
Immunoendocrine Dysregulation during Gestational Diabetes Mellitus: The Central Role of the Placenta

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Role of Placenta in the Endocrine Milieu of GDM
2.1. The Insulin and IGF-I/IGF-II Axis and Molecular Pathways Primarily Disturbed in GDM Placentae
2.2. The Role of Pancreatic β-Cells and Lactotroph Hormones in GDM
3. Hyperglycemia-Induced Metainflammation in GDM Alters Placental Immune Cells Population Favoring an Inflammatory Cytokine Signature and an Imbalance in Adipokines and Defense Peptides
3.1. The Role of Immune Cells in GDM Placentae
3.2. Major Placental Cytokines in GDM
3.3. Altered Production of Adipokines by the GDM Placenta
3.4. Placental Innate Defense-Peptides in GDM
4. Vitamin D Implications in Immune Regulation and Insulin Resistance in GDM
5. Endovascular Changes in GDM: Endothelial Dysfunction and Overstimulation of Placental Angiogenesis
6. GDM and Oxidative Stress
ROS Changes Associated with a Hyperglycemic Environment
7. Adverse Perinatal Outcomes Related to GDM
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- ACOG Practice Bulletin No. 190: Gestational Diabetes Mellitus. Obstet. Gynecol. 2018, 131, e49–e64. [CrossRef]
- Hyperglycemia and Adverse Pregnancy Outcomes. N. Engl. J. Med. 2008, 358. [CrossRef]
- Murray, S.R.; Reynolds, R.M. Short- and Long-Term Outcomes of Gestational Diabetes and Its Treatment on Fetal Development. Prenat. Diagn. 2020, 40. [Google Scholar] [CrossRef]
- Bianco, M.E.; Josefson, J.L. Hyperglycemia During Pregnancy and Long-Term Offspring Outcomes. Curr. Diabetes Rep. 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.T.; Thomas, C.C. Updates in Gestational Diabetes Prevalence, Treatment, and Health Policy. Curr. Diabetes Rep. 2019, 19. [Google Scholar] [CrossRef]
- Brawerman, G.M.; Dolinsky, V.W. Therapies for Gestational Diabetes and Their Implications for Maternal and Offspring Health: Evidence from Human and Animal Studies. Pharmacol. Res. 2018, 130. [Google Scholar] [CrossRef] [PubMed]
- McCance, D.R. Diabetes in Pregnancy: Management from Preconception to the Postnatal Period; NICE Guideline; NICE: Hoboken, NJ, USA, 2015; Volume 29, Available online: https://www.nice.org.uk/guidance/ng3/resources/diabetes-in-pregnancy-management-from-preconception-to-the-postnatal-period-pdf-51038446021 (accessed on 1 June 2021).
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef] [PubMed]
- Moyce, B.L.; Dolinsky, V.W. Maternal β-Cell Adaptations in Pregnancy and Placental Signalling: Implications for Gestational Diabetes. Int. J. Mol. Sci. 2018, 19, 3467. [Google Scholar] [CrossRef] [PubMed]
- Dennison, R.A.; Chen, E.S.; Green, M.E.; Legard, C.; Kotecha, D.; Farmer, G.; Sharp, S.J.; Ward, R.J.; Usher-Smith, J.A.; Griffin, S.J. The Absolute and Relative Risk of Type 2 Diabetes after Gestational Diabetes: A Systematic Review and Meta-Analysis of 129 Studies. Diabetes Res. Clin. Pract. 2021, 171. [Google Scholar] [CrossRef]
- American Diabetes Association. Management of Diabetes in Pregnancy: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43. [Google Scholar] [CrossRef]
- Schäfer-Graf, U.M.; Gembruch, U.; Kainer, F.; Groten, T.; Hummel, S.; Hösli, I.; Grieshop, M.; Kaltheuner, M.; Bührer, C.; Kautzky-Willer, A.; et al. Gestational Diabetes Mellitus (GDM)—Diagnosis, Treatment and Follow-UpGuideline of the DDG and DGGG (S3 Level, AWMF Registry Number 057/008, February 2018). Geburtshilfe Frauenheilkd. 2018, 78. [Google Scholar] [CrossRef]
- Alqudah, A.; McKinley, M.C.; McNally, R.; Graham, U.; Watson, C.J.; Lyons, T.J.; McClements, L. Risk of Pre-Eclampsia in Women Taking Metformin: A Systematic Review and Meta-Analysis. Diabet. Med. 2018, 35. [Google Scholar] [CrossRef]
- Martis, R.; Crowther, C.A.; Shepherd, E.; Alsweiler, J.; Downie, M.R.; Brown, J. Treatments for Women with Gestational Diabetes Mellitus: An Overview of Cochrane Systematic Reviews. Cochrane Database Syst. Rev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yang, H. Metformin–a Potentially Effective Drug for Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. J. Matern. Fetal Neonatal Med. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, W.; Chen, H.; Chen, Q. Comparison of Insulin, Metformin, and Glyburide on Perinatal Complications of Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. Gynecol. Obstet. Investig. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Liu, Q.; Feng, L. Metformin vs Insulin in the Management of Gestational Diabetes: A Meta-Analysis. PLoS ONE 2013, 8, e64585. [Google Scholar] [CrossRef]
- Landi, S.N.; Radke, S.; Engel, S.M.; Boggess, K.; Stürmer, T.; Howe, A.S.; Funk, M.J. Association of Long-Term Child Growth and Developmental Outcomes with Metformin vs Insulin Treatment for Gestational Diabetes. JAMA Pediatr. 2019, 173. [Google Scholar] [CrossRef]
- Wouldes, T.A.; Battin, M.; Coat, S.; Rush, E.C.; Hague, W.M.; Rowan, J.A. Neurodevelopmental Outcome at 2 Years in Offspring of Women Randomised to Metformin or Insulin Treatment for Gestational Diabetes. Arch. Dis. Child. Fetal Neonatal Ed. 2016, 101. [Google Scholar] [CrossRef]
- Carrasco-Wong, I.; Moller, A.; Giachini, F.R.; Lima, V.V.; Toledo, F.; Stojanova, J.; Sobrevia, L.; San Martín, S. Placental Structure in Gestational Diabetes Mellitus. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866. [Google Scholar] [CrossRef]
- Ehlers, E.; Talton, O.O.; Schust, D.J.; Schulz, L.C. Placental Structural Abnormalities in Gestational Diabetes and When They Develop: A Scoping Review. Placenta 2021. [Google Scholar] [CrossRef]
- Lucas, M.J. Diabetes Complicating Pregnancy. Obstet. Gynecol. Clin. N. Am. 2001, 28. [Google Scholar] [CrossRef]
- Huynh, J.; Dawson, D.; Roberts, D.; Bentley-Lewis, R. A Systematic Review of Placental Pathology in Maternal Diabetes Mellitus. Placenta 2015, 36. [Google Scholar] [CrossRef] [PubMed]
- Alqudah, A.; Eastwood, K.A.; Jerotic, D.; Todd, N.; Hoch, D.; McNally, R.; Obradovic, D.; Dugalic, S.; Hunter, A.J.; Holmes, V.A.; et al. FKBPL and SIRT-1 Are Downregulated by Diabetes in Pregnancy Impacting on Angiogenesis and Endothelial Function. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, G.; Marinopoulos, S.; Krielesi, V.; Papapanagiotou, A.; Papantoniou, N.; Mesogitis, S.; Antsaklis, A. Placental Pathology in Women with Gestational Diabetes. Acta Obstet. Gynecol. Scand. 2008, 87. [Google Scholar] [CrossRef]
- Madazli, R.; Tuten, A.; Calay, Z.; Uzun, H.; Uludag, S.; Ocak, V. The Incidence of Placental Abnormalities, Maternal and Cord Plasma Malondialdehyde and Vascular Endothelial Growth Factor Levels in Women with Gestational Diabetes Mellitus and Nondiabetic Controls. Gynecol. Obstet. Investig. 2008, 65. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.J.; Vanderpeet, C.L.; Bartho, L.A.; McKeating, D.R.; Cuffe, J.S.M.; Holland, O.J.; Perkins, A.V. Mitochondrial Dysfunction in Placental Trophoblast Cells Experiencing Gestational Diabetes Mellitus. J. Physiol. 2021, 599. [Google Scholar] [CrossRef]
- Valent, A.M.; Choi, H.; Kolahi, K.S.; Thornburg, K.L. Hyperglycemia and Gestational Diabetes Suppress Placental Glycolysis and Mitochondrial Function and Alter Lipid Processing. FASEB J. 2021, 35. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.S.; Zhao, H.; Hattis, S.; Kumar, S. Elevated Glucose and Insulin Levels Decrease DHA Transfer across Human Trophoblasts via SIRT1-Dependent Mechanism. Nutrients 2020, 12, 1271. [Google Scholar] [CrossRef] [PubMed]
- Hansis, C.; Grifo, J.A.; Tang, Y.X.; Krey, L.C. Assessment of Beta-HCG, Beta-LH MRNA and Ploidy in Individual Human Blastomeres. Reprod. Biomed. Online 2002, 5. [Google Scholar] [CrossRef]
- Butler, S.A.; Luttoo, J.; Freire, M.O.T.; Abban, T.K.; Borrelli, P.T.A.; Iles, R.K. Human Chorionic Gonadotropin (HCG) in the Secretome of Cultured Embryos: Hyperglycosylated HCG and HCG-Free Beta Subunit Are Potential Markers for Infertility Management and Treatment. Reprod. Sci. 2013, 20. [Google Scholar] [CrossRef]
- Feldt-Rasmussen, U.; Mathiesen, E.R. Endocrine Disorders in Pregnancy: Physiological and Hormonal Aspects of Pregnancy. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25. [Google Scholar] [CrossRef]
- Rout, M.; Lulu, S.S. Molecular and Disease Association of Gestational Diabetes Mellitus Affected Mother and Placental Datasets Reveal a Strong Link between Insulin Growth Factor (IGF) Genes in Amino Acid Transport Pathway: A Network Biology Approach. J. Cell. Biochem. 2019, 120. [Google Scholar] [CrossRef]
- Alur, V.; Raju, V.; Vastrad, B.; Tengli, A.; Vastrad, C.; Kotturshetti, S. Integrated Bioinformatics Analysis Reveals Novel Key Biomarkers and Potential Candidate Small Molecule Drugs in Gestational Diabetes Mellitus. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, F.; Peng, Y.; Chen, R.; Zhou, W.; Wang, H.; OuYang, J.; Yu, B.; Xu, Z. Transcriptomic Profiling of Human Placenta in Gestational Diabetes Mellitus at the Single-Cell Level. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Westermeier, F.; Sáez, T.; Arroyo, P.; Toledo, F.; Gutiérrez, J.; Sanhueza, C.; Pardo, F.; Leiva, A.; Sobrevia, L. Insulin Receptor Isoforms: An Integrated View Focused on Gestational Diabetes Mellitus. Diabetes Metab. Res. Rev. 2016, 32. [Google Scholar] [CrossRef]
- Sobrevia, L.; Salsoso, R.; Fuenzalida, B.; Barros, E.; Toledo, L.; Silva, L.; Pizarro, C.; Subiabre, M.; Villalobos, R.; Araos, J.; et al. Insulin Is a Key Modulator of Fetoplacental Endothelium Metabolic Disturbances in Gestational Diabetes Mellitus. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef]
- Larner, J.; Huang, L.C.; Tang, G.; Suzuki, S.; Schwartz, C.F.W.; Romero, G.; Roulidis, Z.; Zeller, K.; Shen, T.Y.; Oswald, A.S.; et al. Insulin Mediators: Structure and Formation. Cold Spring Harb. Symp. Quant. Biol. 1988, 53. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of Glycogen Synthase Kinase-3 by Insulin Mediated by Protein Kinase B. Nature 1995, 378. [Google Scholar] [CrossRef]
- Kohn, A.D.; Kovacina, K.S.; Roth, R.A. Insulin Stimulates the Kinase Activity of RAC-PK, a Pleckstrin Homology Domain Containing Ser/Thr Kinase. EMBO J. 1995, 14. [Google Scholar] [CrossRef]
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the Underlying Defect in Insulin Action in Type 2 Diabetes. Diabetologia 2021, 64. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.T.; Lee, K.Y.; Klaus, K.; Softic, S.; Krumpoch, M.T.; Fentz, J.; Stanford, K.I.; Robinson, M.M.; Cai, W.; Kleinridders, A.; et al. Insulin and IGF-1 Receptors Regulate FoxO-Mediated Signaling in Muscle Proteostasis. J. Clin. Investig. 2016, 126. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the MTORC1 Protein Kinase. Mol. Cell 2007, 25. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex-1 and -2 Gene Products Function Together to Inhibit Mammalian Target of Rapamycin (MTOR)-Mediated Downstream Signaling. Proc. Natl. Acad. Sci. USA 2002, 99. [Google Scholar] [CrossRef]
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-Stimulated Phosphorylation of a Rab GTPase-Activating Protein Regulates GLUT4 Translocation. J. Biol. Chem. 2003, 278. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J.; Khokhlatchev, A.; Kyriakis, J.M.; Luo, Z.; Tzivion, G.; Vavvas, D.; Zhang, X.F. Ras Activation of the Raf Kinase: Tyrosine Kinase Recruitment of the MAP Kinase Cascade. Recent Prog. Horm. Res. 2001, 56. [Google Scholar] [CrossRef]
- Sesti, G.; Tullio, A.N.; D’Alfonso, R.; Napolitano, M.L.; Marini, M.A.; Borboni, P.; Longhi, R.; Albonici, L.; Fusco, A.; Aglianò, A.M.; et al. Tissue-Specific Expression of Two Alternatively Spliced Isoforms of the Human Insulin Receptor Protein. Acta Diabetol. 1994, 31. [Google Scholar] [CrossRef] [PubMed]
