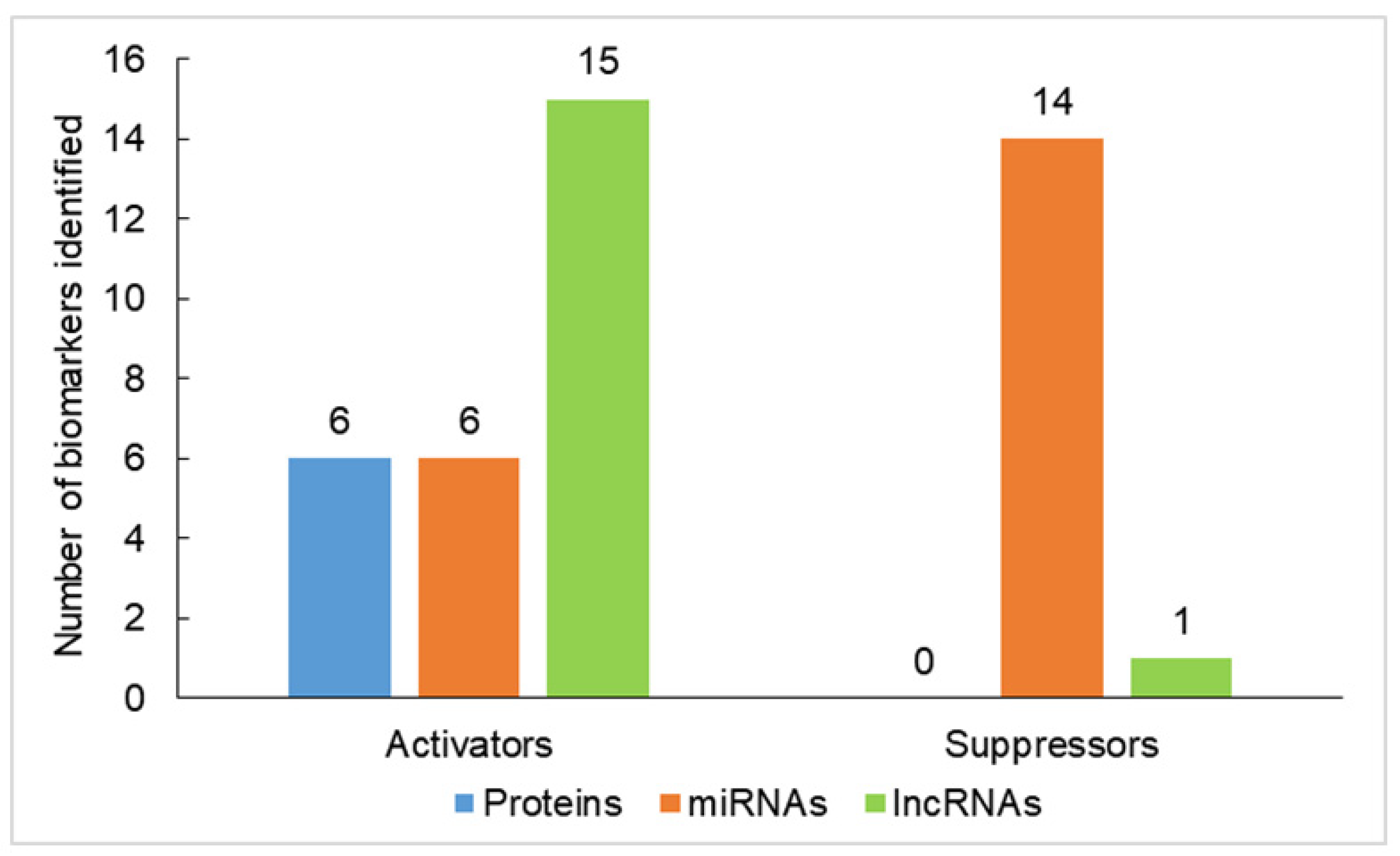

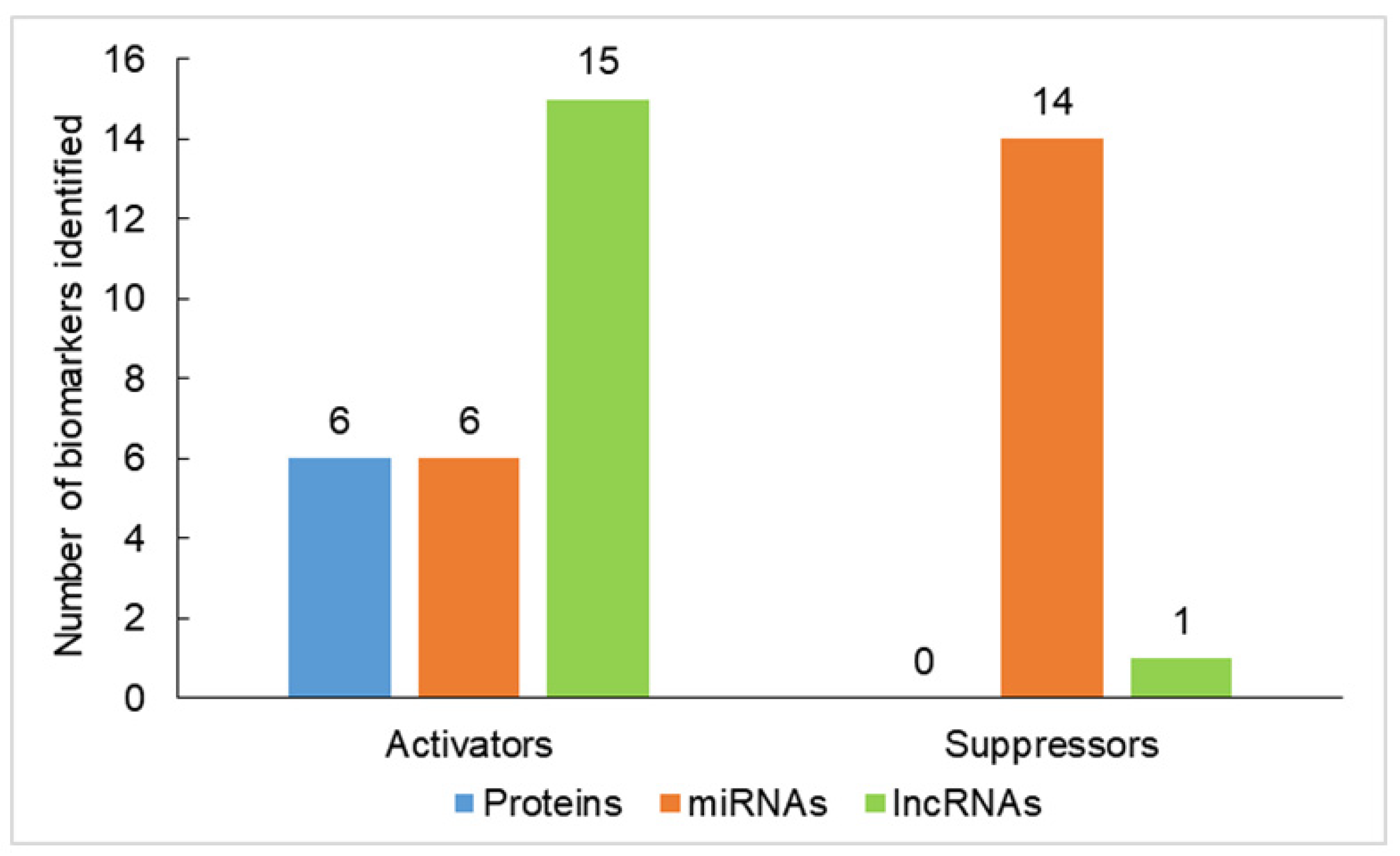{kind=link}
{kind=link}
| Biomarkers | Function | Role in Carcinogenesis | References |
|---|---|---|---|
| Oncogenes or tumor activators | |||
| Proteins | |||
| DNMT1 | Catalyzes hypermethylation of the cytosines and represses gene transcription. | DNMT1 is highly expressed in CSCs in mammospheres and tumorospheres, and DNMT1 deletion suppresses mammary tumorigenesis. ISL1 is hypermethylated and downregulated by DNMT1 in breast cancers and BCSCs. | [76,77,78] |
| DNMT1 silencing reduced MSFE in triple-negative breast cancer (TNBC). | [79] | ||
| BMI1 (B lymphoma Mo-MLV insertion region homolog) | Component of PRC1 that plays a crucial role in epigenetic regulation of various physiological processes, such as cell differentiation, stem cell self-renewal and gene-silencing, through histone modifications. | Increases self-renewal capacity of BCSCs and promotes EMT. | [80,81,82] |
| EZH2 | A PRC2 group protein and a HMT that methylates H3K27 and functions as a transcriptional repressor. | Inhibits the expression of tumor suppressor genes, such as P16 INK4a, E-cadherin, BRCA1 and the adrenergic receptor β2. Activates the Notch1 expression and signaling, leading to stem cell expansion in TNBC. | [83,84,85] |
| SETDB1 (SET Domain Bifurcated Histone Lysine Methyltransferase 1) | HMT catalyzes the di- and tri-methylation of H3K9 to induce gene-silencing. | Promotes breast cancer metastasis through the acquisition of stem-cell-like properties and EMT. | [86,87,88] |
| LSD1 | Removes methyl groups from methylated proteins, including H3K4 and non-histone proteins, such as p53 and DNMT1. | Induces gene expression involved in EMT and elicits the BCSC program. | [89,90] |
| HDAC | Catalyzes the hydrolytic removal of acetyl groups from histone lysine residues. | Plays a significant role in the epigenetic regulation of CSC miRNAs. | [65,91] |
| miRNAs | |||
