
Mitochondrial HSP70 Chaperone System—The Influence of Post-Translational Modifications and Involvement in Human Diseases

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. General Characterization of Heat Shock Proteins
3. The 70-kDa Heat Shock Proteins (HSP70s)
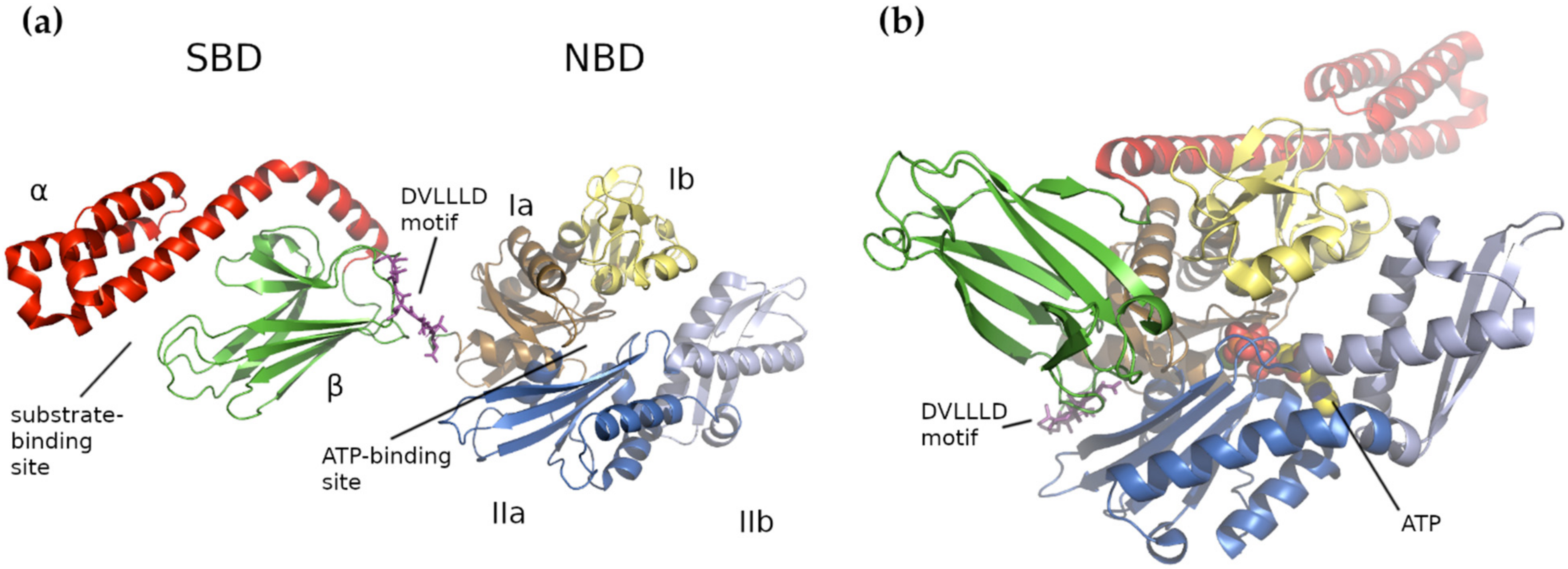
4. Human Mitochondrial HSP70 (mtHSP70)
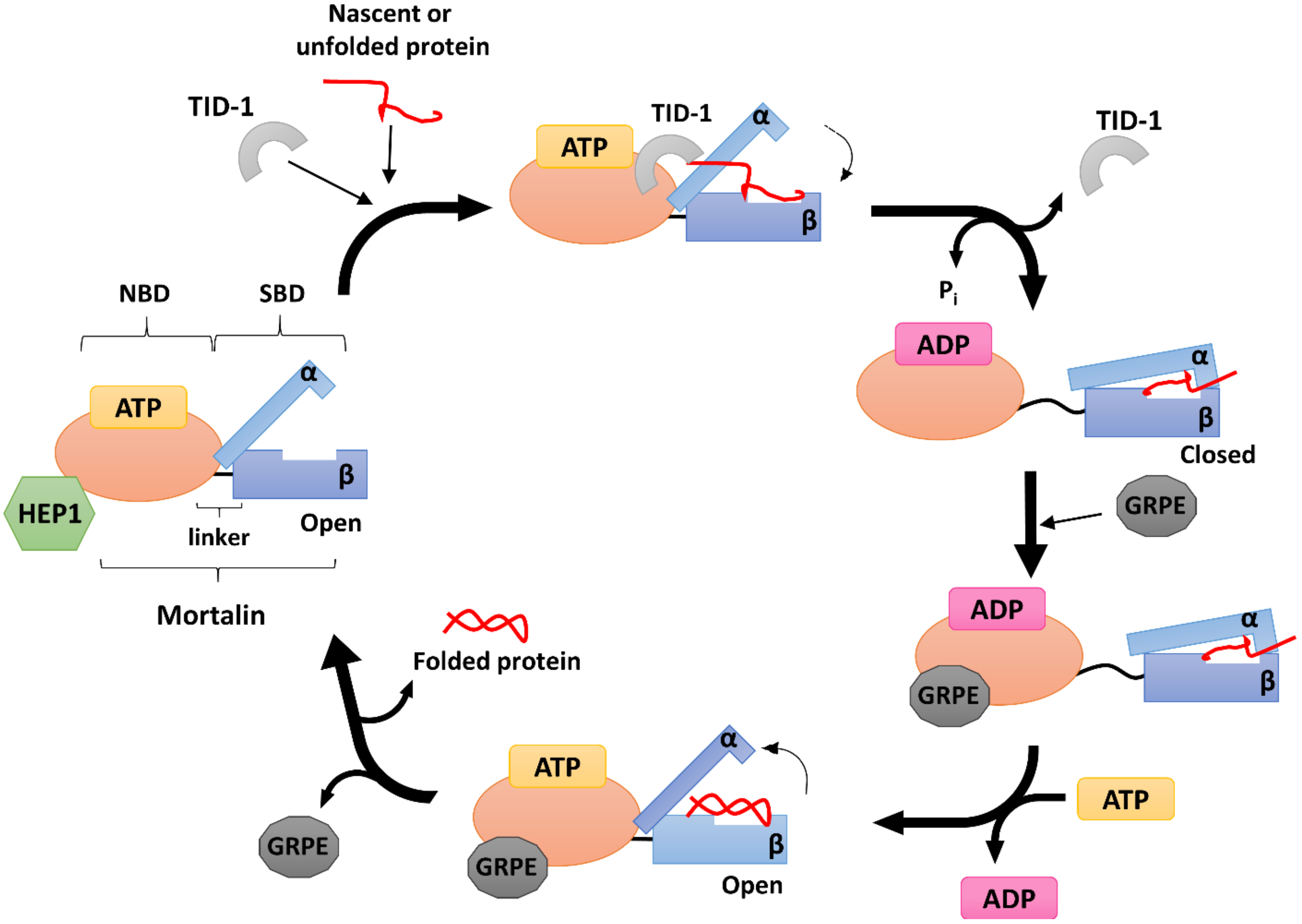
5. Human mtHSP70 Co-Chaperones
5.1. HEP1
5.2. GRPE
5.3. TID-1
6. Mortalin, Mortalin Co-Chaperones, Post-Translational Modifications and Human Diseases
6.1. PTMs
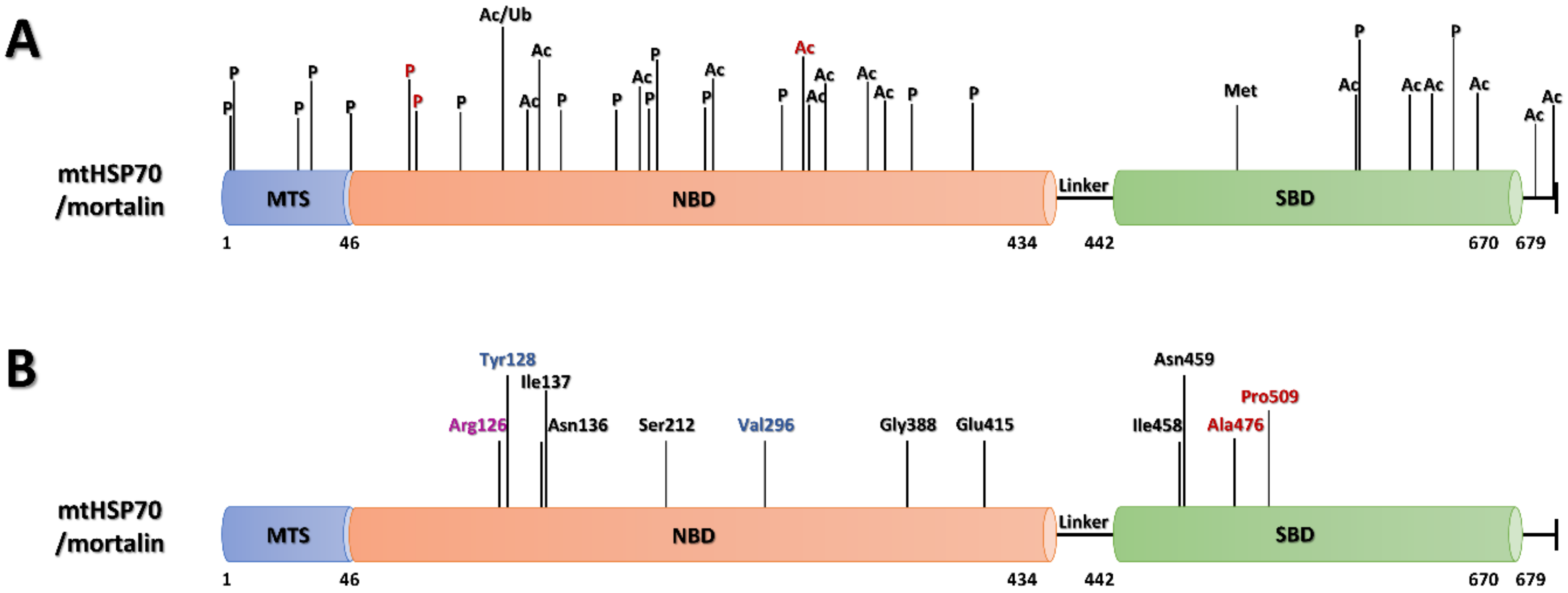
6.1.1. PTMs of Mortalin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Position | Localization | Diseases |
| Phosphorylation | S3 | MTS | breast cancer [228] |
| S5 | MTS | breast cancer [228] | |
| T22 | MTS | breast cancer [228] | |
| S29 | MTS | breast cancer [229] | |
| Y46 | MTS | lung cancer [230] | |
| T87 | NBD | lung cancer [231] | |
| Y128 | NBD | gastric cancer, leukemia, chronic myelogenous leukemia, lung cancer | |
| S148 | NBD | breast cancer [229,232] | |
| S162 | NBD | lung cancer [233] | |
| S164 | NBD | breast cancer [229], lung cancer [233] | |
| S200 | NBD | breast cancer [229] | |
| T271 | NBD | lung cancer | |
| S378 | NBD | erythroid leukemia [234], acute myeloblastic leukemia [234], lung cancer, B cell lymphoma [234], non-Hodgkin’s lymphoma [234], multiple myeloma [234] | |
| S408 | NBD | breast cancer [229] | |
| Y568 | SBD | cervical cancer [235], gastric cancer, leukemia, chronic myelogenous leukemia, liver cancer, hepatocellular carcinoma, lung cancer | |
| S627 | SBD | breast cancer [229], melanoma skin cancer [236] | |
| Mono-Methylation | R513 | SBD | gastric cancer, lung cancer |
| Acetylation | K121 | NBD | lung cancer, B cell lymphoma |
| K135 | NBD | colorectal cancer, leukemia, chronic myelogenous leukemia, liver cancer, hepatocellular carcinoma, lung cancer [221] | |
| K138 | NBD | leukemia, chronic myelogenous leukemia, lung cancer [221] | |
| K159 | NBD | colorectal cancer, gastric cancer, leukemia, chronic myelogenous leukemia, lung cancer | |
| K206 | NBD | not found | |
| K288 | NBD | colorectal cancer, leukemia, chronic myelogenous leukemia, liver cancer, lung cancer [221], ventricular tachycardia | |
| K291 | NBD | ventricular tachycardia | |
| K300 | NBD | colorectal cancer, leukemia, chronic myelogenous leukemia, liver cancer, hepatocellular carcinoma, lung cancer [221] | |
| K345 | NBD | gastric cancer, lung cancer [221] | |
| K567 | SBD | colorectal cancer, leukemia, chronic myelogenous leukemia, lung cancer | |
| K600 | SBD | liver cancer, lung cancer [221] | |
| K612 | SBD | colorectal cancer, leukemia, chronic myelogenous leukemia, lung cancer [221] | |
| K646 | SBD | gastric cancer, lung cancer | |
| K675 | SBD | leukemia, chronic myelogenous leukemia | |
| K678 | SBD | kidney cancer, leukemia, chronic myelogenous leukemia | |
| Ubiquitylation | K121 | NBD | multiple myeloma |
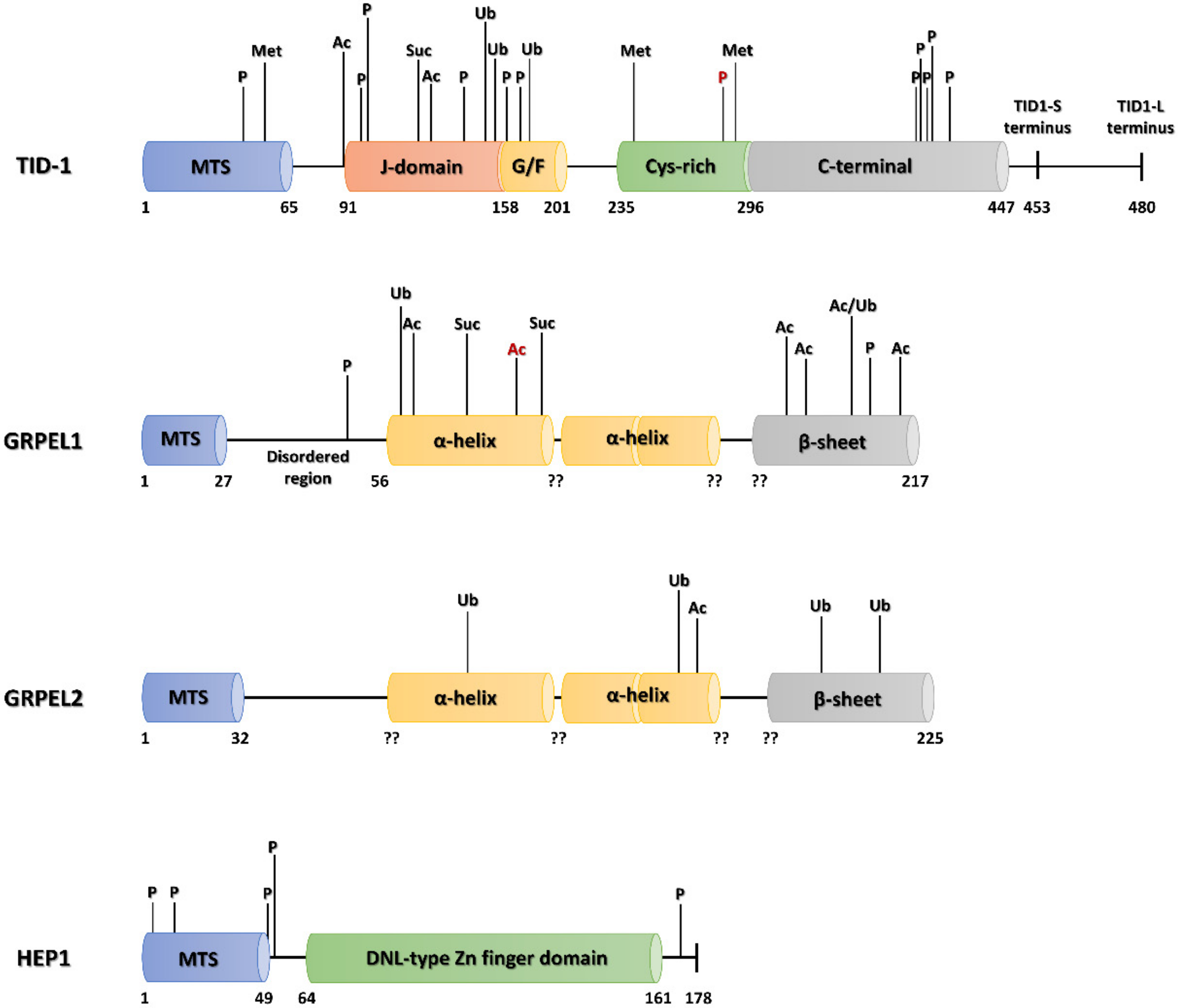
6.1.2. PTMs of Mortalin Co-Chaperones
6.2. Mortalin, Co-Chaperones and Human Diseases
6.2.1. Mortalin in Neurodegenerative Diseases
6.2.2. Mortalin in Cancer
6.2.3. Mortalin in Autosomal Recessive Diseases
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | amino acid |
| AAA+ | ATPases associated with diverse cellular activities |
| AD | Alzheimer’s disease |
| AFG3L2 | AFG3-like matrix AAA+ peptidase subunit 2 |
| ALS | amyotrophic lateral sclerosis |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CLP | caseinolytic protease |
| CSA | congenital sideroblastic anemia |
| CODAS | cerebral, ocular, dental, auricular, and skeletal syndrome |
| DOA | dominant optic atrophy |
| EDTA | ethylenediaminetetraacetic acid |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| ERK1/2 | extracellular-signal regulated kinase 1/2 |
| EVEN-PLUS | epiphyseal, vertebral, ear, nose, plus associated findings syndrome |
| FGF1 | fibroblast growth factor 1 |
| G/F-rich region | glycine-phenylalanine-rich region |
| GRP75 | 75 kDa glucose-regulated protein |
| GRPEL1/2 | Gro-P like protein E1/2 |
| HEP1 | HSP70-escort protein 1 |
| HSCB | heat shock cognate B |
| HSF | heat shock factor |
| HSP | heat shock protein |
| HSPA9 | heat shock 70 kDa protein 9 |
| HPD domain | His-Pro-Asp domain |
| HTRA2 | high temperature requirement protein A2 |
| iAAA | inner membrane-embedded AAA+ protease |
| ISC | iron-sulfur cluster |
| JDP | J-domain protein |
| mAAA | matrix-embedded AAA+ protease |
| MAPK | mitogen activated protein kinase |
| MAPKAPK2 | MAPK activated protein kinase 2 |
| MICOS | mitochondrial contact site and organizing system |
| MPS1 | monopolar spindle 1 |
| MS | mass spectrometry |
| NBD | nucleotide binding domain |
| NEF | nucleotide exchange factor |
| NMR | nuclear magnetic resonance |
| OXPHOS | oxidative phosphorylation |
| PAM | presequence translocase-associated motor |
| PBP74 | peptide-binding protein 74 |
| PD | Parkinson’s disease |
| PQC | protein quality control |
| PTM | post-translational modification |
| pV | peroxovanadate |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SBD | substrate binding domain |
| sHSP | small heat shock protein |
| SPG7 | paraplegin |
| TID-1 | tumorous imaginal disc protein 1 |
| TRAP-1 | tumor necrosis factor receptor-associated protein type 1 |
| VDAC | voltage-dependent anion channel |
| YME1L1 | YME1-like 1 ATPase |
| ZBD | zinc-binding domain |
References
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef]
- Voos, W. Chaperone–protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta 2013, 1833, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Haslbeck, M.; Buchner, J. The Heat Shock Response: Life on the Verge of Death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Patriarca, E.J.; Maresca, B. Acquired thermotolerance following heat shock protein synthesis prevents impairment of mitochondrial ATPase activity at elevated temperatures in Saccharomyces cerevisiae. Exp. Cell Res. 1990, 190, 57–64. [Google Scholar] [CrossRef]
- Söti, C.; Csermely, P. Chaperones and aging: Role in neurodegeneration and in other civilizational diseases. Neurochem. Int. 2002, 41, 383–389. [Google Scholar] [CrossRef]
- Kourtis, N.; Tavernarakis, N. Small heat shock proteins and neurodegeneration: Recent developments. Biomol. Concepts 2018, 9, 94–102. [Google Scholar] [CrossRef]
- Harding, J.J.; Beswick, H.T.; Ajiboye, R.; Huby, R.; Blakytny, R.; Rixon, K.C. Non-enzymic post-translational modification of proteins in aging. A review. Mech. Ageing Dev. 1989, 50, 7–16. [Google Scholar] [CrossRef]