- Illsley, N.P.; Baumann, M.U. Human Placental Glucose Transport in Fetoplacental Growth and Metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Pazura-Turowska, M.; Sawicki, W.; Cendrowski, K. Placental Expression of Glucose Transporter Proteins in Pregnancies Complicated by Gestational and Pregestational Diabetes Mellitus. Can. J. Diabetes 2018, 42. [Google Scholar] [CrossRef]
- Hiden, U.; Maier, A.; Bilban, M.; Ghaffari-Tabrizi, N.; Wadsack, C.; Lang, I.; Dohr, G.; Desoye, G. Insulin Control of Placental Gene Expression Shifts from Mother to Foetus over the Course of Pregnancy. Diabetologia 2006, 49. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; del Rey, C.G.; Navarro, A.; Tolivia, J.; González, C.G. Effects of Gestational Diabetes Mellitus on Proteins Implicated in Insulin Signaling in Human Placenta. Gynecol. Endocrinol. 2006, 22. [Google Scholar] [CrossRef]
- Balachandiran, M.; Bobby, Z.; Dorairajan, G.; Gladwin, V.; Vinayagam, V.; Packirisamy, R.M. Decreased Maternal Serum Adiponectin and Increased Insulin-like Growth Factor-1 Levels along with Increased Placental Glucose Transporter-1 Expression in Gestational Diabetes Mellitus: Possible Role in Fetal Overgrowth: Regulation of Placental GLUT-1 Expression in Gestational Diabetes Mellitus. Placenta 2021, 104. [Google Scholar] [CrossRef]
- Colomiere, M.; Permezel, M.; Riley, C.; Desoye, G.; Lappas, M. Defective Insulin Signaling in Placenta from Pregnancies Complicated by Gestational Diabetes Mellitus. Eur. J. Endocrinol. 2009, 160. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Guadix, P.; Maymó, J.; Dueñas, J.L.; Varone, C.; Fernández-Sánchez, M.; Sánchez-Margalet, V. Insulin and Leptin Signaling in Placenta from Gestational Diabetic Subjects. Horm. Metab. Res. 2015, 48. [Google Scholar] [CrossRef] [PubMed]
- Gęca, T.; Kwaśniewska, A. The Influence of Gestational Diabetes Mellitus upon the Selected Parameters of the Maternal and Fetal System of Insulin-Like Growth Factors (IGF-1, IGF-2, IGFBP1-3)—A Review and a Clinical Study. J. Clin. Med. 2020, 9, 3256. [Google Scholar] [CrossRef]
- Osmond, D.T.D.; King, R.G.; Brennecke, S.P.; Gude, N.M. Placental Glucose Transport and Utilisation Is Altered at Term in Insulin-Treated, Gestational-Diabetic Patients. Diabetologia 2001, 44. [Google Scholar] [CrossRef] [PubMed]
- Osmond, D.T.D.; Nolan, C.J.; King, R.G.; Brennecke, S.P.; Gude, N.M. Effects of Gestational Diabetes on Human Placental Glucose Uptake, Transfer, and Utilisation. Diabetologia 2000, 43. [Google Scholar] [CrossRef]
- Tumminia, A.; Scalisi, N.M.; Milluzzo, A.; Ettore, G.; Vigneri, R.; Sciacca, L. Maternal Diabetes Impairs Insulin and IGF-1 Receptor Expression and Signaling in Human Placenta. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Forbes, B.E.; Blyth, A.J.; Wit, J.M. Disorders of IGFs and IGF-1R Signaling Pathways. Mol. Cell. Endocrinol. 2020, 518. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.R.; Wang, W.J.; Yu, X.; Hua, X.; Ouyang, F.; Luo, Z.C. Insulin-like Growth Factor Axis Biomarkers and Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-Like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell. Biol. 1999, 19. [Google Scholar] [CrossRef]
- Blanquart, C.; Achi, J.; Issad, T. Characterization of IRA/IRB Hybrid Insulin Receptors Using Bioluminescence Resonance Energy Transfer. Biochem. Pharmacol. 2008, 76. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.; Porzio, O.; Zucaro, L.; Fusco, A.; Borboni, P.; Lauro, D.; Sesti, G. Distribution of Insulin/Insulin-like Growth Factor-I Hybrid Receptors in Human Tissues. Mol. Cell. Endocrinol. 1997, 129. [Google Scholar] [CrossRef]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of Insulin/IGF Hybrid Receptors: Contributions of the Insulin Receptor L2 and Fn1 Domains and the Alternatively Spliced Exon 11 Sequence to Ligand Binding and Receptor Activation. Biochem. J. 2007, 403. [Google Scholar] [CrossRef] [PubMed]
- Blyth, A.J.; Kirk, N.S.; Forbes, B.E. Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells 2020, 9, 2276. [Google Scholar] [CrossRef]
- Han, V.K.; Bassett, N.; Walton, J.; Challis, J.R. The Expression of Insulin-like Growth Factor (IGF) and IGF-Binding Protein (IGFBP) Genes in the Human Placenta and Membranes: Evidence for IGF-IGFBP Interactions at the Feto-Maternal Interface. J. Clin. Endocrinol. Metab. 1996, 81. [Google Scholar] [CrossRef]
- Holt, R.I.G.; Simpson, H.L.; Sönksen, P.H. The Role of the Growth Hormone-Insulin-like Growth Factor Axis in Glucose Homeostasis. Diabet. Med. 2003, 20. [Google Scholar] [CrossRef]
- Baumann, M.U.; Schneider, H.; Malek, A.; Palta, V.; Surbek, D.V.; Sager, R.; Zamudio, S.; Illsley, N.P. Regulation of Human Trophoblast GLUT1 Glucose Transporter by Insulin-Like Growth Factor I (IGF-I). PLoS ONE 2014, 9, e106037. [Google Scholar] [CrossRef] [PubMed]
- Scavo, L.M.; Karas, M.; Murray, M.; Leroith, D. Insulin-like Growth Factor-I Stimulates Both Cell Growth and Lipogenesis during Differentiation of Human Mesenchymal Stem Cells into Adipocytes. J. Clin. Endocrinol. Metab. 2004, 89. [Google Scholar] [CrossRef]
- Clemmons, D.R.; Sleevi, M.; Allan, G.; Sommer, A. Effects of Combined Recombinant Insulin-like Growth Factor (IGF)-I and IGF Binding Protein-3 in Type 2 Diabetic Patients on Glycemic Control and Distribution of IGF-I and IGF-II among Serum Binding Protein Complexes. J. Clin. Endocrinol. Metab. 2007, 92. [Google Scholar] [CrossRef]
- Devedjian, J.C.; George, M.; Casellas, A.; Pujol, A.; Visa, J.; Pelegrín, M.; Gros, L.; Bosch, F. Transgenic Mice Overexpressing Insulin-like Growth Factor-II in β Cells Develop Type 2 Diabetes. J. Clin. Investig. 2000, 105. [Google Scholar] [CrossRef]
- Dabelea, D.; Crume, T. Maternal Environment and the Transgenerational Cycle of Obesity and Diabetes. Diabetes 2011, 60. [Google Scholar] [CrossRef] [PubMed]
- Stanirowski, P.J.; Szukiewicz, D.; Pyzlak, M.; Abdalla, N.; Sawicki, W.; Cendrowski, K. Impact of Pre-Gestational and Gestational Diabetes Mellitus on the Expression of Glucose Transporters GLUT-1, GLUT-4 and GLUT-9 in Human Term Placenta. Endocrine 2017, 55. [Google Scholar] [CrossRef]
- Gaither, K.; Quraishi, A.N.; Illsley, N.P. Diabetes Alters the Expression and Activity of the Human Placental GLUT1 Glucose Transporter 1. J. Clin. Endocrinol. Metab. 1999, 84. [Google Scholar] [CrossRef]
- Borges, M.H.; Pullockaran, J.; Catalano, P.M.; Baumann, M.U.; Zamudio, S.; Illsley, N.P. Human Placental GLUT1 Glucose Transporter Expression and the Fetal Insulin-like Growth Factor Axis in Pregnancies Complicated by Diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865. [Google Scholar] [CrossRef]
- Lappas, M. Insulin-like Growth Factor-Binding Protein 1 and 7 Concentrations Are Lower in Obese Pregnant Women, Women with Gestational Diabetes and Their Fetuses. J. Perinatol. 2015, 35. [Google Scholar] [CrossRef]
- Bach, L.A. Current Ideas on the Biology of IGFBP-6: More than an IGF-II Inhibitor? Growth Horm. IGF Res. 2016, 30–31. [Google Scholar] [CrossRef]
- Shang, M.; Wen, Z. Increased Placental IGF-1/MTOR Activity in Macrosomia Born to Women with Gestational Diabetes. Diabetes Res. Clin. Pract. 2018, 146. [Google Scholar] [CrossRef]
- Liu, K.; Wu, H.Y.; Xu, Y.H. Study on the Relationship between the Expression of IGF-1 in Umbilical Cord Blood and Abnormal Glucose Metabolism during Pregnancy. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 647–651. [Google Scholar] [PubMed]
- Federici, M.; Zucaro, L.; Porzio, O.; Massoud, R.; Borboni, P.; Lauro, D.; Sesti, G. Increased Expression of Insulin/Insulin-like Growth Factor-1 Hybrid Receptors in Skeletal Muscle of Noninsulin-Dependent Diabetes Mellitus Subjects. J. Clin. Investig. 1996, 98. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.; Porzio, O.; Zucaro, L.; Giovannone, B.; Borboni, P.; Marini, M.A.; Lauro, D.; Sesti, G. Increased Abundance of Insulin/IGF-I Hybrid Receptors in Adipose Tissue from NIDDM Patients. Mol. Cell. Endocrinol. 1997, 135. [Google Scholar] [CrossRef]
- Valensise, H.; Liu, Y.Y.; Federici, M.; Lauro, D.; Dell’anna, D.; Romanini, C.; Sesti, G. Increased Expression of Low-Affinity Insulin Receptor Isoform and Insulin/Insulin-like Growth Factor-1 Hybrid Receptors in Term Placenta from Insulin-Resistant Women with Gestational Hypertension. Diabetologia 1996, 39. [Google Scholar] [CrossRef]
- Pandini, G.; Vigneri, R.; Costantino, A.; Frasca, F.; Ippolito, A.; Fujita-Yamaguchi, Y.; Siddle, K.; Goldfine, I.D.; Belfiore, A. Insulin and Insulin-like Growth Factor-I (IGF-I) Receptor Overexpression in Breast Cancers Leads to Insulin/IGF-I Hybrid Receptor Overexpression: Evidence for a Second Mechanism of IGF-I Signaling. Clin. Cancer Res. 1999, 5, 1935–1944. [Google Scholar]
- Catalano, P.M.; Tyzbir, E.D.; Roman, N.M.; Amini, S.B.; Sims, E.A.H. Longitudinal Changes in Insulin Release and Insulin Resistance in Nonobese Pregnant Women. Am. J. Obstet. Gynecol. 1991, 165. [Google Scholar] [CrossRef]
- Stefanoska, I.; Jovanović Krivokuća, M.; Vasilijić, S.; Ćujić, D.; Vićovac, L. Prolactin Stimulates Cell Migration and Invasion by Human Trophoblast in Vitro. Placenta 2013, 34. [Google Scholar] [CrossRef]
- Binart, N. Prolactin and Pregnancy in Mice and Humans. Ann. Endocrinol. 2016, 77. [Google Scholar] [CrossRef] [PubMed]
- Garzia, E.; Clauser, R.; Persani, L.; Borgato, S.; Bulfamante, G.; Avagliano, L.; Quadrelli, F.; Marconi, A.M. Prolactin and Proinflammatory Cytokine Expression at the Fetomaternal Interface in First Trimester Miscarriage. Fertil. Steril. 2013, 100. [Google Scholar] [CrossRef]
- Cattini, P.A.; Jin, Y.; Jarmasz, J.S.; Noorjahan, N.; Bock, M.E. Obesity and Regulation of Human Placental Lactogen Production in Pregnancy. J. Neuroendocrinol. 2020, 32, e12859. [Google Scholar] [CrossRef] [PubMed]
- Newbern, D.; Freemark, M. Placental Hormones and the Control of Maternal Metabolism and Fetal Growth. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18. [Google Scholar] [CrossRef]
- Hu, Z.-Z.; Zhuang, L.; Meng, J.; Tsai-Morris, C.-H.; Dufau, M.L. Complex 5′ Genomic Structure of the Human Prolactin Receptor: Multiple Alternative Exons 1 and Promoter Utilization. Endocrinology 2002, 143. [Google Scholar] [CrossRef] [PubMed]
- Abramicheva, P.A.; Smirnova, O.V. Prolactin Receptor Isoforms as the Basis of Tissue-Specific Action of Prolactin in the Norm and Pathology. Biochemistry 2019, 84. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.L.; Brelje, T.C. Prolactin Receptors Are Critical to the Adaptation of Islets to Pregnancy. Endocrinology 2009, 150. [Google Scholar] [CrossRef]
- Yan, L.; Figueroa, D.J.; Austin, C.P.; Liu, Y.; Bugianesi, R.M.; Slaughter, R.S.; Kaczorowski, G.J.; Kohler, M.G. Expression of Voltage-Gated Potassium Channels in Human and Rhesus Pancreatic Islets. Diabetes 2004, 53. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Snider, F.; Cross, J.C. Prolactin Receptor Is Required for Normal Glucose Homeostasis and Modulation of β-Cell Mass during Pregnancy. Endocrinology 2009, 150. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y.; et al. Serotonin Regulates Pancreatic Beta Cell Mass during Pregnancy. Nat. Med. 2010, 16. [Google Scholar] [CrossRef] [PubMed]
- Ben-Jonathan, N.; LaPensee, C.R.; LaPensee, E.W. What Can We Learn from Rodents about Prolactin in Humans? Endocr. Rev. 2008, 29. [Google Scholar] [CrossRef]
- Brelje, T.C.; Scharp, D.W.; Lacy, P.E.; Ogren, L.; Talamantes, F.; Robertson, M.; Friesen, H.G.; Sorenson, R.L. Effect of Homologous Placental Lactogens, Prolactins, and Growth Hormones on Islet b-Cell Division and Insulin Secretion in Rat, Mouse, and Human Islets: Implication for Placental Lactogen Regulation of Islet Function during Pregnancy. Endocrinology 1993, 132. [Google Scholar] [CrossRef]