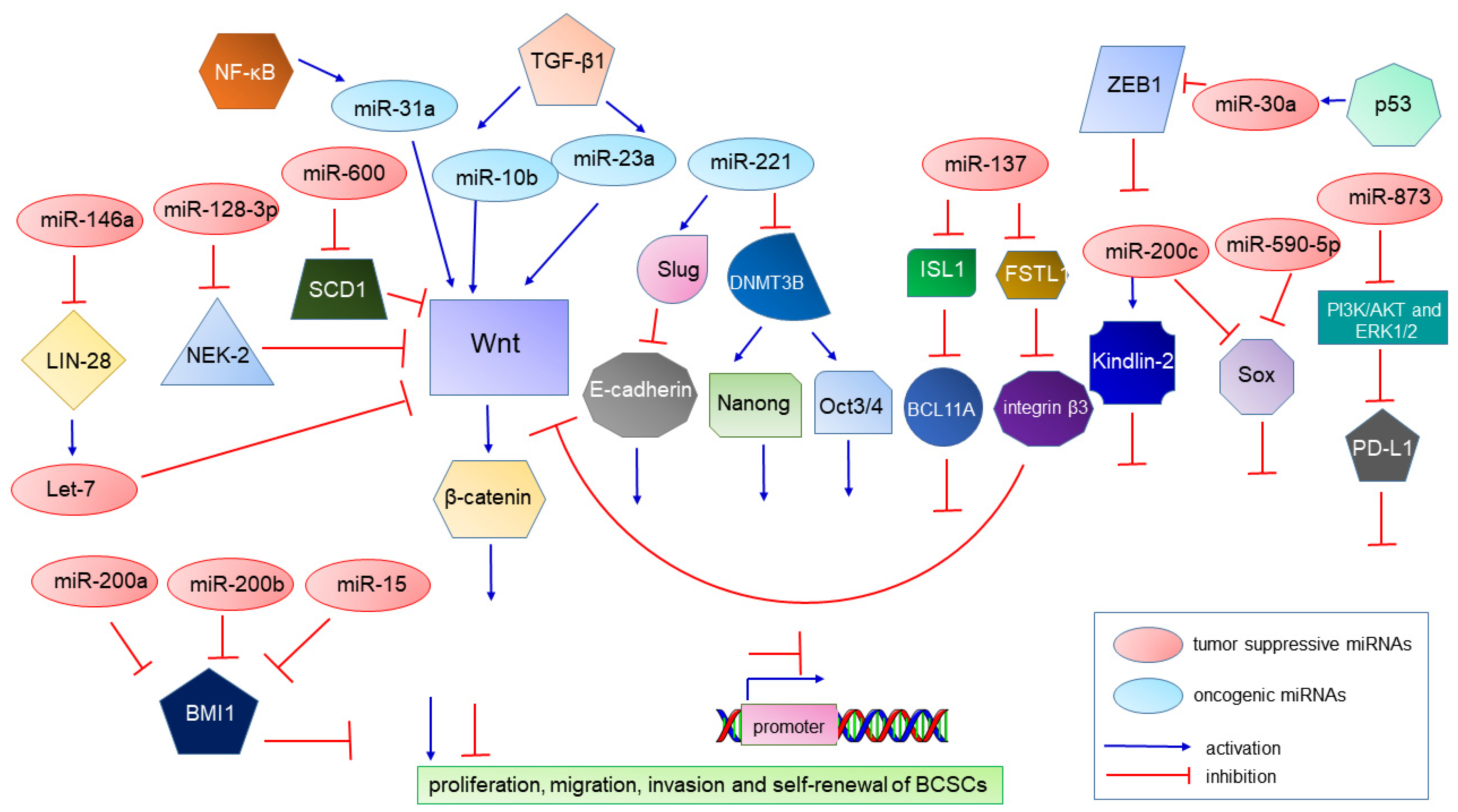
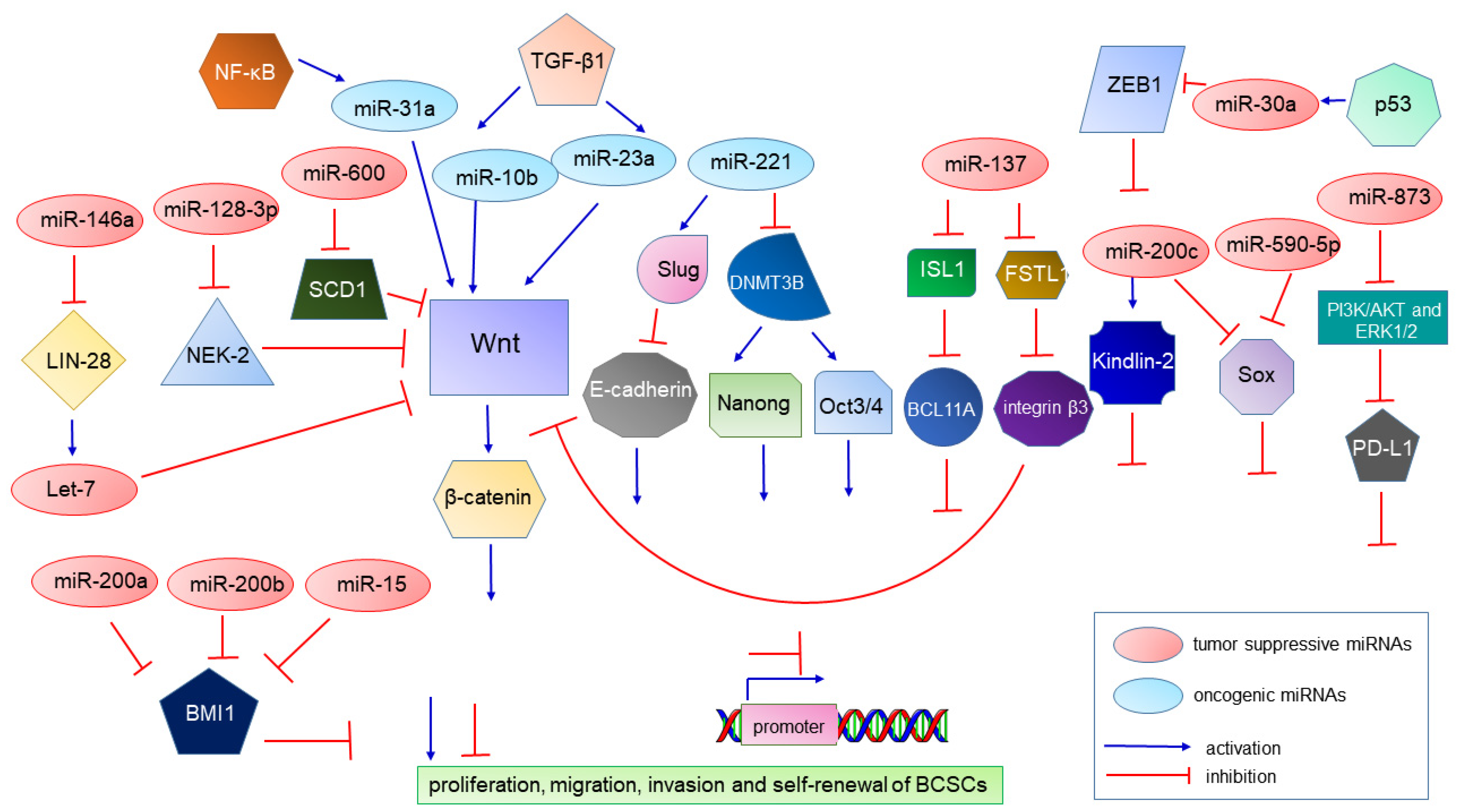
| miR-10b | oncomiR | Contributes to TGF-β1-induced EMT and tumor metastasis. | [92,93] |
| miR-23a | oncomiR | Contributes to TGF-β1-induced EMT and tumor metastasis. | [94] |
| miR-221 | oncomiR | Suppresses CDH1 resulting in E-cadherin suppression. Represses the DNMT3B gene, and this leads to suppression of the NANOG and OCT 3/4 genes and contributes to stemness maintenance in breast cancer. | [95,96] |
| miR-221/222 cluster | oncomiR | Induces the growth, migration, invasion and propagation of BCSCs. | [97,98] |
| miR-31 | oncomiR | Increases the BCSC subpopulation and tumor initiation and metastasis abilities. | [99] |
| miR-520b | oncomiR | Is upregulated in breast cancer tissue and BCSCs and promotes the stemness. | [100] |
| lncRNAs | |||
| HOTAIR | During embryonic development, HOTAIR regulates the silencing of the distant HOXD locus. | Downregulates miRNA-7 associated with EMT and STAT3 activity. | [101,102,103,104] |
| SOX2OT and linc00617 | oncogenes | The stemness factor SOX2 is upregulated in BCSCs by SOX2OT and linc00617. | [105,106] |
| lncRNA-Hh | Oncogene | The self-renewal HH pathway is activated, which promotes CSC maintenance. | [107] |
| H19 | Functions in the epigenetic silencing of the IGF2 gene. | Promotes metastasis through EMT induction. Overexpression of H19 enhances clonogenicity, migration and mammosphere formation. | [108,109,110,111,112,113] |
| LncRNA-HAL | Oncogene | HAL silencing increases cell proliferation and impairs the proportion and function of CSCs. | [114] |
| LINC01133 | Oncogene | Induces the BCSC phenotype. | [115] |
| lncRNA EPIC1 | Oncogene | Overexpression of EPIC1 is correlated to poor survival outcomes in luminal B breast cancer. | [116] |
| lncRNA SOX21-AS1 | Oncogene | Promotes BCSC properties and carcinogenesis via inhibiting Sox2 or the Hippo signaling pathway. | [117,118] |