- Heydari, A.R.; Takahashi, R.; Gutsmann, A.; You, S.; Richardson, A. Hsp70 and aging. Experientia 1994, 50, 1092–1098. [Google Scholar] [CrossRef]
- Conconia, M.; Szweda, L.I.; Levine, R.L.; Stadtman, E.R.; Friguet, B. Age-Related Decline of Rat Liver Multicatalytic Proteinase Activity and Protection from Oxidative Inactivation by Heat-Shock Protein 90. Arch. Biochem. Biophys. 1996, 331, 232–240. [Google Scholar] [CrossRef]
- Sőti, C.; Csermely, P. Molecular chaperones and the aging process. Biogerontology 2000, 1, 225–233. [Google Scholar] [CrossRef]
- Mandell, R.B.; Feldherr, C.M. Identification of two HSP70-related Xenopus oocyte proteins that are capable of recycling across the nuclear envelope. J. Cell Biol. 1990, 111, 1775–1783. [Google Scholar] [CrossRef]
- Kose, S.; Furuta, M.; Koike, M.; Yoneda, Y.; Imamoto, N. The 70-kD heat shock cognate protein (hsc70) facilitates the nuclear export of the import receptors. J. Cell Biol. 2005, 171, 19–25. [Google Scholar] [CrossRef]
- Kodiha, M.; Chu, A.; Lazrak, O.; Stochaj, U. Stress inhibits nucleocytoplasmic shuttling of heat shock protein hsc70. Am. J. Physiol. Physiol. 2005, 289, C1034–C1041. [Google Scholar] [CrossRef]
- Wang, F.; Bonam, S.R.; Schall, N.; Kuhn, L.; Hammann, P.; Chaloin, O.; Madinier, J.-B.; Briand, J.-P.; Page, N.; Muller, S. Blocking nuclear export of HSPA8 after heat shock stress severely alters cell survival. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperon 2005, 10, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.C.; Wadhwa, R. Mortalin Biology: Life, Stress and Death; Springer: Dordrecht, The Netherlands, 2012; p. 342. [Google Scholar] [CrossRef]
- Ananthan, J.; Goldberg, A.L.; Voellmy, R. Abnormal proteins serve as eukaryotic stress signals and trigger the activation of heat shock genes. Science 1986, 232, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Tissiéres, A.; Mitchell, H.K.; Tracy, U.M. Protein synthesis in salivary glands of Drosophila melanogaster: Relation to chromosome puffs. J. Mol. Biol. 1974, 84, 389–398. [Google Scholar] [CrossRef]
- Lindquist, S. The Heat-Shock Response. Annu. Rev. Biochem. 1986, 55, 1151–1191. [Google Scholar] [CrossRef]
- D’Souza, S.M.; Brown, I.R. Constitutive expression of heat shock proteins Hsp90, Hsc70, Hsp70 and Hsp60 in neural and non-neural tissues of the rat during postnatal development. Cell Stress Chaperon 1998, 3, 188–199. [Google Scholar] [CrossRef]
- Walter, S. Structure and function of the GroE chaperone. Cell. Mol. Life Sci. 2002, 59, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Horwich, A.L.; Farr, G.W.; Fenton, W.A. GroEL−GroES-Mediated Protein Folding. Chem. Rev. 2006, 106, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, J.; Horwich, A.L.; Neupert, W.; Hartl, F.-U. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nat. Cell Biol. 1989, 341, 125–130. [Google Scholar] [CrossRef]
- Voos, W. A New Connection: Chaperones Meet a Mitochondrial Receptor. Mol. Cell 2003, 11, 1–3. [Google Scholar] [CrossRef]
- Nielsen, K.L.; Cowan, N.J. A Single Ring Is Sufficient for Productive Chaperonin-Mediated Folding In Vivo. Mol. Cell 1998, 2, 93–99. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Hartl, F.-U.; Martin, J.; Pollock, R.A.; Kalousek, F.; Neupert, W.; Hallberg, E.M.; Hallberg, R.L.; Horwich, A.L. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nat. Cell Biol. 1989, 337, 620–625. [Google Scholar] [CrossRef]
- Hansen, J.; Svenstrup, K.; Ang, D.; Nielsen, M.N.; Christensen, J.H.; Gregersen, N.; Nielsen, J.E.; Georgopoulos, C.; Bross, P. A novel mutation in the HSPD1 gene in a patient with hereditary spastic paraplegia. J. Neurol. 2007, 254, 897–900. [Google Scholar] [CrossRef]
- Hansen, J.J.; Dürr, A.; Cournu-Rebeix, I.; Georgopoulos, C.; Ang, D.; Nielsen, M.N.; Davoine, C.-S.; Brice, A.; Fontaine, B.; Gregersen, N.; et al. Hereditary Spastic Paraplegia SPG13 Is Associated with a Mutation in the Gene Encoding the Mitochondrial Chaperonin Hsp60. Am. J. Hum. Genet. 2002, 70, 1328–1332. [Google Scholar] [CrossRef]
- Magen, D.; Georgopoulos, C.; Bross, P.; Ang, D.; Segev, Y.; Goldsher, D.; Nemirovski, A.; Shahar, E.; Ravid, S.; Luder, A.; et al. Mitochondrial Hsp60 Chaperonopathy Causes an Autosomal-Recessive Neurodegenerative Disorder Linked to Brain Hypomyelination and Leukodystrophy. Am. J. Hum. Genet. 2008, 83, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Pockley, A.G.; Henderson, B.; Multhoff, G. Extracellular cell stress proteins as biomarkers of human disease. Biochem. Soc. Trans. 2014, 42, 1744–1751. [Google Scholar] [CrossRef]
- Cappello, F.; Gammazza, A.M.; Piccionello, A.P.; Campanella, C.; Pace, A.; De Macario, E.C.; Macario, A.J. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin. Ther. Targets 2013, 18, 185–208. [Google Scholar] [CrossRef]
- Doyle, S.M.; Wickner, S. Hsp104 and ClpB: Protein disaggregating machines. Trends Biochem. Sci. 2009, 34, 40–48. [Google Scholar] [CrossRef]
- Abrahão, J.; Mokry, D.Z.; Ramos, C.H.I. Hsp78 (78 kDa Heat Shock Protein), a Representative AAA Family Member Found in the Mitochondrial Matrix of Saccharomyces cerevisiae. Front. Mol. Biosci. 2017, 4, 60. [Google Scholar] [CrossRef]
- Leidhold, C.; von Janowsky, B.; Becker, D.; Bender, T.; Voos, W. Structure and function of Hsp78, the mitochondrial ClpB homolog. J. Struct. Biol. 2006, 156, 149–164. [Google Scholar] [CrossRef]
- A Leonhardt, S.; Fearson, K.; Danese, P.N.; Mason, T.L. HSP78 encodes a yeast mitochondrial heat shock protein in the Clp family of ATP-dependent proteases. Mol. Cell. Biol. 1993, 13, 6304–6313. [Google Scholar] [CrossRef]
- Moczko, M.; Schönfisch, B.; Voos, W.; Pfanner, N.; Rassow, J. The Mitochondrial ClpB Homolog Hsp78 Cooperates with Matrix Hsp70 in Maintenance of Mitochondrial Function. J. Mol. Biol. 1995, 254, 538–543. [Google Scholar] [CrossRef]
- Schmitt, M.; Neupert, W.; Langer, T. Hsp78, a Clp homologue within mitochondria, can substitute for chaperone functions of mt-hsp70. EMBO J. 1995, 14, 3434–3444. [Google Scholar] [CrossRef]
- Schmitt, M.; Neupert, W.; Langer, T. The molecular chaperone Hsp78 confers compartment-specific thermotolerance to mitochondria. J. Cell Biol. 1996, 134, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Rios, P.D.L.; Dietler, G.; Goloubinoff, P. Active Solubilization and Refolding of Stable Protein Aggregates by Cooperative Unfolding Action of Individual Hsp70 Chaperones. J. Biol. Chem. 2004, 279, 37298–37303. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Trifunovic, A. Mitochondrial matrix proteases: Quality control and beyond. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mao, Y. AAA+ ATPases in Protein Degradation: Structures, Functions and Mechanisms. Biomolecules 2020, 10, 629. [Google Scholar] [CrossRef]