- Ernst, S.; Demirci, C.; Valle, S.; Velazquez-Garcia, S.; Garcia-Ocaña, A. Mechanisms in the Adaptation of Maternal β-Cells during Pregnancy. Diabetes Manag. 2011, 1. [Google Scholar] [CrossRef]
- Demirci, C.; Ernst, S.; Alvarez-Perez, J.C.; Rosa, T.; Valle, S.; Shridhar, V.; Casinelli, G.P.; Alonso, L.C.; Vasavada, R.C.; García-Ocana, A. Loss of HGF/c-Met Signaling in Pancreatic β-Cells Leads to Incomplete Maternal β-Cell Adaptation and Gestational Diabetes Mellitus. Diabetes 2012, 61. [Google Scholar] [CrossRef]
- Hakonen, E.; Ustinov, J.; Palgi, J.; Miettinen, P.J.; Otonkoski, T. EGFR Signaling Promotes β-Cell Proliferation and Survivin Expression during Pregnancy. PLoS ONE 2014, 9, e93651. [Google Scholar] [CrossRef]
- Sergeev, I.N.; Rhoten, W.B. 1, 25-Dihydroxyvitamin D3 Evokes Oscillations of Intracellular Calcium in a Pancreatic β-Cell Line. Endocrinology 1995, 136. [Google Scholar] [CrossRef]
- Altuntaś, S.Ç.; Evran, M.; Sert, M.; Tetiker, T. Markers of Metabolic Syndrome in Patients with Pituitary Adenoma: A Case Series of 303 Patients. Horm. Metab. Res. 2019, 51. [Google Scholar] [CrossRef]
- Le, T.N.; Elsea, S.H.; Romero, R.; Chaiworapongsa, T.; Francis, G.L. Prolactin Receptor Gene Polymorphisms Are Associated with Gestational Diabetes. Genet. Test. Mol. Biomark. 2013, 17. [Google Scholar] [CrossRef]
- Ursell, W.; Brudenell, M.; Chard, T. Placental Lactogen Levels in Diabetic Pregnancy. Br. Med. J. 1973, 2, 80–82. [Google Scholar] [CrossRef][Green Version]
- Skouby, S.O.; Kühl, C.; Hornnes, P.J.; Andersen, A.N. Prolactin and Glucose Tolerance in Normal and Gestational Diabetic Pregnancy. Obstet. Gynecol. 1986, 67, 17–20. [Google Scholar]
- Macotela, Y.; Triebel, J.; Clapp, C. Time for a New Perspective on Prolactin in Metabolism. Trends Endocrinol. Metab. 2020, 31. [Google Scholar] [CrossRef]
- Park, S.; Kim, D.S.; Daily, J.W.; Kim, S.H. Serum Prolactin Concentrations Determine Whether They Improve or Impair β-Cell Function and Insulin Sensitivity in Diabetic Rats. Diabetes Metab. Res. Rev. 2011, 27. [Google Scholar] [CrossRef]
- Kawaharada, R.; Masuda, H.; Chen, Z.; Blough, E.; Kohama, T.; Nakamura, A. Intrauterine Hyperglycemia-Induced Inflammatory Signalling via the Receptor for Advanced Glycation End Products in the Cardiac Muscle of the Infants of Diabetic Mother Rats. Eur. J. Nutr. 2018, 57. [Google Scholar] [CrossRef]
- Pantham, P.; Aye, I.L.M.H.; Powell, T.L. Inflammation in Maternal Obesity and Gestational Diabetes Mellitus. Placenta 2015, 36. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29. [Google Scholar] [CrossRef]
- Yessoufou, A.; Moutairou, K. Maternal Diabetes in Pregnancy: Early and Long-Term Outcomes on the Offspring and the Concept of “Metabolic Memory”. Exp. Diabetes Res. 2011, 2011. [Google Scholar] [CrossRef]
- Westermeier, F.; Sáez, P.J.; Villalobos-Labra, R.; Sobrevia, L.; Farías-Jofré, M. Programming of Fetal Insulin Resistance in Pregnancies with Maternal Obesity by ER Stress and Inflammation. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Dudele, A.; Hougaard, K.S.; Kjølby, M.; Hokland, M.; Winther, G.; Elfving, B.; Wegener, G.; Nielsen, A.L.; Larsen, A.; Nøhr, M.K.; et al. Chronic Maternal Inflammation or High-Fat-Feeding Programs Offspring Obesity in a Sex-Dependent Manner. Int. J. Obes. 2017, 41. [Google Scholar] [CrossRef]
- Perrin, E.M.; O’Shea, T.M.; Skinner, A.C.; Bose, C.; Allred, E.N.; Fichorova, R.N.; van der Burg, J.W.; Leviton, A. Elevations of Inflammatory Proteins in Neonatal Blood Are Associated with Obesity and Overweight among 2-Year-Old Children Born Extremely Premature. Pediatr. Res. 2018, 83. [Google Scholar] [CrossRef]
- Tsiotra, P.C.; Halvatsiotis, P.; Patsouras, K.; Maratou, E.; Salamalekis, G.; Raptis, S.A.; Dimitriadis, G.; Boutati, E. Circulating Adipokines and MRNA Expression in Adipose Tissue and the Placenta in Women with Gestational Diabetes Mellitus. Peptides 2018, 101. [Google Scholar] [CrossRef]
- Zhang, J.; Chi, H.; Xiao, H.; Tian, X.; Wang, Y.; Yun, X.; Xu, Y. Interleukin 6 (IL-6) and Tumor Necrosis Factor α (TNF-α) Single Nucleotide Polymorphisms (SNPs), Inflammation and Metabolism in Gestational Diabetes Mellitus in Inner Mongolia. Med. Sci. Monit. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Angelo, A.G.S.; Neves, C.T.C.; Lobo, T.F.; Godoy, R.V.C.; Ono, É.; Mattar, R.; Daher, S. Monocyte Profile in Peripheral Blood of Gestational Diabetes Mellitus Patients. Cytokine 2018, 107. [Google Scholar] [CrossRef] [PubMed]
- Lekva, T.; Michelsen, A.E.; Aukrust, P.; Paasche Roland, M.C.; Henriksen, T.; Bollerslev, J.; Ueland, T. CXC Chemokine Ligand 16 Is Increased in Gestational Diabetes Mellitus and Preeclampsia and Associated with Lipoproteins in Gestational Diabetes Mellitus at 5 Years Follow-Up. Diabetes Vasc. Dis. Res. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Hara, C.D.C.P.; França, E.L.; Fagundes, D.L.G.; de Queiroz, A.A.; Rudge, M.V.C.; Honorio-França, A.C.; Calderon, I.D.M.P. Characterization of Natural Killer Cells and Cytokines in Maternal Placenta and Fetus of Diabetic Mothers. J. Immunol. Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Mrizak, I.; Grissa, O.; Henault, B.; Fekih, M.; Bouslema, A.; Boumaiza, I.; Zaouali, M.; Tabka, Z.; Khan, N.A. Placental Infiltration of Inflammatory Markers in Gestational Diabetic Women. Gen. Physiol. Biophys. 2014, 33. [Google Scholar] [CrossRef]
- Tchirikov, M.; Schlabritz-Loutsevitch, N.; Maher, J.; Buchmann, J.; Naberezhnev, Y.; Winarno, A.S.; Seliger, G. Mid-Trimester Preterm Premature Rupture of Membranes (PPROM): Etiology, Diagnosis, Classification, International Recommendations of Treatment Options and Outcome. J. Perinat. Med. 2018, 46. [Google Scholar] [CrossRef]
- Toniolo, A.; Cassani, G.; Puggioni, A.; Rossi, A.; Colombo, A.; Onodera, T.; Ferrannini, E. The Diabetes Pandemic and Associated Infections: Suggestions for Clinical Microbiology. Rev. Med. Microbiol. 2019, 30. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Y.; Gui, J.; Li, A.Z.; Su, X.L.; Feng, L. Assessment of the Number and Function of Macrophages in the Placenta of Gestational Diabetes Mellitus Patients. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33. [Google Scholar] [CrossRef]
- Zheng, L.; Li, C.; Qi, W.; Qiao, B.; Zhao, H.; Zhou, Y.; Lu, C. Expression of Macrophage Migration Inhibitory Factor Gene in Placenta Tissue and Its Correlation with Gestational Diabetes Mellitus. Natl. Med. J. China 2017, 97. [Google Scholar] [CrossRef]
- Sisino, G.; Bouckenooghe, T.; Aurientis, S.; Fontaine, P.; Storme, L.; Vambergue, A. Diabetes during Pregnancy Influences Hofbauer Cells, a Subtype of Placental Macrophages, to Acquire a pro-Inflammatory Phenotype. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Jialal, I. Free Fatty Acids in the Presence of High Glucose Amplify Monocyte Inflammation via Toll-like Receptors. Am. J. Physiol. Endocrinol. Metab. 2011, 300. [Google Scholar] [CrossRef]
- Schliefsteiner, C.; Peinhaupt, M.; Kopp, S.; Lögl, J.; Lang-Olip, I.; Hiden, U.; Heinemann, A.; Desoye, G.; Wadsack, C. Human Placental Hofbauer Cells Maintain an Anti-Inflammatory M2 Phenotype despite the Presence of Gestational Diabetes Mellitus. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Stoikou, M.; Grimolizzi, F.; Giaglis, S.; Schäfer, G.; van Breda, S.V.; Hoesli, I.M.; Lapaire, O.; Huhn, E.A.; Hasler, P.; Rossi, S.W.; et al. Gestational Diabetes Mellitus Is Associated with Altered Neutrophil Activity. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis Is Induced by High Glucose and Associated with Type 2 Diabetes. Acta Diabetol. 2015, 52. [Google Scholar] [CrossRef]
- Sun, T.; Meng, F.; Zhao, H.; Yang, M.; Zhang, R.; Yu, Z.; Huang, X.; Ding, H.; Liu, J.; Zang, S. Elevated First-Trimester Neutrophil Count Is Closely Associated with the Development of Maternal Gestational Diabetes Mellitus and Adverse Pregnancy Outcomes. Diabetes 2020, 69. [Google Scholar] [CrossRef]
- Vorobjeva, N.V.; Pinegin, B.V. Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Health and Disease. Biochemistry 2014, 79. [Google Scholar] [CrossRef]
- Olmos-Ortiz, A.; Flores-Espinosa, P.; Mancilla-Herrera, I.; Vega-Sánchez, R.; Díaz, L.; Zaga-Clavellina, V. Innate Immune Cells and Toll-like Receptor–Dependent Responses at the Maternal–Fetal Interface. Int. J. Mol. Sci. 2019, 20, 3654. [Google Scholar] [CrossRef]
- Shmeleva, E.V.; Colucci, F. Maternal Natural Killer Cells at the Intersection between Reproduction and Mucosal Immunity. Mucosal Immunol. 2021. [Google Scholar] [CrossRef]
- Lobo, T.F.; Borges, C.D.M.; Mattar, R.; Gomes, C.P.; de Angelo, A.G.S.; Pendeloski, K.P.T.; Daher, S. Impaired Treg and NK Cells Profile in Overweight Women with Gestational Diabetes Mellitus. Am. J. Reprod. Immunol. 2018, 79. [Google Scholar] [CrossRef]
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal Changes in Glucose Metabolism during Pregnancy in Obese Women with Normal Glucose Tolerance and Gestational Diabetes Mellitus. Am. J. Obstet. Gynecol. 1999, 180. [Google Scholar] [CrossRef]
- Atègbo, J.M.; Grissa, O.; Yessoufou, A.; Hichami, A.; Dramane, K.L.; Moutairou, K.; Miled, A.; Grissa, A.; Jerbi, M.; Tabka, Z.; et al. Modulation of Adipokines and Cytokines in Gestational Diabetes and Macrosomia. J. Clin. Endocrinol. Metab. 2006, 91. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.F.; Weickert, M.O.; Sivakumar, K.; James, S.G.; Snead, D.R.J.; Tan, B.K.; Randeva, H.S.; Bastie, C.C.; Vatish, M. Elevated Soluble CD163 in Gestational Diabetes Mellitus: Secretion from Human Placenta and Adipose Tissue. PLoS ONE 2014, 9, e101327. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Lizotte, F.; Hivert, M.-F.; Geraldes, P.; Perron, P. Calcifediol Decreases Interleukin-6 Secretion by Cultured Human Trophoblasts from GDM Pregnancies. J. Endocr. Soc. 2019, 3. [Google Scholar] [CrossRef]
- Radaelli, T.; Varastehpour, A.; Catalano, P.; Hauguel-De Mouzon, S. Gestational Diabetes Induces Placental Genes for Chronic Stress and Inflammatory Pathways. Diabetes 2003, 52. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yin, Q.; Wu, H. Structural basis of signal transduction in the TNF receptor superfamily. Adv. Immunol. 2013, 119. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB System. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N. Innate Immune Activation in Obesity. Mol. Asp. Med. 2013, 34. [Google Scholar] [CrossRef]
- Feng, H.; Su, R.; Song, Y.; Wang, C.; Lin, L.; Ma, J.; Yang, H. Positive Correlation between Enhanced Expression of TLR4/MyD88/NF-ΚB with Insulin Resistance in Placentae of Gestational Diabetes Mellitus. PLoS ONE 2016, 11, e0157185. [Google Scholar] [CrossRef]
- Corrêa-Silva, S.; Alencar, A.P.; Moreli, J.B.; Borbely, A.U.; Lima, L.D.S.; Scavone, C.; Damasceno, D.C.; Rudge, M.V.C.; Bevilacqua, E.; Calderon, I.M.P. Hyperglycemia Induces Inflammatory Mediators in the Human Chorionic Villous. Cytokine 2018, 111. [Google Scholar] [CrossRef]
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-α Is a Predictor of Insulin Resistance in Human Pregnancy. Diabetes 2002, 51. [Google Scholar] [CrossRef]
- Friis, C.M.; Roland, M.C.P.; Godang, K.; Ueland, T.; Tanbo, T.; Bollerslev, J.; Henriksen, T. Adiposity-Related Inflammation: Effects of Pregnancy. Obesity 2013, 21. [Google Scholar] [CrossRef]