| lncRNA THOR | Oncogene | Silencing of THOR induces reductions in mammosphere formation, stemness marker expression and ALDH1 activity of BCSCs. | [119] |
| LncCCAT1 | Oncogene | Is significantly upregulated in breast cancer tissue and BCSCs, leading to poor patient outcomes. | [120] |
| LncRNA LUCAT1 | Oncogene | Is expressed in breast cancer tissue and highly expressed in BCSCs, and regulates stemness features. | [121] |
| MALAT1 | Has vital biological implications. | Is overexpressed in BCSC MCF7, and its knockdown decreases the proportion of BCSC MCF7 and mammosphere formation. | [110,122] |
| lncRNA FEZF1-AS | Oncogene | Knockdown of LncRNA FEZF1-AS reduces the ability of BCSC to form mammospheres, the expression of stem cell markers and the rate of CD44+/CD24− production. | [123] |
| lncRNA LINC00511 | Oncogene | Is highly expressed in breast cancer, which is correlated with the poor prognosis of patients. | [124] |
| Tumor suppressors | |||
| miRNAs | |||
| miR-200c | A tumor suppressor | Targeting the self-renewal gene Bmi-1 represses tumorigenicity of human BCSCs in vivo, and also targets Pin1 to regulate stemness of human primary BCSCs. | [125,126] |
| miR-200a, miR-200b and miR-15 | Tumor suppressors | Overexpression of miR-200a, miR-200b and miR-15 decreased BMI1 and Ub-H2A protein expression in the CD44+ CSC population of MDA-MB-231 cells. | [127] |
| miR-200b | A tumor suppressor | Inactivates FERMT2 and results in the inhibition of EMT and metastasis. | [128] |
| let-7 | A tumor suppressor | Plays a significant role in BCSC, which is controlled via DNA methylation. | [129,130] |
| miR-30a | A tumor suppressor | Suppresses the ZEB2 expression and controls aggressiveness. | [131] |
| miR-590-5p | A tumor suppressor | Inhibits stemness by targeting the SOX2 gene and leads to a decrease in the BCSC population. | [132] |
| miR-140 | A tumor suppressor | Inhibits stemness by targeting the SOX2 gene and leads to a decrease in the BCSC population. | [133] |