- Van Dyck, L.; Dembowski, M.; Neupert, W.; Langer, T. Mcx1p, a ClpX homologue in mitochondria of Saccharomyces cerevisiae. FEBS Lett. 1998, 438, 250–254. [Google Scholar] [CrossRef]
- Yu, A.Y.H.; Houry, W.A. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007, 581, 3749–3757. [Google Scholar] [CrossRef]
- Abdrakhmanova, A.; Zickermann, V.; Bostina, M.; Radermacher, M.; Schägger, H.; Kerscher, S.; Brandt, U. Subunit composition of mitochondrial complex I from the yeast Yarrowia lipolytica. Biochim. Biophys. Acta 2004, 1658, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar]
- Ambro, L.; Pevala, V.; Bauer, J.; Kutejová, E. The influence of ATP-dependent proteases on a variety of nucleoid-associated processes. J. Struct. Biol. 2012, 179, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Gala, M.G.; Vögtle, F. Mitochondrial proteases in human diseases. FEBS Lett. 2021, 595, 1205–1222. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, D.; Lazzaro, F.; Brusco, A.; Plumari, M.; Battaglia, G.; Pastore, A.; Finardi, A.; Cagnoli, C.; Tempia, F.; Frontali, M.; et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010, 42, 313–321. [Google Scholar] [CrossRef]
- Pierson, T.M.; Adams, D.; Bonn, F.; Martinelli, P.; Cherukuri, P.F.; Teer, J.K.; Hansen, N.F.; Cruz, P.; Mullikin for the NISC Comparative Sequencing Program, J.C.; Blakesley, R.W.; et al. Whole-Exome Sequencing Identifies Homozygous AFG3L2 Mutations in a Spastic Ataxia-Neuropathy Syndrome Linked to Mitochondrial m-AAA Proteases. PLoS Genet. 2011, 7, e1002325. [Google Scholar] [CrossRef]
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic Paraplegia and OXPHOS Impairment Caused by Mutations in Paraplegin, a Nuclear-Encoded Mitochondrial Metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef]
- Pfeffer, G.; Gorman, G.; Griffin, H.R.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef]
- Charif, M.; Chevrollier, A.; Gueguen, N.; Bris, C.; Goudenège, D.; Desquiret-Dumas, V.; Leruez, S.; Colin, E.; Meunier, A.; Vignal, C.; et al. Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy. Neurol. Genet. 2020, 6, e428. [Google Scholar] [CrossRef]
- Hartmann, B.; Wai, T.; Hu, H.; MacVicar, T.; Musante, L.; Fischer-Zirnsak, B.; Stenzel, W.; Gräf, R.; Heuvel, L.V.D.; Ropers, H.-H.; et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife 2016, 5, e16078. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.G.; et al. CODAS Syndrome Is Associated with Mutations of LONP1, Encoding Mitochondrial AAA+ Lon Protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Dikoglu, E.; Alfaiz, A.; Górna, M.; Bertola, D.; Chae, J.H.; Cho, T.-J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.-H.; et al. Mutations inLONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. Part A 2015, 167, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault Syndrome Is Caused by Recessive Mutations in CLPP, Encoding a Mitochondrial ATP-Dependent Chambered Protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef]
- Brodie, E.; Zhan, H.; Saiyed, T.; Truscott, K.N.; Dougan, D.A. Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Yien, Y.Y.; Ducamp, S.; van der Vorm, L.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in humanCLPXelevates levels ofδ-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc. Natl. Acad. Sci. USA 2017, 114, E8045–E8052. [Google Scholar] [CrossRef]
- Kriehuber, T.; Rattei, T.; Weinmaier, T.; Bepperling, A.; Haslbeck, M.; Buchner, J. Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J. 2010, 24, 3633–3642. [Google Scholar] [CrossRef]
- Horwitz, J. Alpha-crystallin. Exp. Eye Res. 2003, 76, 145–153. [Google Scholar] [CrossRef]
- Ingolia, T.D.; Craig, E.A. Four small Drosophila heat shock proteins are related to each other and to mammalian alpha-crystallin. Proc. Natl. Acad. Sci. USA 1982, 79, 2360–2364. [Google Scholar] [CrossRef]
- Van Montfort, R.L.; Basha, E.; Friedrich, K.L.; Slingsby, C.; Vierling, E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Genet. 2001, 8, 1025–1030. [Google Scholar] [CrossRef]
- Simon, S.; Aissat, A.; Degrugillier, F.; Simonneau, B.; Fanen, P.; Arrigo, A.-P. Small Hsps as Therapeutic Targets of Cystic Fibrosis Transmembrane Conductance Regulator Protein. Int. J. Mol. Sci. 2021, 22, 4252. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J.; Bova, M.P.; Ding, L.L.; A Haley, D.; Stewart, P.L. Lens α-crystallin: Function and structure. Eye 1999, 13, 403–408. [Google Scholar] [CrossRef]
- Andley, U.P. Effects of α-Crystallin on Lens Cell Function and Cataract Pathology. Curr. Mol. Med. 2009, 9, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Luo, Z.; Zhang, L.; Wang, L.; Nie, Q.; Wang, Z.-F.; Huang, Z.; Hu, X.; Gong, L.; Arrigo, A.-P.; et al. The small heat shock protein αA-crystallin negatively regulates pancreatic tumorigenesis. Oncotarget 2016, 7, 65808–65824. [Google Scholar] [CrossRef]
- Zeng, L.; Tan, J.; Lu, W.; Lu, T.; Hu, Z. The potential role of small heat shock proteins in mitochondria. Cell. Signal. 2013, 25, 2312–2319. [Google Scholar] [CrossRef]
- Kappé, G.; Verschuure, P.; Philipsen, R.L.; A Staalduinen, A.; Van de Boogaart, P.; Boelens, W.C.; De Jong, W.W. Characterization of two novel human small heat shock proteins: Protein kinase-related HspB8 and testis-specific HspB9. Biochim. Biophys. Acta 2001, 1520, 1–6. [Google Scholar] [CrossRef]
- Depre, C.; Hase, M.; Gaussin, V.; Zajac, A.; Wang, L.; Hittinger, L.; Ghaleh, B.; Yu, X.; Kudej, R.K.; Wagner, T.; et al. H11 Kinase Is a Novel Mediator of Myocardial Hypertrophy In Vivo. Circ. Res. 2002, 91, 1007–1014. [Google Scholar] [CrossRef]
- Kumarapeli, A.R.K.; Su, H.; Huang, W.; Tang, M.; Zheng, H.; Horak, K.M.; Li, M.; Wang, X. B-Crystallin Suppresses Pressure Overload Cardiac Hypertrophy. Circ. Res. 2008, 103, 1473–1482. [Google Scholar] [CrossRef]
- Marunouchi, T.; Abe, Y.; Murata, M.; Inomata, S.; Sanbe, A.; Takagi, N.; Tanonaka, K. Changes in Small Heat Shock Proteins HSPB1, HSPB5 and HSPB8 in Mitochondria of the Failing Heart Following Myocardial Infarction in Rats. Biol. Pharm. Bull. 2013, 36, 529–539. [Google Scholar] [CrossRef]
- Xu, X.; Sarbeng, E.B.; Vorvis, C.; Kumar, D.P.; Zhou, L.; Liu, Q. Unique Peptide Substrate Binding Properties of 110-kDa Heat-shock Protein (Hsp110) Determine Its Distinct Chaperone Activity. J. Biol. Chem. 2012, 287, 5661–5672. [Google Scholar] [CrossRef]