- Vega-Sanchez, R.; Barajas-Vega, H.A.; Rozada, G.; Espejel-Nuñez, A.; Beltran-Montoya, J.; Vadillo-Ortega, F. Association between Adiposity and Inflammatory Markers in Maternal and Fetal Blood in a Group of Mexican Pregnant Women. Br. J. Nutr. 2010, 104. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T.L. Increasing Maternal Body Mass Index Is Associated with Systemic Inflammation in the Mother and the Activation of Distinct Placental Inflammatory Pathways. Biol. Reprod. 2014, 90. [Google Scholar] [CrossRef]
- Mohammed, A.; Aliyu, I.S. Maternal Serum Level of TNF-α in Nigerian Women with Gestational Diabetes Mellitus. Pan Afr. Med. J. 2018, 31. [Google Scholar] [CrossRef]
- Catalano, P.M. Obesity, Insulin Resistance, and Pregnancy Outcome. Reproduction 2010, 140. [Google Scholar] [CrossRef]
- Carey, A.L.; Febbraio, M.A. Interleukin-6 and Insulin Sensitivity: Friend or Foe? Diabetologia 2004, 47. [Google Scholar] [CrossRef]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in Pregnancy Stimulates Macrophage Accumulation and Inflammation in the Placenta. Placenta 2008, 29. [Google Scholar] [CrossRef]
- Stirm, L.; Kovářová, M.; Perschbacher, S.; Michlmaier, R.; Fritsche, L.; Siegel-Axel, D.; Schleicher, E.; Peter, A.; Pauluschke-Fröhlich, J.; Brucker, S.; et al. BMI-Independent Effects of Gestational Diabetes on Human Placenta. J. Clin. Endocrinol. Metab. 2018, 103. [Google Scholar] [CrossRef] [PubMed]
- Han, C.S.; Herrin, M.A.; Pitruzzello, M.C.; Mulla, M.J.; Werner, E.F.; Pettker, C.M.; Flannery, C.A.; Abrahams, V.M. Glucose and Metformin Modulate Human First Trimester Trophoblast Function: A Model and Potential Therapy for Diabetes-Associated Uteroplacental Insufficiency. Am. J. Reprod. Immunol. 2015, 73. [Google Scholar] [CrossRef] [PubMed]
- Schulze, F.; Wehner, J.; Kratschmar, D.V.; Makshana, V.; Meier, D.T.; Häuselmann, S.P.; Dalmas, E.; Thienel, C.; Dror, E.; Wiedemann, S.J.; et al. Inhibition of IL-1beta Improves Glycaemia in a Mouse Model for Gestational Diabetes. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Peng, H.Y.; Li, M.Q.; Li, H.P. High Glucose Suppresses the Viability and Proliferation of HTR-8/SVneo Cells through Regulation of the MiR-137/PRKAA1/IL-6 Axis. Int. J. Mol. Med. 2018, 42. [Google Scholar] [CrossRef]
- Cawyer, C.; Afroze, S.H.; Drever, N.; Allen, S.; Jones, R.; Zawieja, D.C.; Kuehl, T.; Uddin, M.N. Attenuation of Hyperglycemia-Induced Apoptotic Signaling and Anti-Angiogenic Milieu in Cultured Cytotrophoblast Cells. Hypertens. Pregnancy 2016, 35. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.E.; Scholz-Romero, K.; Sweeney, E.; Peiris, H.; Kobayashi, M.; Duncombe, G.; Mitchell, M.D.; Salomon, C. The Effect of Glucose on the Release and Bioactivity of Exosomes from First Trimester Trophoblast Cells. J. Clin. Endocrinol. Metab. 2015, 100. [Google Scholar] [CrossRef]
- Troncoso, F.; Acurio, J.; Herlitz, K.; Aguayo, C.; Bertoglia, P.; Guzman-Gutierrez, E.; Loyola, M.; Gonzalez, M.; Rezgaoui, M.; Desoye, G.; et al. Gestational Diabetes Mellitus Is Associated with Increased Pro-Migratory Activation of Vascular Endothelial Growth Factor Receptor 2 and Reduced Expression of Vascular Endothelial Growth Factor Receptor 1. PLoS ONE 2017, 12, e0182509. [Google Scholar] [CrossRef]
- Loegl, J.; Nussbaumer, E.; Cvitic, S.; Huppertz, B.; Desoye, G.; Hiden, U. GDM Alters Paracrine Regulation of Feto-Placental Angiogenesis via the Trophoblast. Lab. Investig. 2017, 97. [Google Scholar] [CrossRef]
- Landecho, M.F.; Tuero, C.; Valentí, V.; Bilbao, I.; de la Higuera, M.; Frühbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Maymó, J.; Gambino, Y.; Guadix, P.; Dueñas, J.L.; Varone, C.; Sánchez-Margalet, V. Insulin Enhances Leptin Expression in Human Trophoblastic Cells. Biol. Reprod. 2013, 89. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin Action in Normal and Pathological Pregnancies. J. Cell. Mol. Med. 2018, 22. [Google Scholar] [CrossRef]
- Maymó, J.L.; Pérez Pérez, A.; Maskin, B.; Dueñas, J.L.; Calvo, J.C.; Sánchez Margalet, V.; Varone, C.L. The Alternative Epac/CAMP Pathway and the MAPK Pathway Mediate HCG Induction of Leptin in Placental Cells. PLoS ONE 2012, 7, e46216. [Google Scholar] [CrossRef]
- Gambino, Y.P.; Pérez Pérez, A.; Dueñas, J.L.; Calvo, J.C.; Sánchez-Margalet, V.; Varone, C.L. Regulation of Leptin Expression by 17beta-Estradiol in Human Placental Cells Involves Membrane Associated Estrogen Receptor Alpha. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823. [Google Scholar] [CrossRef] [PubMed]
- Magariños, M.P.; Sánchez-Margalet, V.; Kotler, M.; Calvo, J.C.; Varone, C.L. Leptin Promotes Cell Proliferation and Survival of Trophoblastic Cells. Biol. Reprod. 2007, 76. [Google Scholar] [CrossRef] [PubMed]
- White, V.; González, E.; Capobianco, E.; Pustovrh, C.; Martínez, N.; Higa, R.; Baier, M.; Jawerbaum, A. Leptin Modulates Nitric Oxide Production and Lipid Metabolism in Human Placenta. Reprod. Fertil. Dev. 2006, 18. [Google Scholar] [CrossRef] [PubMed]
- Côté, S.; Gagné-Ouellet, V.; Guay, S.P.; Allard, C.; Houde, A.A.; Perron, P.; Baillargeon, J.P.; Gaudet, D.; Guérin, R.; Brisson, D.; et al. PPARGC1α Gene DNA Methylation Variations in Human Placenta Mediate the Link between Maternal Hyperglycemia and Leptin Levels in Newborns. Clin. Epigenet. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Soheilykhah, S.; Mojibian, M.; Rahimi-Saghand, S.; Rashidi, M.; Hadinedoushan, H. Maternal Serum Leptin Concentration in Gestational Diabetes. Taiwan. J. Obstet. Gynecol. 2011, 50. [Google Scholar] [CrossRef]
- Chen, D.; Xia, G.; Xu, P.; Dong, M. Peripartum Serum Leptin and Soluble Leptin Receptor Levels in Women with Gestational Diabetes. Acta Obstet. Gynecol. Scand. 2010, 89. [Google Scholar] [CrossRef]
- Mokhtari, M.; Hashemi, M.; Yaghmaei, M.; Naderi, M.; Shikhzadeh, A.; Ghavami, S. Evaluation of the Serum Leptin in Normal Pregnancy and Gestational Diabetes Mellitus in Zahedan, Southeast Iran. Arch. Gynecol. Obstet. 2011, 284. [Google Scholar] [CrossRef]
- Lepercq, J.; Cauzac, M.; Lahlou, N.; Timsit, J.; Girard, J.; Auwerx, J.; de Mouzon, S.H. Overexpression of Placental Leptin in Diabetic Pregnancy: A Critical Role for Insulin. Diabetes 1998, 47. [Google Scholar] [CrossRef]
- Lappas, M.; Permezel, M.; Rice, G.E. Leptin and Adiponectin Stimulate the Release of Proinflammatory Cytokines and Prostaglandins from Human Placenta and Maternal Adipose Tissue via Nuclear Factor-ΚB, Peroxisomal Proliferator-Activated Receptor-γ and Extracellularly Regulated Kinase 1/2. Endocrinology 2005, 146. [Google Scholar] [CrossRef]
- Cameo, P.; Bischof, P.; Calvo, J.C. Effect of Leptin on Progesterone, Human Chorionic Gonadotropin, and Interleukin-6 Secretion by Human Term Trophoblast Cells in Culture. Biol. Reprod. 2003, 68. [Google Scholar] [CrossRef] [PubMed]
- Soh, E.B.E.; Mitchell, M.D.; Keelan, J.A. Does Leptin Exhibit Cytokine-like Properties in Tissues of Pregnancy? Am. J. Reprod. Immunol. 2000, 43. [Google Scholar] [CrossRef]
- Miehle, K.; Stepan, H.; Fasshauer, M. Leptin, Adiponectin and Other Adipokines in Gestational Diabetes Mellitus and Pre-Eclampsia. Clin. Endocrinol. 2012, 76. [Google Scholar] [CrossRef] [PubMed]
- De Gennaro, G.; Palla, G.; Battini, L.; Simoncini, T.; del Prato, S.; Bertolotto, A.; Bianchi, C. The Role of Adipokines in the Pathogenesis of Gestational Diabetes Mellitus. Gynecol. Endocrinol. 2019, 35. [Google Scholar] [CrossRef]
- Retnakaran, R.; Qi, Y.; Connelly, P.W.; Sermer, M.; Hanley, A.J.; Zinman, B. Low Adiponectin Concentration during Pregnancy Predicts Postpartum Insulin Resistance, Beta Cell Dysfunction and Fasting Glycaemia. Diabetologia 2010, 53. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Qiu, C.; Muy-Rivera, M.; Vadachkoria, S.; Song, T.; Luthy, D.A. Plasma Adiponectin Concentrations in Early Pregnancy and Subsequent Risk of Gestational Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2004, 89. [Google Scholar] [CrossRef]
- Ramachandrayya, S.A.; D’Cunha, P.; Rebeiro, C. Maternal Circulating Levels of Adipocytokines and Insulin Resistance as Predictors of Gestational Diabetes Mellitus: Preliminary Findings of a Longitudinal Descriptive Study. J. Diabetes Metab. Disord. 2020, 19. [Google Scholar] [CrossRef]
- Chen, J.; Tan, B.; Karteris, E.; Zervou, S.; Digby, J.; Hillhouse, E.W.; Vatish, M.; Randeva, H.S. Secretion of Adiponectin by Human Placenta: Differential Modulation of Adiponectin and Its Receptors by Cytokines. Diabetologia 2006, 49. [Google Scholar] [CrossRef]
- Benaitreau, D.; dos Santos, E.; Leneveu, M.C.; Alfaidy, N.; Feige, J.J.; de Mazancourt, P.; Pecquery, R.; Dieudonné, M.N. Effects of Adiponectin on Human Trophoblast Invasion. J. Endocrinol. 2010, 207. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, L.; Hivert, M.F.; Guay, S.P.; St-Pierre, J.; Perron, P.; Brisson, D. Placental Adiponectin Gene DNA Methylation Levels Are Associated with Mothers’ Blood Glucose Concentration. Diabetes 2012, 61. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.; Adam, S.; Abrahams, Y.; Rheeder, P.; Pheiffer, C. Adiponectin DNA Methylation in South African Women with Gestational Diabetes Mellitus: Effects of HIV Infection. PLoS ONE 2021, 16, e0248694. [Google Scholar] [CrossRef] [PubMed]
- King, A.E.; Paltoo, A.; Kelly, R.W.; Sallenave, J.M.; Bocking, A.D.; Challis, J.R.G. Expression of Natural Antimicrobials by Human Placenta and Fetal Membranes. Placenta 2007, 28. [Google Scholar] [CrossRef]
- Yarbrough, V.L.; Winkle, S.; Herbst-Kralovetz, M.M. Antimicrobial Peptides in the Female Reproductive Tract: A Critical Component of the Mucosal Immune Barrier with Physiological and Clinical Implications. Hum. Reprod. Update 2015, 21. [Google Scholar] [CrossRef]
- Zaga-Clavellina, V.; Garcia-Lopez, G.; Flores-Espinosa, P. Evidence of in Vitro Differential Secretion of Human Beta-Defensins-1, -2, and -3 after Selective Exposure to Streptococcus Agalactiae in Human Fetal Membranes. J. Matern. Fetal Neonatal Med. 2012, 25. [Google Scholar] [CrossRef]
- Zaga-Clavellina, V.; Martha, R.V.M.; Flores-Espinosa, P. In Vitro Secretion Profile of Pro-Inflammatory Cytokines IL-1β, TNF-α, IL-6, and of Human Beta-Defensins (HBD)-1, HBD-2, and HBD-3 from Human Chorioamniotic Membranes after Selective Stimulation with Gardnerella Vaginalis. Am. J. Reprod. Immunol. 2012, 67. [Google Scholar] [CrossRef]
- Liu, N.; Kaplan, A.T.; Low, J.; Nguyen, L.; Liu, G.Y.; Equils, O.; Hewison, M. Vitamin D Induces Innate Antibacterial Responses in Human Trophoblasts via an Intracrine Pathway 1. Biol. Reprod. 2009, 80. [Google Scholar] [CrossRef]
- Klaffenbach, D.; Friedrich, D.; Strick, R.; Strissel, P.L.; Beckmann, M.W.; Rascher, W.; Gessner, A.; Dötsch, J.; Meißner, U.; Schnare, M. Contribution of Different Placental Cells to the Expression and Stimulation of Antimicrobial Proteins (AMPs). Placenta 2011, 32. [Google Scholar] [CrossRef]
- Svinarich, D.M.; Gomez, R.; Romero, R. Detection of Human Defensins in the Placenta. Am. J. Reprod. Immunol. 1997, 38. [Google Scholar] [CrossRef]
- Kim, H.S.; Cho, J.H.; Park, H.W.; Yoon, H.; Kim, M.S.; Kim, S.C. Endotoxin-Neutralizing Antimicrobial Proteins of the Human Placenta. J. Immunol. 2002, 168. [Google Scholar] [CrossRef]
- Oliveira-Bravo, M.; Sangiorgi, B.B.; Schiavinato, J.L.D.S.; Carvalho, J.L.; Covas, D.T.; Panepucci, R.A.; Neves, F.D.A.R.; Franco, O.L.; Pereira, R.W.; Saldanha-Araujo, F. LL-37 Boosts Immunosuppressive Function of Placenta-Derived Mesenchymal Stromal Cells. Stem Cell Res. Ther. 2016, 7. [Google Scholar] [CrossRef]
- Szukiewicz, D.; Alkhalayla, H.; Pyzlak, M.; Szewczyk, G. High Glucose Culture Medium Downregulates Production of Human β-Defensin-2 (HBD-2) in Human Amniotic Epithelial Cells (HAEC). FASEB J. 2016, 30, 307. Available online: https://faseb.onlinelibrary.wiley.com/doi/abs/10.1096/fasebj.30.1_supplement.307.1?cited-by=yes&legid=fasebj%3B30%2F1_Supplement%2F307.1 (accessed on 1 June 2021).