| miR-146a | A tumor suppressor | Plays a role in mediating the induction and maintenance of BCSCs during EMT. | [134,135,136] |
| miR-600 | A tumor suppressor | Reduces BCSC self-renewal through the inhibition of Wnt. | [137] |
| miR-128-3p | A tumor suppressor | Inhibits cell proliferation, migration, invasion and self-renewal of BCSCs. | [138] |
| miR-137 | A tumor suppressor | Inhibits stemness and chemoresistance. | [139] |
| miR-873 | A tumor suppressor | Inactivates PI3K/AKT and ERK1/2 signaling and attenuates the stemness and chemoresistance abilities of BCSCs. | [140] |
| lncRNAs | |||
| lncRNA FGF13-AS1 | A tumor suppressor | Is downregulated in breast cancer and inhibits glycolysis and stemness properties of breast cancer cells. | [141] |
| Targets | Mechanism of Treatment | Treatment | Status | References |
|---|---|---|---|---|
| DNMT | Lead to hypomethylation and gene de-repression, resulting in preventing EMT. | DNMT inhibitors: 5-AzaC (Vidaza) and 5-aza-20-deoxycytidine 5-AzaDC (decitabine) | The single use of 5-AzaDC or HDACi has been approved by the FDA for hematologic malignancies. | [200,201,202] |
| Guadecitabine (SGI-110) | Phase 2 clinical trial | [202] | ||
| JMJD2 | Attenuates the growth of breast cancer cells. | NCDM-32B, a JMJD2 inhibitor | Tested on the breast cancer cells | [203] |
| EZH2 | Demonstrated a strong anti-cancer effect by inhibiting breast tumor growth and metastasis. | ZLD1039, an EZH2 inhibitor | Tested on xenograft-bearing mice | [204] |
| G9a | Induces apoptosis and impairs cell migration, cell cycle and anchorage-dependent growth in breast cancer cells. | BIX-01294, a G9a inhibitor | Tested on cells | [205] |
| HDAC | Induces PTEN membrane translocation through PTEN acetylation at K163 by inhibiting HDAC6, and this PTEN activation leads to inhibition of tumor growth. | HDACi, such as Trichostatin A (TSA) or suberoylanilide hydroxamic acid (SAHA) | Tested on xenograft tumor model | [206] |