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J. Hsp70—A master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef]
- Mayer, M.P. Recruitment of Hsp70 chaperones: A crucial part of viral survival strategies. Rev. Physiol. Biochem. Pharmacol. 2005, 153, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.; Morimoto, R.I. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proc. Natl. Acad. Sci. USA 1985, 82, 6455–6459. [Google Scholar] [CrossRef]
- Karlin, S.; Brocchieri, L. Heat Shock Protein 70 Family: Multiple Sequence Comparisons, Function, and Evolution. J. Mol. Evol. 1998, 47, 565–577. [Google Scholar] [CrossRef]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Dores-Silva, P.R.; Nishimura, L.S.; Kiraly, V.T.; Borges, J.C. Structural and functional studies of the Leishmania braziliensis mitochondrial Hsp70: Similarities and dissimilarities to human orthologues. Arch. Biochem. Biophys. 2017, 613, 43–52. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef]
- Vickery, L.E.; Silberg, J.J.; Ta, D.T. Hsc66 and Hsc20, a new heat shock cognate molecular chaperone system from Escherichia coli. Protein Sci. 1997, 6, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Silberg, J.J.; Hoff, K.G.; Vickery, L.E. The Hsc66-Hsc20 Chaperone System in Escherichia coli: Chaperone Activity and Interactions with the DnaK-DnaJ-GrpE System. J. Bacteriol. 1998, 180, 6617–6624. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tokumoto, U.; Takahashi, Y. Genetic Analysis of the isc Operon in Escherichia coli Involved in the Biogenesis of Cellular Iron-Sulfur Proteins. J. Biochem. 2001, 130, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nakamura, M. Functional Assignment of the ORF2-iscS-iscU-iscA-hscB-hscA-fdx-0RF3 Gene Cluster Involved in the Assembly of Fe-S Clusters in Escherichia coli. J. Biochem. 1999, 126, 917–926. [Google Scholar] [CrossRef]
- Lelivelt, M.J.; Kawula, T.H. Hsc66, an Hsp70 homolog in Escherichia coli, is induced by cold shock but not by heat shock. J. Bacteriol. 1995, 177, 4900–4907. [Google Scholar] [CrossRef]
- Yoshimune, K.; Yoshimura, T.; Esaki, N. Hsc62, a New DnaK Homologue ofEscherichia coli. Biochem. Biophys. Res. Commun. 1998, 250, 115–118. [Google Scholar] [CrossRef]
- Kluck, C.J.; Patzelt, H.; Genevaux, P.; Brehmer, D.; Rist, W.; Schneider-Mergener, J.; Bukau, B.; Mayer, M. Structure-Function Analysis of HscC, theEscherichia coli Member of a Novel Subfamily of Specialized Hsp70 Chaperones. J. Biol. Chem. 2002, 277, 41060–41069. [Google Scholar] [CrossRef]
- Kominek, J.; Marszalek, J.; Neuvéglise, C.; Craig, E.A.; Williams, B.L. The Complex Evolutionary Dynamics of Hsp70s: A Genomic and Functional Perspective. Genome Biol. Evol. 2013, 5, 2460–2477. [Google Scholar] [CrossRef]
- Takakuwa, J.E.; Nitika; Knighton, L.E.; Truman, A.W. Oligomerization of Hsp70: Current Perspectives on Regulation and Function. Front. Mol. Biosci. 2019, 6, 81. [Google Scholar] [CrossRef]
- Kabani, M.; Martineau, C.N. Multiple Hsp70 Isoforms in the Eukaryotic Cytosol: Mere Redundancy or Functional Specificity? Curr. Genom. 2008, 9, 338-248. [Google Scholar] [CrossRef]
- Bonam, S.R.; Ruff, M.; Muller, S. Ruff HSPA8/HSC70 in Immune Disorders: A Molecular Rheostat that Adjusts Chaperone-Mediated Autophagy Substrates. Cells 2019, 8, 849. [Google Scholar] [CrossRef] [PubMed]
- Velasco, L.; Dublang, L.; Moro, F.; Muga, A. The Complex Phosphorylation Patterns that Regulate the Activity of Hsp70 and Its Cochaperones. Int. J. Mol. Sci. 2019, 20, 4122. [Google Scholar] [CrossRef] [PubMed]
- Minder, A.C.; Narberhaus, F.; Babst, M.; Hennecke, H.; Fischer, H.-M. The dnaKJ operon belongs to the σ32-dependent class of heat shock genes in Bradyrhizobium japonicum. Mol. Genet. Genom. 1997, 254, 195–206. [Google Scholar] [CrossRef]
- Susin, M.F.; Baldini, R.L.; Gueiros-Filho, F.; Gomes, S.L. GroES/GroEL and DnaK/DnaJ Have Distinct Roles in Stress Responses and during Cell Cycle Progression in Caulobacter crescentus. J. Bacteriol. 2006, 188, 8044–8053. [Google Scholar] [CrossRef] [PubMed]
- Keith, L.M.W.; Partridge, J.E.; Bender, C.L. dnaK and the Heat Stress Response of Pseudomonas syringae pv. glycinea. Mol. Plant-Microbe Interact. 1999, 12, 563–574. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rupprecht, E.; Gathmann, S.; Fuhrmann, E.; Schneider, D. Three different DnaK proteins are functionally expressed in the cyanobacterium Synechocystis sp. PCC 6803. Microbiology 2007, 153, 1828–1841. [Google Scholar] [CrossRef] [PubMed]
- Fay, A.; Glickman, M.S. An Essential Nonredundant Role for Mycobacterial DnaK in Native Protein Folding. PLoS Genet. 2014, 10, e1004516. [Google Scholar] [CrossRef] [PubMed]
- Schramm, F.D.; Heinrich, K.; Thüring, M.; Bernhardt, J.; Jonas, K. An essential regulatory function of the DnaK chaperone dictates the decision between proliferation and maintenance in Caulobacter crescentus. PLoS Genet. 2017, 13, e1007148. [Google Scholar] [CrossRef] [PubMed]
- Genevaux, P.; Keppel, F.; Schwager, F.; Langendijk-Genevaux, P.; Hartl, F.U.; Georgopoulos, C. In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 2004, 5, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Werner-Washburne, M.; E Stone, D.; A Craig, E. Complex interactions among members of an essential subfamily of hsp70 genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7. [Google Scholar] [CrossRef]
- Daugaard, M.; Jäättelä, M.; Rohde, M. Hsp70-2 is Required for Tumor Cell Growth and Survival. Cell Cycle 2005, 4, 877–880. [Google Scholar] [CrossRef]
- General, I.J.; Liu, Y.; Blackburn, M.E.; Mao, W.; Gierasch, L.M.; Bahar, I. ATPase Subdomain IA Is a Mediator of Interdomain Allostery in Hsp70 Molecular Chaperones. PLoS Comput. Biol. 2014, 10, e1003624. [Google Scholar] [CrossRef]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef]
- Morshauser, R.C.; Wang, H.; Flynn, G.C.; Zuiderweg, E.R.P. The Peptide-Binding Domain of the Chaperone Protein Hsc70 Has an Unusual Secondary Structure Topology. Biochemistry 1995, 34, 6261–6266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, X.; Burkholder, W.F.; Gragerov, A.; Ogata, C.M.; Gottesman, M.E.; Hendrickson, W.A. Structural Analysis of Substrate Binding by the Molecular Chaperone DnaK. Science 1996, 272, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, E.B.; Chang, C.Y.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M. Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Zuiderweg, E.R.P.; Bertelsen, E.B.; Rousaki, A.; Mayer, M.; Gestwicki, J.E.; Ahmad, A. Allostery in the Hsp70 Chaperone Proteins. Top. Curr. Chem. 2012, 328, 99–153. [Google Scholar] [CrossRef]