- Montoya-Rosales, A.; Castro-Garcia, P.; Torres-Juarez, F.; Enciso-Moreno, J.A.; Rivas-Santiago, B. Glucose Levels Affect LL-37 Expression in Monocyte-Derived Macrophages Altering the Mycobacterium Tuberculosis Intracellular Growth Control. Microb. Pathog. 2016, 97. [Google Scholar] [CrossRef] [PubMed]
- Froy, O.; Hananel, A.; Chapnik, N.; Madar, Z. Differential Effect of Insulin Treatment on Decreased Levels of Beta-Defensins and Toll-like Receptors in Diabetic Rats. Mol. Immunol. 2007, 44. [Google Scholar] [CrossRef]
- Rivas-Santiago, B.; Trujillo, V.; Montoya, A.; Gonzalez-Curiel, I.; Castañeda-Delgado, J.; Cardenas, A.; Rincon, K.; Hernandez, M.L.; Hernández-Pando, R. Expression of Antimicrobial Peptides in Diabetic Foot Ulcer. J. Dermatol. Sci. 2012, 65. [Google Scholar] [CrossRef]
- Olmos-Ortiz, A.; García-Quiroz, J.; Avila, E.; Caldiño-Soto, F.; Halhali, A.; Larrea, F.; Díaz, L. Lipopolysaccharide and CAMP Modify Placental Calcitriol Biosynthesis Reducing Antimicrobial Peptides Gene Expression. Am. J. Reprod. Immunol. 2018, 79. [Google Scholar] [CrossRef]
- Barrera, D.; Díaz, L.; Noyola-Martínez, N.; Halhali, A. Vitamin D and Inflammatory Cytokines in Healthy and Preeclamptic Pregnancies. Nutrients 2015, 7, 5293. [Google Scholar] [CrossRef]
- Díaz, L.; Noyola-Martínez, N.; Barrera, D.; Hernández, G.; Avila, E.; Halhali, A.; Larrea, F. Calcitriol Inhibits TNF-α-Induced Inflammatory Cytokines in Human Trophoblasts. J. Reprod. Immunol. 2009, 81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, T.; Huo, Y.; Liu, L.; Liu, S.; Yin, X.; Wang, R.; Gao, X. Placenta Expression of Vitamin D and Related Genes in Pregnant Women with Gestational Diabetes Mellitus. J. Steroid Biochem. Mol. Biol. 2020, 204. [Google Scholar] [CrossRef]
- Walker, V.P.; Zhang, X.; Rastegar, I.; Liu, P.T.; Hollis, B.W.; Adams, J.S.; Modlin, R.L. Cord Blood Vitamin D Status Impacts Innate Immune Responses. J. Clin. Endocrinol. Metab. 2011, 96. [Google Scholar] [CrossRef]
- Nargesi, S.; Ghorbani, A.; Shirzadpour, E.; Mohamadpour, M.; Mousavi, S.F.; Amraei, M. A Systematic Review and Meta-Analysis of the Association between Vitamin D Deficiency and Gestational Diabetes Mellitus. Biomed. Res. Ther. 2018, 5. [Google Scholar] [CrossRef]
- Maghbooli, Z.; Hossein-Nezhad, A.; Karimi, F.; Shafaei, A.R.; Larijani, B. Correlation between Vitamin D3 Deficiency and Insulin Resistance in Pregnancy. Diabetes Metab. Res. Rev. 2008, 24. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Ortiz, A.; Avila, E.; Durand-Carbajal, M.; Díaz, L. Regulation of Calcitriol Biosynthesis and Activity: Focus on Gestational Vitamin D Deficiency and Adverse Pregnancy Outcomes. Nutrients 2015, 7, 443. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.J.; Hong, S.C.; Oh, M.J.; Kim, H.J. Vitamin D Deficiency in Gestational Diabetes Mellitus and the Role of the Placenta. Am. J. Obstet. Gynecol. 2013, 209. [Google Scholar] [CrossRef] [PubMed]
- Akoh, C.C.; Pressman, E.K.; Whisner, C.M.; Thomas, C.; Cao, C.; Kent, T.; Cooper, E.; O’Brien, K.O. Vitamin D Mediates the Relationship between Placental Cathelicidin and Group B Streptococcus Colonization during Pregnancy. J. Reprod. Immunol. 2017, 121. [Google Scholar] [CrossRef]
- Akoh, C.C.; Pressman, E.K.; Cooper, E.; Queenan, R.A.; Pillittere, J.; O’Brien, K.O. Low Vitamin D Is Associated with Infections and Proinflammatory Cytokines During Pregnancy. Reprod. Sci. 2018, 25. [Google Scholar] [CrossRef]
- Norman, A.W.; Frankel, B.J.; Heldt, A.M.; Grodsky, G.M. Vitamin D Deficiency Inhibits Pancreatic Secretion of Insulin. Science 1980, 209. [Google Scholar] [CrossRef]
- Maestro, B.; Dávila, N.; Carranza, M.C.; Calle, C. Identification of a Vitamin D Response Element in the Human Insulin Receptor Gene Promoter. J. Steroid Biochem. Mol. Biol. 2003, 84, 223–230. [Google Scholar] [CrossRef]
- Maestro, B.; Molero, S.; Bajo, S.; Dávila, N.; Calle, C. Transcriptional Activation of the Human Insulin Receptor Gene by 1,25-Dihydroxyvitamin D3. Cell Biochem. Funct. 2002, 20, 227–232. [Google Scholar] [CrossRef]
- Li, G.; Lin, L.; Wang, Y.L.; Yang, H. 1,25(OH)2D3 Protects Trophoblasts Against Insulin Resistance and Inflammation Via Suppressing MTOR Signaling. Reprod. Sci. 2019, 26. [Google Scholar] [CrossRef]
- Vimpeli, T.; Huhtala, H.; Wilsgaard, T.; Acharya, G. Fetal Cardiac Output and Its Distribution to the Placenta at 11–20 Weeks of Gestation. Ultrasound Obstet. Gynecol. 2009, 33. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of Acute Kidney Injury. In Seldin and Giebisch’s the Kidney, 4th ed.; Academic Press: San Diego, CA, USA, 2008. [Google Scholar]
- Lautt, W.W. Hepatic Circulation Physiology and Pathophysiology; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2009; Volume 1. [Google Scholar]
- Zucchelli, E.; Majid, Q.A.; Foldes, G. New Artery of Knowledge: 3D Models of Angiogenesis. Vasc. Biol. 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Kolte, D.; McClung, J.A.; Aronow, W.S. Vasculogenesis and Angiogenesis. In Translational Research in Coronary Artery Disease: Pathophysiology to Treatment; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Clapp, C.; Thebault, S.; Jeziorski, M.C.; Martínez De La Escalera, G. Peptide Hormone Regulation of Angiogenesis. Physiol. Rev. 2009, 89. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; le Boeuf, F.; Huot, J. Endothelial Cell Migration during Angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.P.; Levy, N.S.; Wegner, S.; Goldberg, M.A. Transcriptional Regulation of the Rat Vascular Endothelial Growth Factor Gene by Hypoxia. J. Biol. Chem. 1995, 270. [Google Scholar] [CrossRef]
- Gleadle, J.M.; Ratcliffe, P.J. Induction of Hypoxia-Inducible Factor-1, Erythropoietin, Vascular Endothelial Growth Factor, and Glucose Transporter-1 by Hypoxia: Evidence against a Regulatory Role for Src Kinase. Blood 1997, 89. [Google Scholar] [CrossRef]
- Leach, L.; Babawale, M.O.; Anderson, M.; Lammiman, M. Vasculogenesis, Angiogenesis and the Molecular Organisation of Endothelial Junctions in the Early Human Placenta. J. Vasc. Res. 2002, 39. [Google Scholar] [CrossRef] [PubMed]
- Demir, R.; Kayisli, U.A.; Cayli, S.; Huppertz, B. Sequential Steps During Vasculogenesis and Angiogenesis in the Very Early Human Placenta. Placenta 2006, 27. [Google Scholar] [CrossRef]
- Omorphos, N.P.; Gao, C.; Tan, S.S.; Sangha, M.S. Understanding Angiogenesis and the Role of Angiogenic Growth Factors in the Vascularisation of Engineered Tissues. Mol. Biol. Rep. 2021, 48. [Google Scholar] [CrossRef]
- Edatt, L.; Poyyakkara, A.; Raji, G.R.; Ramachandran, V.; Shankar, S.S.; Kumar, V.B.S. Role of Sirtuins in Tumor Angiogenesis. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A. Endothelial Cells and the IGF System. J. Mol. Endocrinol. 2015, 54. [Google Scholar] [CrossRef] [PubMed]
- Slater, T.; Haywood, N.J.; Matthews, C.; Cheema, H.; Wheatcroft, S.B. Insulin-like Growth Factor Binding Proteins and Angiogenesis: From Cancer to Cardiovascular Disease. Cytokine Growth Factor Rev. 2019, 46. [Google Scholar] [CrossRef]
- Albonici, L.; Benvenuto, M.; Focaccetti, C.; Cifaldi, L.; Miele, M.T.; Limana, F.; Manzari, V.; Bei, R. Plgf Immunological Impact during Pregnancy. Int. J. Mol. Sci. 2020, 21, 8714. [Google Scholar] [CrossRef]
- Gobble, R.M.; Groesch, K.A.; Chang, M.; Torry, R.J.; Torry, D.S. Differential Regulation of Human PlGF Gene Expression in Trophoblast and Nontrophoblast Cells by Oxygen Tension. Placenta 2009, 30. [Google Scholar] [CrossRef] [PubMed]
- Yinon, Y.; Nevo, O.; Xu, J.; Many, A.; Rolfo, A.; Todros, T.; Post, M.; Caniggia, I. Severe Intrauterine Growth Restriction Pregnancies Have Increased Placental Endoglin Levels: Hypoxic Regulation via Transforming Growth Factor-Β3. Am. J. Pathol. 2008, 172. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Roque, L.; Núñez-Gómez, E.; Rodríguez-Barbero, A.; Bernabéu, C.; López-Novoa, J.M.; Pericacho, M. Pregnancy-Induced High Plasma Levels of Soluble Endoglin in Mice Lead to Preeclampsia Symptoms and Placental Abnormalities. Int. J. Mol. Sci. 2021, 22, 165. [Google Scholar] [CrossRef] [PubMed]
- Ettelaie, C.; Su, S.; Li, C.; Collier, M.E.W. Tissue Factor-Containing Microparticles Released from Mesangial Cells in Response to High Glucose and AGE Induce Tube Formation in Microvascular Cells. Microvasc. Res. 2008, 76. [Google Scholar] [CrossRef]
- Chen, Y.L.; Rosa, R.H.; Kuo, L.; Hein, T.W. Hyperglycemia Augments Endothelin-1–Induced Constriction of Human Retinal Venules. Transl. Vis. Sci. Technol. 2020, 9. [Google Scholar] [CrossRef]
- Martin-Aragon Baudel, M.; Espinosa-Tanguma, R.; Nieves-Cintron, M.; Navedo, M.F. Purinergic Signaling During Hyperglycemia in Vascular Smooth Muscle Cells. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef]