| Inhibits HDAC1 and HDAC7, that may therefore modify the epigenetic markers that characterize CSCs. | HDACi, TSA, a pan HDACi | Tested on the mouse model | [207] | |
| Inhibits HDAC3. | A pan-HDAC inhibitor (HDACi), AR-42 | Tested on the mouse model | [208] | |
| LSD1 | Alter promoter activity of multiple genes in breast cancer cells. | LSD1 inhibitors: bizine, the tranylcypromine derivatives NCL1 and GSK2879552, biguanide, bisguanidine polyamine analogs and GSK287 | Under clinical evaluation for cancer treatment | [209,210] |
| Alter promoter activity of multiple genes in breast cancer cells. | polyamine analog inhibitors of LSD1, 2d or PG11144 | Tested on cells | [211] | |
| Partially inhibited CSC formation. | LSD1 inhibitor pargyline | Tested on mouse model | [90] | |
| Reduced tumor growth of patient-derived CSCs. | LSD1 inhibitor QC6352 | Tested on the xenograft model | [212] | |
| Induces significant growth arrest and apoptosis | A dual HDM inhibitor (MC3324) | Tested on both xenograft mice and chicken embryo models | [213] | |
| Could reduce colony formation and a decrease in SOX2 expression | LSD1 inhibitor iadademstat (ORY-100) | Tested on the patient-derived xenograft model | [214] | |
| Targeting BCSCs in vitro and in vivo | KDM1A inhibitor NCD38 | Tested on in orthotopic xenograft models | [215] | |
| miR-34 | Epigenetic restoration of miR-34 could sensitize cancer cells to drugs and suppress stem cell features. | miR-34-based drug MRX34 | Passed phase I clinical studies | [216] |
| miR-148a | Inhibits the BCSC properties via miR-148a-mediated inhibition of the TGF-β-SMAD2 signaling pathway. | CaA | Tested on mouse xenograft model | [217] |
| glabridin | Tested on mouse xenograft models | [218] | ||
| let-7 | The suppressive effects exerted by let-7 on stem-like cells involved let-7c/ER/Wnt signaling. | Tamoxifen (ER modulator) | Tested on xenografted tumor model | [219] |