- Alvira-De-Celis, S.; Cuéllar, J.; Röhl, A.; Yamamoto, S.; Itoh, H.; Alfonso, C.; Rivas, G.; Buchner, J.; Valpuesta, J. Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nat. Commun. 2014, 5, 5484. [Google Scholar] [CrossRef]
- Allan, R.K.; Ratajczak, T. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperon 2010, 16, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H.R. The PDB and protein homeostasis: From chaperones to degradation and disaggregase machines. J. Biol. Chem. 2021, 296, 100744. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, K.P. The Molecular Chaperone Hsp70 Family Members Function by a Bidirectional Heterotrophic Allosteric Mechanism. Protein Pept. Lett. 2011, 18, 132–142. [Google Scholar] [CrossRef]
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Dores-Silva, P.R.; Barbosa, L.; Ramos, C.; Borges, J.C. Human Mitochondrial Hsp70 (Mortalin): Shedding Light on ATPase Activity, Interaction with Adenosine Nucleotides, Solution Structure and Domain Organization. PLoS ONE 2015, 10, e0117170. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Kaul, S.; Ikawa, Y.; Sugimoto, Y. Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J. Biol. Chem. 1993, 268, 6615–6621. [Google Scholar] [CrossRef]
- Kaul, S.C.; Wadhwa, R.; Matsuda, Y.; Hensler, P.J.; Pereira-Smith, O.M.; Komatsu, Y.; Mitsui, Y. Mouse and human chromosomal assignments of mortalin, a novel member of the murine hsp70 family of proteins. FEBS Lett. 1995, 361, 269–272. [Google Scholar] [CrossRef][Green Version]
- Wadhwa, R.; Akiyama, S.; Sugihara, T.; Reddel, R.R.; Mitsui, Y.; Kaul, S.C. Genetic Differences between the Pancytosolic and Perinuclear Forms of Murine Mortalin. Exp. Cell Res. 1996, 226, 381–386. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Yamasaki, K.; Kaul, S.C.; Wadhwa, R. Structural and Functional Differences between Mouse Mot-1 and Mot-2 Proteins That Differ in Two Amino Acids. Ann. N. Y. Acad. Sci. 2006, 1067, 220–223. [Google Scholar] [CrossRef]
- Gestl, E.E.; Böttger, S.A. Cytoplasmic sequestration of the tumor suppressor p53 by a heat shock protein 70 family member, mortalin, in human colorectal adenocarcinoma cell lines. Biochem. Biophys. Res. Commun. 2012, 423, 411–416. [Google Scholar] [CrossRef]
- Wadhwa, R.; Takano, S.; Robert, M.; Yoshida, A.; Nomura, H.; Reddel, R.; Mitsui, Y.; Kaul, S. Inactivation of Tumor Suppressor p53 by Mot-2, a hsp70 Family Member. J. Biol. Chem. 1998, 273, 29586–29591. [Google Scholar] [CrossRef]
- Cappello, F.; De Macario, E.C.; Marasà, L.; Zummo, G.; Macario, A.J.L. Hsp60 expression, new locations, functions, and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef]
- Wadhwa, R.; Taira, K.; Kaul, S.C. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: What, when, and where? Cell Stress Chaperones 2002, 7, 309–316. [Google Scholar] [CrossRef]
- Wadhwa, R.; Takano, S.; Kaur, K.; Aida, S.; Yaguchi, T.; Kaul, Z.; Hirano, T.; Taira, K.; Kaul, S.C. Identification and characterization of molecular interactions between mortalin/mtHsp70 and HSP60. Biochem. J. 2005, 391, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Leustek, T.; Dalie, B.; Amir-Shapira, D.; Brot, N.; Weissbach, H. A member of the Hsp70 family is localized in mitochondria and resembles Escherichia coli DnaK. Proc. Natl. Acad. Sci. USA 1989, 86, 7805–7808. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.J.; Hoogenraad, N.J.; Høj, P.B. Isolation and characterisation of a cDNA encoding rat mitochondrial GrpE, a stress-inducible nucleotide-exchange factor of ubiquitous appearance in mammalian organs. FEBS Lett. 1996, 396, 181–188. [Google Scholar] [CrossRef]
- Bouvard, V.; Zaitchouk, T.; Vacher, M.; Duthu, A.; Canivet, M.; Choisy-Rossi, C.; Nieruchalski, M.; May, E. Tissue and cell-specific expression of the p53-target genes: Bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene 2000, 19, 649–660. [Google Scholar] [CrossRef]
- Kaul, S.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar] [CrossRef]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef]
- Amick, J.; Schlanger, S.E.; Wachnowsky, C.; Moseng, M.A.; Emerson, C.C.; Dare, M.; Luo, W.-I.; Ithychanda, S.S.; Nix, J.C.; Cowan, J.A.; et al. Crystal structure of the nucleotide-binding domain of mortalin, the mitochondrial Hsp70 chaperone. Protein Sci. 2014, 23, 833–842. [Google Scholar] [CrossRef]
- Giometti, C.S.; Tollaksen, S.L.; Chubb, C.; Williams, C.; Huberman, E. Analysis of proteins from human breast epithelial cells using two-dimensional gel electrophoresis. Electrophoresis 1995, 16, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, D.F.; Frutiger, S.; Paquet, N.; Bairoch, A.; Ravier, F.; Pasquali, C.; Sanchez, J.-C.; Tissot, J.-D.; Bjellqvist, B.; Vargas, R.; et al. Human liver protein map: A reference database established by microsequencing and gel comparison. Electrophoresis 1992, 13, 992–1001. [Google Scholar] [CrossRef]
- Ji, H.; Reid, G.E.; Moritz, R.L.; Eddes, J.S.; Burgess, A.W.; Simpson, R.J. A two-dimensional gel database of human colon carcinoma proteins. Electrophoresis 1997, 18, 605–613. [Google Scholar] [CrossRef]
- Sun, J.; Che, S.-L.; Piao, J.-J.; Xu, M.; Chen, L.-Y.; Lin, Z.-H. Mortalin overexpression predicts poor prognosis in early stage of non–small cell lung cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Dores-Silva, P.; Minari, K.; Ramos, C.; Barbosa, L.; Borges, J. Structural and stability studies of the human mtHsp70-escort protein 1: An essential mortalin co-chaperone. Int. J. Biol. Macromol. 2013, 56, 140–148. [Google Scholar] [CrossRef]
- Harrison, C.J.; Hayer-Hartl, M.; Di Liberto, M.; Hartl, F.-U.; Kuriyan, J. Crystal Structure of the Nucleotide Exchange Factor GrpE Bound to the ATPase Domain of the Molecular Chaperone DnaK. Science 1997, 276, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M.; Pfanner, N.; van der Laan, M. A dynamic machinery for import of mitochondrial precursor proteins. FEBS Lett. 2007, 581, 2802–2810. [Google Scholar] [CrossRef]
- Dolezal, P.; Likic, V.; Tachezy, J.; Lithgow, T. Evolution of the Molecular Machines for Protein Import into Mitochondria. Science 2006, 313, 314–318. [Google Scholar] [CrossRef]
- Mokranjac, D.; Neupert, W. Thirty years of protein translocation into mitochondria: Unexpectedly complex and still puzzling. Biochim. Biophys. Acta 2009, 1793, 33–41. [Google Scholar] [CrossRef]
- Kang, P.-J.; Ostermann, J.; Shilling, J.; Neupert, W.; Craig, E.A.; Pfanner, N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nat. Cell Biol. 1990, 348, 137–143. [Google Scholar] [CrossRef]