- Clyne, A.M. Endothelial Response to Glucose: Dysfunction, Metabolism, and Transport. Biochem. Soc. Trans. 2021, 49. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Dong, L.; Gong, A.; Zhang, J.; Jing, L.; Ding, T.; Li, P.-A.; Zhang, J.-Z. Damage to the Blood-brain Barrier and Activation of Neuroinflammation by Focal Cerebral Ischemia under Hyperglycemic Condition. Int. J. Mol. Med. 2021, 48. [Google Scholar] [CrossRef] [PubMed]
- Babawale, M.O.; Lovat, S.; Mayhew, T.M.; Lammiman, M.J.; James, D.K.; Leach, L. Effects of Gestational Diabetes on Junctional Adhesion Molecules in Human Term Placental Vasculature. Diabetologia 2000, 43. [Google Scholar] [CrossRef] [PubMed]
- Leach, L. Placental Vascular Dysfunction in Diabetic Pregnancies: Intimations of Fetal Cardiovascular Disease? Microcirculation 2011, 18. [Google Scholar] [CrossRef]
- Chang, S.C.; Vivian Yang, W.C. Hyperglycemia Induces Altered Expressions of Angiogenesis Associated Molecules in the Trophoblast. Evid. Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef]
- Li, H.P.; Chen, X.; Li, M.Q. Gestational Diabetes Induces Chronic Hypoxia Stress and Excessive Inflammatory Response in Murine Placenta. Int. J. Clin. Exp. Pathol. 2013, 6, 650–659. [Google Scholar] [PubMed]
- Cawyer, C.R.; Horvat, D.; Leonard, D.; Allen, S.R.; Jones, R.O.; Zawieja, D.C.; Kuehl, T.J.; Uddin, M.N. Hyperglycemia Impairs Cytotrophoblast Function via Stress Signaling. Am. J. Obstet. Gynecol. 2014, 211. [Google Scholar] [CrossRef]
- Meng, Q.; Shao, L.; Luo, X.; Mu, Y.; Xu, W.; Gao, L.; Xu, H.; Cui, Y. Expressions of VEGF-A and VEGFR-2 in Placentae from GDM Pregnancies. Reprod. Biol. Endocrinol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Pietro, L.; Daher, S.; Rudge, M.V.C.; Calderon, I.M.P.; Damasceno, D.C.; Sinzato, Y.K.; Bandeira, C.; Bevilacqua, E. Vascular Endothelial Growth Factor (VEGF) and VEGF-Receptor Expression in Placenta of Hyperglycemic Pregnant Women. Placenta 2010, 31. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.Y.; Li, H.P.; Li, M.Q. High Glucose Induces Dysfunction of Human Umbilical Vein Endothelial Cells by Upregulating MiR-137 in Gestational Diabetes Mellitus. Microvasc. Res. 2018, 118. [Google Scholar] [CrossRef]
- Korbecki, J.; Kojder, K.; Kapczuk, P.; Kupnicka, P.; Gawrońska-Szklarz, B.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors-A Review of Literature. Int. J. Mol. Sci. 2021, 22, 843. [Google Scholar] [CrossRef] [PubMed]
- Bassand, K.; Metzinger, L.; Naïm, M.; Mouhoubi, N.; Haddad, O.; Assoun, V.; Zaïdi, N.; Sainte-Catherine, O.; Butt, A.; Guyot, E.; et al. MiR-126-3p Is Essential for CXCL12-Induced Angiogenesis. J. Cell. Mol. Med. 2021, 25, 6032–6045. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Liu, G.; Liu, W.; Xu, W.; Li, H.; Piao, S.; Sui, Y.; Feng, W. CXCL1 Stimulates Decidual Angiogenesis via the VEGF-A Pathway during the First Trimester of Pregnancy. Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef]
- Darakhshan, S.; Fatehi, A.; Hassanshahi, G.; Mahmoodi, S.; Hashemi, M.S.; Karimabad, M.N. Serum Concentration of Angiogenic (CXCL1, CXCL12) and Angiostasis (CXCL9, CXCL10) CXC Chemokines Are Differentially Altered in Normal and Gestational Diabetes Mellitus Associated Pregnancies. J. Diabetes Metab. Disord. 2019, 18. [Google Scholar] [CrossRef]
- Hiden, U.; Lassance, L.; Ghaffari Tabrizi, N.; Miedl, H.; Tam-Amersdorfer, C.; Cetin, I.; Lang, U.; Desoye, G. Fetal Insulin and IGF-II Contribute to Gestational Diabetes Mellitus (GDM)-Associated up-Regulation of Membrane-Type Matrix Metalloproteinase 1 (MT1-MMP) in the Human Feto-Placental Endothelium. J. Clin. Endocrinol. Metab. 2012, 97. [Google Scholar] [CrossRef] [PubMed]
- Majali-Martinez, A.; Hiden, U.; Ghaffari-Tabrizi-Wizsy, N.; Lang, U.; Desoye, G.; Dieber-Rotheneder, M. Placental Membrane-Type Metalloproteinases (MT-MMPs): Key Players in Pregnancy. Cell Adhes. Migr. 2016, 10. [Google Scholar] [CrossRef]
- Abdullah, B.; Deveci, K.; Atilgan, R.; Kiliçli, F.; Söylemez, M.S. Serum Angiopoietin-Related Growth Factor (AGF) Levels Are Elevated in Gestational Diabetes Mellitus and Associated with Insulin Resistance. Ginekol. Pol. 2012, 83, 749–753. [Google Scholar] [PubMed]
- Calderon, I.M.P.; Damasceno, D.C.; Amorin, R.L.; Costa, R.A.A.; Brasil, M.A.M.; Rudge, M.V.C. Morphometric Study of Placental Villi and Vessels in Women with Mild Hyperglycemia or Gestational or Overt Diabetes. Diabetes Res. Clin. Pract. 2007, 78. [Google Scholar] [CrossRef] [PubMed]
- Baumüller, S.; Lehnen, H.; Schmitz, J.; Fimmers, R.; Müller, A.M. The Impact of Insulin Treatment on the Expression of Vascular Endothelial Cadherin and Beta-Catenin in Human Fetoplacental Vessels. Pediatric Dev. Pathol. 2015, 18. [Google Scholar] [CrossRef]
- Rao, R.; Sen, S.; Han, B.; Ramadoss, S.; Chaudhuri, G. Gestational Diabetes, Preeclampsia and Cytokine Release: Similarities and Differences in Endothelial Cell Function. Adv. Exp. Med. Biol. 2014, 814. [Google Scholar] [CrossRef]
- Yang, Z.; Laubach, V.E.; French, B.A.; Kron, I.L. Acute Hyperglycemia Enhances Oxidative Stress and Exacerbates Myocardial Infarction by Activating Nicotinamide Adenine Dinucleotide Phosphate Oxidase during Reperfusion. J. Thorac. Cardiovasc. Surg. 2009, 137. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107. [Google Scholar] [CrossRef]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial Dysfunction in Diabetes Mellitus: Molecular Mechanisms and Clinical Implications. Rev. Endocr. Metab. Disord. 2010, 11. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine. J. Free Radic. Biol. Med. 1985, 1. [Google Scholar] [CrossRef]
- Ward, J.P.T. From physiological redox signalling to oxidant stress. In Pulmonary Vasculature Redox Signaling in Health and Disease; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2017; Volume 967. [Google Scholar] [CrossRef]
- Burton, G.J.; Cindrova-Davies, T.; Yung, H.W.; Jauniaux, E. Oxygen and Development of the Human Placenta. Reproduction 2021, 161. [Google Scholar] [CrossRef]
- Saghian, R.; Bogle, G.; James, J.L.; Clark, A.R. Establishment of Maternal Blood Supply to the Placenta: Insights into Plugging, Unplugging and Trophoblast Behaviour from an Agent-Based Model. Interface Focus 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative Stress in Placental Pathology. Placenta 2018, 69. [Google Scholar] [CrossRef]
- Holland, O.J.; Hickey, A.J.R.; Alvsaker, A.; Moran, S.; Hedges, C.; Chamley, L.W.; Perkins, A.V. Changes in Mitochondrial Respiration in the Human Placenta over Gestation. Placenta 2017, 57. [Google Scholar] [CrossRef]
- Wang, Y.; Walsh, S.W. Placental Mitochondria as a Source of Oxidative Stress in Pre-Eclampsia. Placenta 1998, 19. [Google Scholar] [CrossRef]
- Pereira, R.D.; de Long, N.E.; Wang, R.C.; Yazdi, F.T.; Holloway, A.C.; Raha, S. Angiogenesis in the Placenta: The Role of Reactive Oxygen Species Signaling. BioMed Res. Int. 2015, 2015, 814543. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Skepper, J.N.; Burton, G.J. In Vitro Ischemia-Reperfusion Injury in Term Human Placenta as a Model for Oxidative Stress in Pathological Pregnancies. Am. J. Pathol. 2001, 159. [Google Scholar] [CrossRef]
- Jauniaux, E.; Hempstock, J.; Greenwold, N.; Burton, G.J. Trophoblastic Oxidative Stress in Relation to Temporal and Regional Differences in Maternal Placental Blood Flow in Normal and Abnormal Early Pregnancies. Am. J. Pathol. 2003, 162. [Google Scholar] [CrossRef]
- Cuffe, J.S.; Xu, Z.C.; Perkins, A.V. Biomarkers of Oxidative Stress in Pregnancy Complications. Biomark. Med. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Transcriptional Regulation of NADPH Oxidase Isoforms, Nox1 and Nox4, by Nuclear Factor-ΚB in Human Aortic Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2010, 396. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, H.; Geng, Q.; Ma, Q.; Long, Y.; Zhou, C.; Chen, M. Association of Oxidative Stress Biomarkers with Gestational Diabetes Mellitus in Pregnant Women: A Case-Control Study. PLoS ONE 2015, 10, e0126490. [Google Scholar] [CrossRef]
- Eriksson, U.J. Congenital Anomalies in Diabetic Pregnancy. Semin. Fetal Neonatal Med. 2009, 14. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Vervaart, P.P.; Permezel, M.; Georgiou, H.M.; Rice, G.E. Altered Placental Oxidative Stress Status in Gestational Diabetes Mellitus. Placenta 2004, 25. [Google Scholar] [CrossRef]
- Lappas, M.; Permezel, M.; Rice, G.E. Release of Proinflammatory Cytokines and 8-Isoprostane from Placenta, Adipose Tissue, and Skeletal Muscle from Normal Pregnant Women and Women with Gestational Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2004, 89. [Google Scholar] [CrossRef]
- Biri, A.; Onan, A.; Devrim, E.; Babacan, F.; Kavutcu, M.; Durak, I. Oxidant Status in Maternal and Cord Plasma and Placental Tissue in Gestational Diabetes. Placenta 2006, 27. [Google Scholar] [CrossRef]
- Pessler, D.; Rudich, A.; Bashan, N. Oxidative Stress Impairs Nuclear Proteins Binding to the Insulin Responsive Element in the GLUT4 Promoter. Diabetologia 2001, 44. [Google Scholar] [CrossRef] [PubMed]