| Efficient liposomal delivery system for the combination of miRNA and siRNA to target the BCSCs. | Herceptin-conjugated cationic immuno-liposome with hyaluronic acid and protamine | Tested on cells | [220] | |
| miR-26b | Suppresses BCSC metastasis via the miR-26b/YAF2 axis. | TV-circRGPD6 nanoparticle | Tested on orthotopic xenograft models | [221] |
| Combination therapy | ||||
| DNMT and HDAC | Reprogram aggressive TNBC cells that have undergone EMT into a less aggressive phenotype. Suppressed TNBC cell proliferation, colony formation, motility and stemness of the cancer cells in vitro and in vivo. | DNMTi, guadecitabine, in combination with HDACi, entinostat | XtMCF cells in CB17/SCID mice | [222] |
| DNMT and PARP | Enhanced tight binding of talazoparib to DNA and increased DSB formation and cytotoxicity. | DNMTi combined with PARPi, talazoparib | Tested on xenograft tumors | [223] |
| DNMT and PARP | DNMTi increased PARP trapping and reprogramed the DNA damage response to cause HRD, sensitizing BRCA-proficient cancer cells to PARPi. | DNMTi and PARPi | Will be tested in a phase I/II TNBC clinical trial | [224] |
| EZH2 and PARP | Combination showed increased sensitivity to PARP inhibition. | Combined PARP inhibition and EZH2 inhibition | Tested on xenograft model | [225] |
| LSD1 and HDAC | LSD1 interacts with HDACs to control breast cancer cell growth. | Combined treatment of LSD1 inhibitor, pargyline and HDACi, SAHA | Tested on cells | [226] |
| LSD1 | Reduced stem cell potential and increased chemo-sensitivity. | LSD1 inhibitor combined with doxorubicin | Tested on xenograft model | [227] |
| LSD1 and PD-1 | Suppressed tumor growth and pulmonary metastasis. | LSD1 inhibition in combination with anti-PD1 antibodies | Tested on xenograft tumors | [228] |