- Rehling, P.; Brandner, K.; Pfanner, N. Mitochondrial import and the twin-pore translocase. Nat. Rev. Mol. Cell Biol. 2004, 5, 519–530. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Van Wilpe, S.; Ryan, M.; Hill, K.; Maarse, A.C.; Meisinger, C.; Brix, J.; Dekker, P.; Moczko, M.; Wagner, R.; Meijer, M.; et al. Tom22 is a multifunctional organizer of the mitochondrial preprotein translocase. Nat. Cell Biol. 1999, 401, 485–489. [Google Scholar] [CrossRef]
- Abe, Y.; Shodai, T.; Muto, T.; Mihara, K.; Torii, H.; Nishikawa, S.-I.; Endo, T.; Kohda, D. Structural Basis of Presequence Recognition by the Mitochondrial Protein Import Receptor Tom20. Cell 2000, 100, 551–560. [Google Scholar] [CrossRef]
- Dekker, P.J.; Keil, P.; Rassow, J.; Maarse, A.C.; Pfanner, N.; Meijer, M. Identification of MIM23, a putative component of the protein import machinery of the mitochondrial inner membrane. FEBS Lett. 1993, 330, 66–70. [Google Scholar] [CrossRef]
- Lohret, T.A.; Jensen, R.E.; Kinnally, K.W. Tim23, a Protein Import Component of the Mitochondrial Inner Membrane, Is Required for Normal Activity of the Multiple Conductance Channel, MCC. J. Cell Biol. 1997, 137, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Dores-Silva, P.R.; Cauvi, D.M.; Kiraly, V.T.; Borges, J.C.; De Maio, A. Human HSPA9 (mtHsp70, mortalin) interacts with lipid bilayers containing cardiolipin, a major component of the inner mitochondrial membrane. Biochim. Biophys. Acta 2020, 1862, 183436. [Google Scholar] [CrossRef]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 Chaperone Machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef]
- Dutkiewicz, R.; Schilke, B.; Knieszner, H.; Walter, W.; Craig, E.A.; Marszalek, J.; Oinuma, K.-I.; Hashimoto, Y.; Konishi, K.; Goda, M.; et al. Ssq1, a Mitochondrial Hsp70 Involved in Iron-Sulfur (Fe/S) Center Biogenesis. J. Biol. Chem. 2003, 278, 29719–29727. [Google Scholar] [CrossRef]
- Schilke, B.; Williams, B.; Knieszner, H.; Pukszta, S.; D’Silva, P.; Craig, E.A.; Marszalek, J. Evolution of Mitochondrial Chaperones Utilized in Fe-S Cluster Biogenesis. Curr. Biol. 2006, 16, 1660–1665. [Google Scholar] [CrossRef]
- Shan, Y.; Cortopassi, G. Mitochondrial Hspa9/Mortalin regulates erythroid differentiation via iron-sulfur cluster assembly. Mitochondrion 2016, 26, 94–103. [Google Scholar] [CrossRef]
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta 2012, 1823, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Felts, S.J.; Owen, B.A.L.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related Protein TRAP1 Is a Mitochondrial Protein with Distinct Functional Properties. J. Biol. Chem. 2000, 275, 3305–3312. [Google Scholar] [CrossRef]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, W.; Song, X.-D.; Zuo, J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol. Cell. Biochem. 2005, 268, 45–51. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Schelling, C.; Kato, H.; Rapaport, D.; Woitalla, D.; Schiesling, C.; Schulte, C.; Sharma, M.; Illig, T.; Bauer, P.; et al. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: Functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 2010, 19, 4437–4452. [Google Scholar] [CrossRef] [PubMed]
- Alkhaja, A.K.; Jans, D.C.; Nikolov, M.; Vukotic, M.; Lytovchenko, O.; Ludewig, F.; Schliebs, W.; Riedel, D.; Urlaub, H.; Jakobs, S.; et al. MINOS1 is a conserved component of mitofilin complexes and required for mitochondrial function and cristae organization. Mol. Biol. Cell 2012, 23, 247–257. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. From proliferative to neurological role of an hsp70 stress chaperone, mortalin. Biogerontology 2008, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Deocaris, C.C.; Widodo, N.; Ishii, T.; Kaul, S.C.; Wadhwa, R. Functional Significance of Minor Structural and Expression Changes in Stress Chaperone Mortalin. Ann. N. Y. Acad. Sci. 2007, 1119, 165–175. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Widodo, N.; Shrestha, B.G.; Kaur, K.; Ohtaka, M.; Yamasaki, K.; Kaul, S.C.; Wadhwa, R. Mortalin sensitizes human cancer cells to MKT-077-induced senescence. Cancer Lett. 2007, 252, 259–269. [Google Scholar] [CrossRef]
- Wadhwa, R.; Yaguchi, T.; Hasan, K.; Taira, K.; Kaul, S.C. Mortalin–MPD (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem. Biophys. Res. Commun. 2003, 302, 735–742. [Google Scholar] [CrossRef]
- Takano, S.; Wadhwa, R.; Mitsui, Y.; Kaul, S.C. Identification and characterization of molecular interactions between glucose-regulated proteins (GRPs) mortalin/GRP75/peptide-binding protein 74 (PBP74) and GRP94. Biochem. J. 2001, 357, 393–398. [Google Scholar] [CrossRef]
- Lewrenz, I.; Rietzschel, N.; Guiard, B.; Lill, R.; van der Laan, M.; Voos, W. The Functional Interaction of Mitochondrial Hsp70s with the Escort Protein Zim17 Is Critical for Fe/S Biogenesis and Substrate Interaction at the Inner Membrane Preprotein Translocase. J. Biol. Chem. 2013, 288, 30931–30943. [Google Scholar] [CrossRef]
- Sichting, M.; Mokranjac, D.; Azem, A.; Neupert, W.; Hell, K. Maintenance of structure and function of mitochondrial Hsp70 chaperones requires the chaperone Hep1. EMBO J. 2005, 24, 1046–1056. [Google Scholar] [CrossRef]
- Vu, M.T.; Zhai, P.; Lee, J.; Guerra, C.; Liu, S.; Gustin, M.C.; Silberg, J.J. The DNLZ/HEP zinc-binding subdomain is critical for regulation of the mitochondrial chaperone HSPA9. Protein Sci. 2012, 21, 258–267. [Google Scholar] [CrossRef]
- Goswami, A.V.; Chittoor, B.; D’Silva, P. Understanding the Functional Interplay between Mammalian Mitochondrial Hsp70 Chaperone Machine Components. J. Biol. Chem. 2010, 285, 19472–19482. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Vu, M.T.; Hoff, K.G.; Silberg, J.J. A conserved histidine in human DNLZ/HEP is required for stimulation of HSPA9 ATPase activity. Biochem. Biophys. Res. Commun. 2011, 408, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Blamowska, M.; Sichting, M.; Mapa, K.; Mokranjac, D.; Neupert, W.; Hell, K. ATPase Domain and Interdomain Linker Play a Key Role in Aggregation of Mitochondrial Hsp70 Chaperone Ssc1. J. Biol. Chem. 2010, 285, 4423–4431. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Stanworth, C.; Liu, S.; Silberg, J.J. The Human Escort Protein Hep Binds to the ATPase Domain of Mitochondrial Hsp70 and Regulates ATP Hydrolysis. J. Biol. Chem. 2008, 283, 26098–26106. [Google Scholar] [CrossRef] [PubMed]