- Trocino, R.A.; Akazawa, S.; Ishibashi, M.; Matsumoto, K.; Matsuo, H.; Yamamoto, H.; Goto, S.; Urata, Y.; Kondo, T.; Nagataki, S. Significance of Glutathione Depletion and Oxidative Stress in Early Embryogenesis in Glucose-Induced Rat Embryo Culture. Diabetes 1995, 44. [Google Scholar] [CrossRef] [PubMed]
- Kinalski, M.; Śledziewski, A.; Telejko, B.; Kowalska, I.; Krȩtowski, A.; Zarzycki, W.; Kinalska, I. Lipid Peroxidation, Antioxidant Defence and Acid-Base Status in Cord Blood at Birth: The Influence of Diabetes. Horm. Metab. Res. 2001, 33. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, L.; Tandon, O.P.; Vaney, N.; Agarwal, N. Lipid Peroxidation and Antioxidant Enzymes in Gestational Diabetics. Indian J. Physiol. Pharmacol. 2003, 47, 441–446. [Google Scholar]
- Manoharan, B.; Bobby, Z.; Dorairajan, G.; Jacob, S.E.; Gladwin, V.; Vinayagam, V.; Packirisamy, R.M. Increased Placental Expressions of Nuclear Factor Erythroid 2–Related Factor 2 and Antioxidant Enzymes in Gestational Diabetes: Protective Mechanisms against the Placental Oxidative Stress? Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 238. [Google Scholar] [CrossRef]
- Navarro, A.; Alonso, A.; Garrido, P.; González, C.; González del Rey, C.; Ordoñez, C.; Tolivia, J. Increase in Placental Apolipoprotein D as an Adaptation to Human Gestational Diabetes. Placenta 2010, 31. [Google Scholar] [CrossRef]
- Gelisgen, R.; Genc, H.; Kayali, R.; Oncul, M.; Benian, A.; Guralp, O.; Uludag, S.; Cakatay, U.; Albayrak, M.; Uzun, H. Protein Oxidation Markers in Women with and without Gestational Diabetes Mellitus: A Possible Relation with Paraoxonase Activity. Diabetes Res. Clin. Pract. 2011, 94. [Google Scholar] [CrossRef]
- Pasternak, Y.; Biron-Shental, T.; Ohana, M.; Einbinder, Y.; Arbib, N.; Benchetrit, S.; Zitman-Gal, T. Gestational Diabetes Type 2: Variation in High-Density Lipoproteins Composition and Function. Int. J. Mol. Sci. 2020, 21, 6281. [Google Scholar] [CrossRef]
- Kapustin, R.; Chepanov, S.; Kopteeva, E.; Arzhanova, O. Maternal Serum Nitrotyrosine, 8-Isoprostane and Total Antioxidant Capacity Levels in Pre-Gestational or Gestational Diabetes Mellitus. Gynecol. Endocrinol. 2020, 36. [Google Scholar] [CrossRef] [PubMed]
- Bartakova, V.; Kollarova, R.; Kuricova, K.; Sebekova, K.; Belobradkova, J.; Kankova, K. Serum Carboxymethyl-Lysine, a Dominant Advanced Glycation End Product, Is Increased in Women with Gestational Diabetes Mellitus. Biomed. Pap. 2016, 160. [Google Scholar] [CrossRef]
- Sisay, M.; Edessa, D.; Ali, T.; Mekuria, A.N.; Gebrie, A. The Relationship between Advanced Glycation End Products and Gestational Diabetes: A Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0240382. [Google Scholar] [CrossRef]
- Li, S.; Yang, H. Relationship between Advanced Glycation End Products and Gestational Diabetes Mellitus. J. Matern. Fetal Neonatal Med. 2019, 32. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, A.; Lv, X. Advanced Glycation End Products and Adipocytokines and Oxidative Stress in Placental Tissues of Pregnant Women with Gestational Diabetes Mellitus. Exp. Ther. Med. 2019. [Google Scholar] [CrossRef]
- Ghaneei, A.; Yassini, S.; Ghanei, M.E.; Shojaoddiny-Ardekani, A. Increased Serum Oxidized Low-Density Lipoprotein Levels in Pregnancies Complicated by Gestational Diabetes Mellitus. Iran. J. Reprod. Med. 2015, 13, 421–424. [Google Scholar] [PubMed]
- Aydemir, B.; Baykara, O.; Cinemre, F.B.S.; Cinemre, H.; Tuten, A.; Kiziler, A.R.; Akdemir, N.; Oncul, M.; Kaya, B.; Sozer, V.; et al. LOX-1 Gene Variants and Maternal Levels of Plasma Oxidized LDL and Malondialdehyde in Patients with Gestational Diabetes Mellitus. Arch. Gynecol. Obstet. 2016, 293. [Google Scholar] [CrossRef]
- Kopylov, A.T.; Papysheva, O.; Gribova, I.; Kotaysch, G.; Kharitonova, L.; Mayatskaya, T.; Sokerina, E.; Kaysheva, A.L.; Morozov, S.G. Molecular Pathophysiology of Diabetes Mellitus during Pregnancy with Antenatal Complications. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Qiu, C.; Hevner, K.; Abetew, D.; Enquobahrie, D.A.; Williams, M.A. Oxidative DNA Damage in Early Pregnancy and Risk of Gestational Diabetes Mellitus: A Pilot Study. Clin. Biochem. 2011, 44. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, S.K.; Boguszewska, K.; Szewczuk, M.; Kázmierczak-Barańska, J.; Karwowski, B.T. 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine (8-OxodG) and 8-Hydroxy-2′-Deoxyguanosine (8-OHdG) as a Potential Biomarker for Gestational Diabetes Mellitus (GDM) Development. Molecules 2020, 25, 202. [Google Scholar] [CrossRef]
- Kharb, S. Low Whole Blood Glutathione Levels in Pregnancies Complicated by Preeclampsia and Diabetes. Clin. Chim. Acta 2000, 294. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Chen, R.; Wei, Y.; Feng, Y.; Zheng, W.; Liao, H.; Zhang, Z. Aberrant Expression of Oxidative Stress Related Proteins Affects the Pregnancy Outcome of Gestational Diabetes Mellitus Patients. Am. J. Transl. Res. 2019, 11, 269–279. [Google Scholar]
- Peuchant, E.; Brun, J.L.; Rigalleau, V.; Dubourg, L.; Thomas, M.J.; Daniel, J.Y.; Leng, J.J.; Gin, H. Oxidative and Antioxidative Status in Pregnant Women with Either Gestational or Type 1 Diabetes. Clin. Biochem. 2004, 37. [Google Scholar] [CrossRef]
- Huerta-Cervantes, M.; Peña-Montes, D.J.; Montoya-Pérez, R.; Trujillo, X.; Huerta, M.; López-Vázquez, M.Á.; Olvera-Cortés, M.E.; Saavedra-Molina, A. Gestational Diabetes Triggers Oxidative Stress in Hippocampus and Cerebral Cortex and Cognitive Behavior Modifications in Rat Offspring: Age-and Sex-Dependent Effects. Nutrients 2020, 12, 376. [Google Scholar] [CrossRef]
- Gujski, M.; Szukiewicz, D.; Chołuj, M.; Sawicki, W.; Bojar, I. Fetal and Placental Weight in Pre-Gestational Maternal Obesity (PGMO) vs. Excessive Gestational Weight Gain (EGWG)—A Preliminary Approach to the Perinatal Outcomes in Diet-Controlled Gestational Diabetes Mellitus. J. Clin. Med. 2020, 9, 3530. [Google Scholar] [CrossRef] [PubMed]
- Zehravi, M.; Maqbool, M.; Ara, I. Correlation between Obesity, Gestational Diabetes Mellitus, and Pregnancy Outcomes: An Overview. Int. J. Adolesc. Med. Health 2021. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Becker, M.; Weinstein-Fudim, L.; Ergaz, Z. Diabetes during Pregnancy: A Maternal Disease Complicating the Course of Pregnancy with Long-Term Deleterious Effects on the Offspring. A Clinical Review. Int. J. Mol. Sci. 2021, 22, 2965. [Google Scholar] [CrossRef] [PubMed]
- Karasneh, R.A.; Migdady, F.H.; Alzoubi, K.H.; Al-Azzam, S.I.; Khader, Y.S.; Nusair, M.B. Trends in Maternal Characteristics, and Maternal and Neonatal Outcomes of Women with Gestational Diabetes: A Study from Jordan. Ann. Med. Surg. 2021, 67. [Google Scholar] [CrossRef]
- Riskin, A.; Itzchaki, O.; Bader, D.; Lofe, A.; Toropine, A.; Riskin-Mashiah, S. Perinatal Outcomes in Infants of Mothers with Diabetes in Pregnancy. Isr. Med. Assoc. J. 2020, 22, 569–575. [Google Scholar] [PubMed]
- Schwartz, N.; Nachum, Z.; Green, M.S. The Prevalence of Gestational Diabetes Mellitus Recurrence—Effect of Ethnicity and Parity: A Metaanalysis. Am. J. Obstet. Gynecol. 2015, 213. [Google Scholar] [CrossRef]
- Liang, X.; Zheng, W.; Liu, C.; Zhang, L.; Zhang, L.; Tian, Z.; Li, G. Clinical Characteristics, Gestational Weight Gain and Pregnancy Outcomes in Women with a History of Gestational Diabetes Mellitus. Diabetol. Metab. Syndr. 2021, 13, 73. [Google Scholar] [CrossRef]
- Herath, H.; Herath, R.; Wickremasinghe, R. Gestational Diabetes Mellitus and Risk of Type 2 Diabetes 10 Years after the Index Pregnancy in Sri Lankan Women—A Community Based Retrospective Cohort Study. PLoS ONE 2017, 12, e0179647. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D. Type 2 Diabetes Mellitus after Gestational Diabetes: A Systematic Review and Meta-Analysis. Lancet 2009, 373. [Google Scholar] [CrossRef]
- Retnakaran, R. Hyperglycemia in Pregnancy and Its Implications for a Woman’s Future Risk of Cardiovascular Disease. Diabetes Res. Clin. Pract. 2018, 145. [Google Scholar] [CrossRef]
- Tobias, D.K.; Stuart, J.J.; Li, S.; Chavarro, J.; Rimm, E.B.; Rich-Edwards, J.; Hu, F.B.; Manson, J.E.; Zhang, C. Association of History of Gestational Diabetes with Long-Term Cardiovascular Disease Risk in a Large Prospective Cohort of US Women. JAMA Intern. Med. 2017, 177. [Google Scholar] [CrossRef] [PubMed]
- Franzago, M.; Fraticelli, F.; di Nicola, M.; Bianco, F.; Marchetti, D.; Celentano, C.; Liberati, M.; de Caterina, R.; Stuppia, L.; Vitacolonna, E. Early Subclinical Atherosclerosis in Gestational Diabetes: The Predictive Role of Routine Biomarkers and Nutrigenetic Variants. J. Diabetes Res. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Nobile De Santis, M.S.; Taricco, E.; Radaelli, T.; Spada, E.; Rigano, S.; Ferrazzi, E.; Milani, S.; Cetin, I. Growth of Fetal Lean Mass and Fetal Fat Mass in Gestational Diabetes. Ultrasound Obstet. Gynecol. 2010, 36. [Google Scholar] [CrossRef]