| lysine-specific histone demethylase-1A/HDAC, PARP and PD-1/PD-L1 | Targeting BCSC and overcoming resistance and recurrence. | Lysine-specific histone demethylase-1A inhibitors or HDACi and PARPi/anti-PD-1/PD-L1 | - | [229] |
| HDAC and PD-L1 | Inhibits tumor growth and increases survival. | HDACi and immune checkpoint inhibitors | Tested on breast cancer mouse model | [230] |
| HDAC and JAK/BRD4 | JAK/BRD4 inhibition sensitizes TNBC cells to HDAC inhibitions. | The combination of HDACi and JAK/BRD4 inhibitors | Tested on mouse models | [231] |
| HDAC | The objective response rates were comparable to those achieved with the previously approved ixabepilon monotherapy or combination with capecitabine. | Combination of the cytostatic drug ixabepilon with HDACi vorinostat | A phase Ib study | [232] |
| HDAC | Enhanced toxicity and increasing autophage induction. | The combination therapy of HDACi YCW1 with ionizing radiation. This led to induction of cell death in TBNC cell lines in vitro and in mouse models. | Tested on orthotopic mouse model | [233] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.-J.; Chu, P.-Y. Epigenetic Regulation of Breast Cancer Stem Cells Contributing to Carcinogenesis and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 8113. https://doi.org/10.3390/ijms22158113
Wu H-J, Chu P-Y. Epigenetic Regulation of Breast Cancer Stem Cells Contributing to Carcinogenesis and Therapeutic Implications. International Journal of Molecular Sciences. 2021; 22(15):8113. https://doi.org/10.3390/ijms22158113
Chicago/Turabian StyleWu, Hsing-Ju, and Pei-Yi Chu. 2021. "Epigenetic Regulation of Breast Cancer Stem Cells Contributing to Carcinogenesis and Therapeutic Implications" International Journal of Molecular Sciences 22, no. 15: 8113. https://doi.org/10.3390/ijms22158113
APA StyleWu, H.-J., & Chu, P.-Y. (2021). Epigenetic Regulation of Breast Cancer Stem Cells Contributing to Carcinogenesis and Therapeutic Implications. International Journal of Molecular Sciences, 22(15), 8113. https://doi.org/10.3390/ijms22158113