- Silberg, J.J.; Tapley, T.L.; Hoff, K.G.; Vickery, L.E. Regulation of the HscA ATPase Reaction Cycle by the Co-chaperone HscB and the Iron-Sulfur Cluster Assembly Protein IscU. J. Biol. Chem. 2004, 279, 53924–53931. [Google Scholar] [CrossRef] [PubMed]
- Misselwitz, B.; Staeck, O.; A Rapoport, T. J Proteins Catalytically Activate Hsp70 Molecules to Trap a Wide Range of Peptide Sequences. Mol. Cell 1998, 2, 593–603. [Google Scholar] [CrossRef]
- Liberek, K.; Marszalek, J.; Ang, D.; Georgopoulos, C.; Zylicz, M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl. Acad. Sci. USA 1991, 88, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C. GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperon 2003, 8, 218–224. [Google Scholar] [CrossRef]
- Packschies, L.; Theyssen, H.; Buchberger, A.; Bukau, B.; Goody, R.; Reinstein, J. GrpE Accelerates Nucleotide Exchange of the Molecular Chaperone DnaK with an Associative Displacement Mechanism. Biochemistry 1997, 36, 3417–3422. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Muga, A. Thermal Adaptation of the Yeast Mitochondrial Hsp70 System is Regulated by the Reversible Unfolding of its Nucleotide Exchange Factor. J. Mol. Biol. 2006, 358, 1367–1377. [Google Scholar] [CrossRef]
- Naylor, D.J.; Stines, A.P.; Hoogenraad, N.J.; Høj, P.B. Evidence for the Existence of Distinct Mammalian Cytosolic, Microsomal, and Two Mitochondrial GrpE-like Proteins, the Co-chaperones of Specific Hsp70 Members. J. Biol. Chem. 1998, 273, 21169–21177. [Google Scholar] [CrossRef]
- Takayama, S.; Bimston, D.N.; Matsuzawa, S.; Freeman, B.C.; Aimé-Sempé, C.; Xie, Z.; Morimoto, R.I.; Reed, J.C. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 1997, 16, 4887–4896. [Google Scholar] [CrossRef]
- Ikeda, E.; Yoshida, S.; Mitsuzawa, H.; Uno, I.; Toh-E, A. YGE1is a yeast homologue ofEscherichia coli grpEand is required for maintenance of mitochondrial functions. FEBS Lett. 1994, 339, 265–268. [Google Scholar] [CrossRef]
- Srivastava, S.; Savanur, M.A.; Sinha, D.; Birje, A.; Vigneshwaran, R.; Saha, P.P.; D’Silva, P. Regulation of mitochondrial protein import by the nucleotide exchange factors GrpEL1 and GrpEL2 in human cells. J. Biol. Chem. 2017, 292, 18075–18090. [Google Scholar] [CrossRef] [PubMed]
- Konovalova, S.; Liu, X.; Manjunath, P.; Baral, S.; Neupane, N.; Hilander, T.; Yang, Y.; Balboa, D.; Terzioglu, M.; Euro, L.; et al. Redox regulation of GRPEL2 nucleotide exchange factor for mitochondrial HSP70 chaperone. Redox Biol. 2018, 19, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Naveen, V.; Chien, C.-H.; Chang, Y.-W.; Hsiao, C.-D. Crystal Structure of DnaK Protein Complexed with Nucleotide Exchange Factor GrpE in DnaK Chaperone System. J. Biol. Chem. 2012, 287, 21461–21470. [Google Scholar] [CrossRef]
- Grimshaw, J.P.; Jelesarov, I.; Schönfeld, H.-J.; Christen, P. Reversible Thermal Transition in GrpE, the Nucleotide Exchange Factor of the DnaK Heat-Shock System. J. Biol. Chem. 2001, 276, 6098–6104. [Google Scholar] [CrossRef]
- Melero, R.; Moro, F.; Pérez, F.M.; Perales-Calvo, J.; Quintana-Gallardo, L.; Llorca, O.; Muga, A.; Valpuesta, J.M. Modulation of the Chaperone DnaK Allosterism by the Nucleotide Exchange Factor GrpE. J. Biol. Chem. 2015, 290, 10083–10092. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Taneva, S.G.; Velazquez-Campoy, A.; Muga, A. GrpE N-terminal Domain Contributes to the Interaction with DnaK and Modulates the Dynamics of the Chaperone Substrate Binding Domain. J. Mol. Biol. 2007, 374, 1054–1064. [Google Scholar] [CrossRef]
- Gelinas, A.D.; Langsetmo, K.; Toth, J.; Bethoney, K.A.; Stafford, W.F.; Harrison, C.J. A Structure-based Interpretation of E. coli GrpE Thermodynamic Properties. J. Mol. Biol. 2002, 323, 131–142. [Google Scholar] [CrossRef]
- Schilling, B.; De-Medina, T.; Syken, J.; Vidal, M.; Munger, K. A Novel Human DnaJ Protein, hTid-1, a Homolog of the Drosophila Tumor Suppressor Protein Tid56, Can Interact with the Human Papillomavirus Type 16 E7 Oncoprotein. Virology 1998, 247, 74–85. [Google Scholar] [CrossRef]
- Wang, T.-H.; Lin, Y.-H.; Yang, S.-C.; Chang, P.-C.; Wang, T.-C.; Chen, C.-Y. Tid1-S regulates the mitochondrial localization of EGFR in non-small cell lung carcinoma. Oncogenesis 2017, 6, e361. [Google Scholar] [CrossRef]
- Iosefson, O.; Sharon, S.; Goloubinoff, P.; Azem, A. Reactivation of protein aggregates by mortalin and Tid1—The human mitochondrial Hsp70 chaperone system. Cell Stress Chaperon 2011, 17, 57–66. [Google Scholar] [CrossRef]
- Syken, J.; De-Medina, T.; Münger, K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc. Natl. Acad. Sci. USA 1999, 96, 8499–8504. [Google Scholar] [CrossRef] [PubMed]
- Cyr, D.M.; Langer, T.; Douglas, M.G. DnaJ-like proteins: Molecular chaperones and specific regulators of Hsp70. Trends Biochem. Sci. 1994, 19, 176–181. [Google Scholar] [CrossRef]
- Georgopoulos, C.; Welch, W.J. Role of the Major Heat Shock Proteins as Molecular Chaperones. Annu. Rev. Cell Biol. 1993, 9, 601–634. [Google Scholar] [CrossRef]
- Silver, P.A.; Way, J.C. Eukaryotic DnaJ homologs and the specificity of Hsp70 activity. Cell 1993, 74, 5–6. [Google Scholar] [CrossRef]
- Genevaux, P.; Schwager, F.; Georgopoulos, C.; Kelley, W.L. Scanning Mutagenesis Identifies Amino Acid Residues Essential for the in Vivo Activity of the Escherichia coli DnaJ (Hsp40) J-Domain. Genetics 2002, 162, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Genevaux, P.; Georgopoulos, C.P.; Kelley, W.L. The Hsp70 chaperone machines of Escherichia coli: A paradigm for the repartition of chaperone functions. Mol. Microbiol. 2007, 66, 840–857. [Google Scholar] [CrossRef]
- Szabo, A.; Korszun, R.; Hartl, F.U.; Flanagan, J. A zinc finger-like domain of the molecular chaperone DnaJ is involved in binding to denatured protein substrates. EMBO J. 1996, 15, 408–417. [Google Scholar] [CrossRef]
- Liu, T.; Daniels, C.K.; Cao, S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374. [Google Scholar] [CrossRef]
- Ng, A.C.-H.; Baird, S.D.; Screaton, R.A. Essential Role of TID1 in Maintaining Mitochondrial Membrane Potential Homogeneity and Mitochondrial DNA Integrity. Mol. Cell. Biol. 2014, 34, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Garrido, N.; Spelbrink, J.N.; Suzuki, C.K. Tid1 Isoforms Are Mitochondrial DnaJ-like Chaperones with Unique Carboxyl Termini That Determine Cytosolic Fate. J. Biol. Chem. 2006, 281, 13150–13158. [Google Scholar] [CrossRef] [PubMed]
- Tarunina, M.; Alger, L.; Chu, G.; Munger, K.; Gudkov, A.; Jat, P.S. Functional Genetic Screen for Genes Involved in Senescence: Role of Tid1, a Homologue of the Drosophila Tumor Suppressor l(2)tid, in Senescence and Cell Survival. Mol. Cell. Biol. 2004, 24, 10792–10801. [Google Scholar] [CrossRef]