- Elessawy, M.; Harders, C.; Kleinwechter, H.; Demandt, N.; Sheasha, G.A.; Maass, N.; Pecks, U.; Eckmann-Scholz, C. Measurement and Evaluation of Fetal Fat Layer in the Prediction of Fetal Macrosomia in Pregnancies Complicated by Gestational Diabetes. Arch. Gynecol. Obstet. 2017, 296. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Fatihoglu, E. Fetal Epicardial Fat Thickness: Can It Serve as a Sonographic Screening Marker for Gestational Diabetes Mellitus? J. Med. Ultrasound 2020, 28. [Google Scholar] [CrossRef] [PubMed]
- Blumer, I.; Hadar, E.; Hadden, D.R.; Jovanovič, L.; Mestman, J.H.; Murad, M.H.; Yogev, Y. Diabetes and Pregnancy: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2013, 98. [Google Scholar] [CrossRef]
- Yildiz Atar, H.; Baatz, J.E.; Ryan, R.M. Molecular Mechanisms of Maternal Diabetes Effects on Fetal and Neonatal Surfactant. Children 2021, 8, 281. [Google Scholar] [CrossRef]
- Lee, I.L.; Barr, E.L.M.; Longmore, D.; Barzi, F.; Brown, A.D.H.; Connors, C.; Boyle, J.A.; Kirkwood, M.; Hampton, V.; Lynch, M.; et al. Cord Blood Metabolic Markers Are Strong Mediators of the Effect of Maternal Adiposity on Fetal Growth in Pregnancies across the Glucose Tolerance Spectrum: The PANDORA Study. Diabetologia 2020, 63. [Google Scholar] [CrossRef]
- Tarvonen, M.; Hovi, P.; Sainio, S.; Vuorela, P.; Andersson, S.; Teramo, K. Intrapartal Cardiotocographic Patterns and Hypoxia-Related Perinatal Outcomes in Pregnancies Complicated by Gestational Diabetes Mellitus. Acta Diabetol. 2021. [Google Scholar] [CrossRef]
- Basu, M.; Garg, V. Maternal Hyperglycemia and Fetal Cardiac Development: Clinical Impact and Underlying Mechanisms. Birth Defects Res. 2018, 110, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Balsells, M.; García-Patterson, A.; Gich, I.; Corcoy, R. Major Congenital Malformations in Women with Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. Diabetes Metab. Res. Rev. 2012, 28. [Google Scholar] [CrossRef] [PubMed]
- Mitanchez, D. Foetal and Neonatal Complications in Gestational Diabetes: Perinatal Mortality, Congenital Malformations, Macrosomia, Shoulder Dystocia, Birth Injuries, Neonatal Complications. Diabetes Metab. 2010, 36. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Frías, M.L.; Frías, J.P.; Bermejo, E.; Rodríguez-Pinilla, E.; Prieto, L.; Frías, J.L. Pre-Gestational Maternal Body Mass Index Predicts an Increased Risk of Congenital Malformations in Infants of Mothers with Gestational Diabetes. Diabet. Med. 2005, 22. [Google Scholar] [CrossRef]
- Lowe, W.L.; Scholtens, D.M.; Kuang, A.; Linder, B.; Lawrence, J.M.; Lebenthal, Y.; McCance, D.; Hamilton, J.; Nodzenski, M.; Talbot, O.; et al. Hyperglycemia and Adverse Pregnancy Outcome Follow-up Study (HAPO FUS): Maternal Gestational Diabetes Mellitus and Childhood Glucose Metabolism. Diabetes Care 2019, 42. [Google Scholar] [CrossRef]
- Briana, D.D.; Germanou, K.; Boutsikou, M.; Boutsikou, T.; Athanasopoulos, N.; Marmarinos, A.; Gourgiotis, D.; Malamitsi-Puchner, A. Potential Prognostic Biomarkers of Cardiovascular Disease in Fetal Macrosomia: The Impact of Gestational Diabetes. J. Matern. Fetal Neonatal Med. 2018, 31. [Google Scholar] [CrossRef]
- Fetita, L.S.; Sobngwi, E.; Serradas, P.; Calvo, F.; Gautier, J.F. Review: Consequences of Fetal Exposure to Maternal Diabetes in Offspring. J. Clin. Endocrinol. Metab. 2006, 91. [Google Scholar] [CrossRef]
- Ekelund, M.; Shaat, N.; Almgren, P.; Groop, L.; Berntorp, K. Prediction of Postpartum Diabetes in Women with Gestational Diabetes Mellitus. Diabetologia 2010, 53. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, Å.; Laivuori, H.; Loft, A.; Oldereid, N.B.; Pinborg, A.; Petzold, M.; Romundstad, L.B.; Söderström-Anttila, V.; Bergh, C. The Association Between High Birth Weight and Long-Term Outcomes—Implications for Assisted Reproductive Technologies: A Systematic Review and Meta-Analysis. Front. Pediatr. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Crume, T.L.; Ogden, L.; Daniels, S.; Hamman, R.F.; Norris, J.M.; Dabelea, D. The Impact of in Utero Exposure to Diabetes on Childhood Body Mass Index Growth Trajectories: The EPOCH Study. J. Pediatr. 2011, 158. [Google Scholar] [CrossRef] [PubMed]



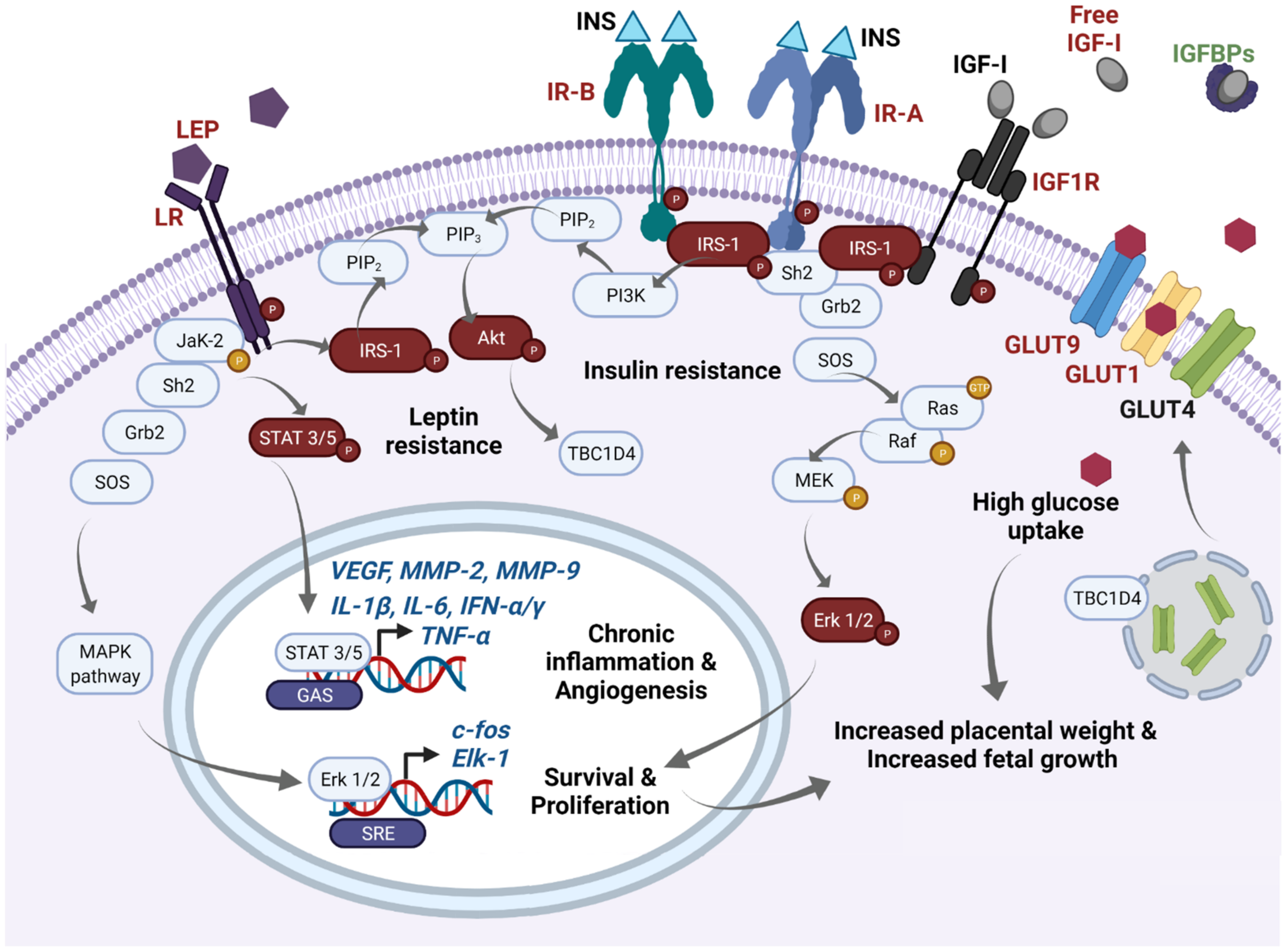{kind=link}
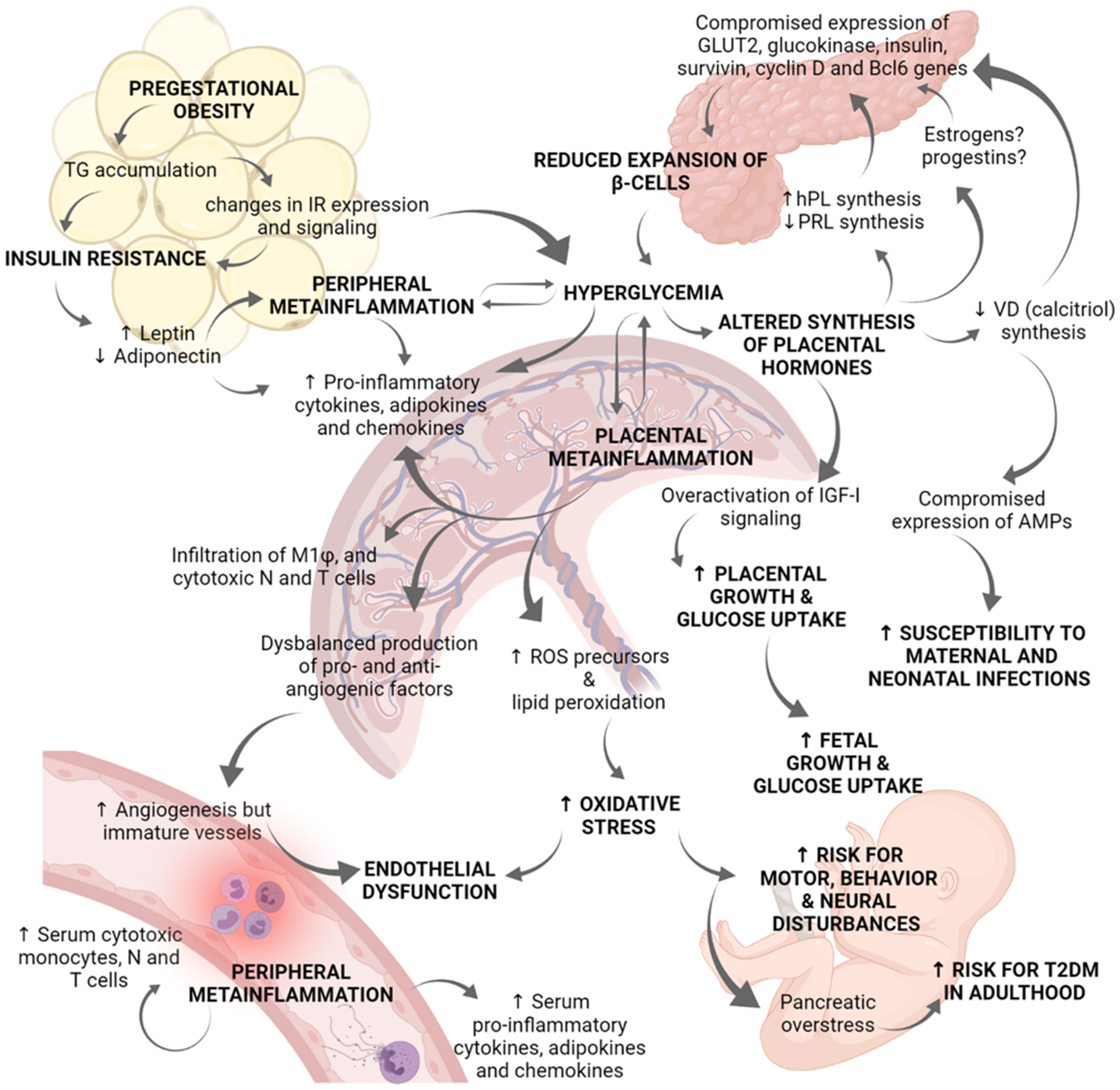{kind=link}
| Metabolite | Description | GDM Status | Reference |
|---|---|---|---|
| Protein carbonyls | Created in response to oxidative stress and are produced in the process of protein carbonylation. | Increased in plasma at 24–28 WOG | [284,285] |
| Nitrotyrosine | Superoxide O2−• is produced by the electroreduction of molecular oxygen, is highly reactive, with a short half-life. It is metabolized to hydrogen peroxide by the high activity of superoxide dismutase. In highly vascularized tissues such as the human placenta, superoxide can combine with nitric oxide to form the very reactive and damaging ROS, peroxynitrite (ONOO−•). ONOO−• reacts with hydroxyl groups on serine, tyrosine, and threonine residues to form nitrotyrosine adducts. | Increased in the blood (24–28 WOG) | [284,286] |
| Advanced glycation end products (AGEs) | AGEs are formed by a combination of glycation and oxidation reaction. AGEs are strongly associated with diabetic complications of pregnancy that have increased levels of glucose as well as ROS and are associated with complications of diabetes such as retinopathy, nephropathy, and neuropathy. AGEs are formed due to the non-enzymatic glycation of proteins, lipids, and nucleic acids during hyperglycemia. It has the potential to damage vasculature by modifying the substrate or utilizing AGEs and Receptor of AGE (RAGE) interaction. AGEs such as Ne-carboxymethyl lysine (CML) are produced under oxidative conditions. | AGEs are elevated in maternal serum. CML is elevated in plasma (24–30 WOG) | [287,288,289,290] |
| Oxidized low-density lipoproteins (Ox-LDL) | The formation of ox-LDL involves the oxidation of both protein and lipid components. | Elevated in maternal plasma | [291,292] |
| 4 -hydroxynonenal (4-HNE) | A product of lipid peroxidation that can modify proteins and have effects on tissue survival. | Elevated in placental and uterine tissues | [283,293] |
| Malondialdehyde (MDA) | MDA is a marker of lipid peroxidation and oxidative stress and is formed as the result of lipid peroxidation of polyunsaturated fatty acids. | Elevated maternal plasma levels at 28 WOG | [292] |
| Oxidized DNA | DNA is vulnerable to oxidation and undergoes a continuous cycle of damage and repair in living cells. The nucleoside oxidation marker 8-oxo-7,8 dihydro-2 deoxyguanosine (8OH-dG) is used as a biomarker of oxidative stress excreted in urine. | 8OH-dG level in the urine was 26% higher in women who subsequently developed GDM | [294,295] |
| Glutathione (GSH) and Glutathione peroxidase (GPX) | Under healthy conditions, cells have up to 98% of total GSH in their reduced form. GSH acts as an electron donor to reduce unstable ROS in a process that permits the formation of oxidized GSH. An altered ratio reduced GSH/oxidized GSH is an indicator of oxidative stress. While reduced GSH can directly prevent non-enzymatic oxidation of thiol groups; it also acts as a co-substrate for the antioxidant enzyme GPX. GPX can remove active peroxides, but this results in the conversion of reduced GSH to oxidized GSH. | Reduced GSH in blood at term. Diminution of GPX in blood | [296,297,298] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olmos-Ortiz, A.; Flores-Espinosa, P.; Díaz, L.; Velázquez, P.; Ramírez-Isarraraz, C.; Zaga-Clavellina, V. Immunoendocrine Dysregulation during Gestational Diabetes Mellitus: The Central Role of the Placenta. Int. J. Mol. Sci. 2021, 22, 8087. https://doi.org/10.3390/ijms22158087
Olmos-Ortiz A, Flores-Espinosa P, Díaz L, Velázquez P, Ramírez-Isarraraz C, Zaga-Clavellina V. Immunoendocrine Dysregulation during Gestational Diabetes Mellitus: The Central Role of the Placenta. International Journal of Molecular Sciences. 2021; 22(15):8087. https://doi.org/10.3390/ijms22158087
Chicago/Turabian StyleOlmos-Ortiz, Andrea, Pilar Flores-Espinosa, Lorenza Díaz, Pilar Velázquez, Carlos Ramírez-Isarraraz, and Verónica Zaga-Clavellina. 2021. "Immunoendocrine Dysregulation during Gestational Diabetes Mellitus: The Central Role of the Placenta" International Journal of Molecular Sciences 22, no. 15: 8087. https://doi.org/10.3390/ijms22158087
APA StyleOlmos-Ortiz, A., Flores-Espinosa, P., Díaz, L., Velázquez, P., Ramírez-Isarraraz, C., & Zaga-Clavellina, V. (2021). Immunoendocrine Dysregulation during Gestational Diabetes Mellitus: The Central Role of the Placenta. International Journal of Molecular Sciences, 22(15), 8087. https://doi.org/10.3390/ijms22158087