- Elwi, A.N.; Lee, B.; Meijndert, H.C.; Braun, J.E.; Kim, S.-W. Mitochondrial chaperone DnaJA3 induces Drp1-dependent mitochondrial fragmentation. Int. J. Biochem. Cell Biol. 2012, 44, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.L.; Elwi, A.N.; Kim, S.-W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010, 1, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.Y.; Trinh, D.L.N.; Zajchowski, L.D.; Lee, B.; Elwi, A.N.; Kim, S.-W. Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 2009, 29, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.-F.; Hayashi, M.; Woo-Kim, S.; Tian, B.; Huang, J.-F.; Fearns, C.; Takayama, S.; Zapata, J.M.; Yang, Y.; Lee, J.-D. Tid1, a Cochaperone of the Heat Shock 70 Protein and the Mammalian Counterpart of the Drosophila Tumor Suppressor l(2)tid, Is Critical for Early Embryonic Development and Cell Survival. Mol. Cell. Biol. 2004, 24, 2226–2236. [Google Scholar] [CrossRef]
- Choi, J.H.; Choi, D.-K.; Sohn, K.-C.; Kwak, S.S.; Suk, J.; Lim, J.-S.; Shin, I.; Kim, S.-W.; Lee, J.-H.; Joe, C.O. Absence of a Human DnaJ Protein hTid-1S Correlates with Aberrant Actin Cytoskeleton Organization in Lesional Psoriatic Skin. J. Biol. Chem. 2012, 287, 25954–25963. [Google Scholar] [CrossRef]
- Cheng, L.-H.; Hung, K.-F.; Lee, T.-C.; Huang, C.-Y.; Chiu, W.-T.; Lo, J.-F.; Huang, T.-F. Mitochondrial co-chaperone protein Tid1 is required for energy homeostasis during skeletal myogenesis. Stem Cell Res. Ther. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Imanaka-Yoshida, K.; Yoshida, T.; Wood, M.; Fearns, C.; Tatake, R.J.; Lee, J.-D. A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat. Med. 2005, 12, 128–132. [Google Scholar] [CrossRef]
- Kim, S.J.; Kwon, M.-C.; Ryu, M.J.; Chung, H.K.; Tadi, S.; Kim, Y.K.; Kim, J.M.; Lee, S.H.; Park, J.H.; Kweon, G.R.; et al. CRIF1 Is Essential for the Synthesis and Insertion of Oxidative Phosphorylation Polypeptides in the Mammalian Mitochondrial Membrane. Cell Metab. 2012, 16, 274–283. [Google Scholar] [CrossRef]
- Lee, B.; Ahn, Y.; Kang, S.-M.; Park, Y.; Jeon, Y.-J.; Rho, J.M.; Kim, S.-W. Stoichiometric expression of mtHsp40 and mtHsp70 modulates mitochondrial morphology and cristae structure via Opa1L cleavage. Mol. Biol. Cell 2015, 26, 2156–2167. [Google Scholar] [CrossRef]
- Patra, M.; Weiss, C.; Abu-Libdeh, B.; Ashhab, M.; Abuzer, S.; Elpeleg, O.; Mahajnah, M.; Kessel, A.; Azem, A. A novel variant of the human mitochondrial DnaJ protein, Tid1, associates with a human disease exhibiting developmental delay and polyneuropathy. Eur. J. Hum. Genet. 2019, 27, 1072–1080. [Google Scholar] [CrossRef]
- Hernando, R.; Manso, R. Muscle Fibre Stress in Response to Exercise. Synthesis, Accumulation and Isoform Transitions of 70-kDa Heat-Shock Proteins. JBIC J. Biol. Inorg. Chem. 1997, 243, 460–467. [Google Scholar] [CrossRef] [PubMed]
- González, B.; Manso, R. Induction, modification and accumulation of HSP70s in the rat liver after acute exercise: Early and late responses. J. Physiol. 2004, 556, 369–385. [Google Scholar] [CrossRef]
- Wong, T.W.; Goldberg, A.R. Tyrosyl protein kinases in normal rat liver: Identification and partial characterization. Proc. Natl. Acad. Sci. USA 1983, 80, 2529–2533. [Google Scholar] [CrossRef]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell. Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Kotrasová, V.; Keresztesová, B.; Ondrovičová, G.; Bauer, J.; Havalová, H.; Pevala, V.; Kutejová, E.; Kunová, N. Mitochondrial Kinases and the Role of Mitochondrial Protein Phosphorylation in Health and Disease. Life 2021, 11, 82. [Google Scholar] [CrossRef]
- Doulias, P.-T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric Oxide Regulates Mitochondrial Fatty Acid Metabolism Through Reversible Protein S-Nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef]
- Hadari, Y.R.; Haring, H.U.; Zick, Y. p75, a Member of the Heat Shock Protein Family, Undergoes Tyrosine Phosphorylation in Response to Oxidative Stress. J. Biol. Chem. 1997, 272, 657–662. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Cheng, Z.; Zhu, J.; Xu, W.; Peng, X.; Chen, C.; Li, W.; Wang, F.; Cao, L.; Yi, X.; et al. Suberoylanilide Hydroxamic Acid Treatment Reveals Crosstalks among Proteome, Ubiquitylome and Acetylome in Non-Small Cell Lung Cancer A549 Cell Line. Sci. Rep. 2015, 5, 9520. [Google Scholar] [CrossRef]
- Kanai, M.; Ma, Z.; Izumi, H.; Kim, S.-H.; Mattison, C.P.; Winey, M.; Fukasawa, K. Physical and functional interaction between mortalin and Mps1 kinase. Genes Cells 2007, 12, 797–810. [Google Scholar] [CrossRef]
- Mattison, C.P.; Old, W.; Steiner, E.; Huneycutt, B.J.; Resing, K.A.; Ahn, N.G.; Winey, M. Mps1 Activation Loop Autophosphorylation Enhances Kinase Activity. J. Biol. Chem. 2007, 282, 30553–30561. [Google Scholar] [CrossRef]
- Mizukoshiab, E.; Suzuki, M.; Misonoa, T.; Loupatova, A.; Munekatab, E.; Kaul, S.; Wadhwac, R.; Imamura, T. Cell-Cycle Dependent Tyrosine Phosphorylation on Mortalin Regulates Its Interaction with Fibroblast Growth Factor-1. Biochem. Biophys. Res. Commun. 2001, 280, 1203–1209. [Google Scholar] [CrossRef]
- De Mena, L.; Coto, E.; Sánchez-Ferrero, E.; Ribacoba, R.; Guisasola, L.M.M.; Salvador, C.; Blázquez, M.; Álvarez, L.D.M. Mutational screening of the mortalin gene (HSPA9) in Parkinson’s disease. J. Neural Transm. 2009, 116, 1289–1293. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Castillo-Taucher, S.; Moreno-Salinas, R.; Cho, T.-J.; Chae, J.-H.; Choi, M.; Kim, O.-H.; Dikoglu, E.; Campos-Xavier, B.; Girardi, E.; et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci. Rep. 2015, 5, 17154. [Google Scholar] [CrossRef]
- Schmitz-Abe, K.; Ciesielski, S.; Schmidt, P.J.; Campagna, D.R.; Rahimov, F.; Schilke, B.A.; Cuijpers, M.; Rieneck, K.; Lausen, B.; Linenberger, M.L.; et al. Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9. Blood 2015, 126, 2734–2738. [Google Scholar] [CrossRef]
- Chen, Y.; Choong, L.-Y.; Lin, Q.; Philp, R.; Wong, C.-H.; Ang, B.-K.; Tan, Y.-L.; Loh, M.-C.; Hew, C.-L.; Shah, N.; et al. Differential Expression of Novel Tyrosine Kinase Substrates during Breast Cancer Development. Mol. Cell. Proteom. 2007, 6, 2072–2087. [Google Scholar] [CrossRef]
- Mertins, P.; Cptac, N.; Mani, D.R.; Ruggles, K.; Gillette, M.A.; Clauser, K.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nat. Cell Biol. 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y.; Tseng, V.S.-M.; Chen, L.-C.; Chang, Y.-C.; Ping, P.; Liao, C.-C.; Tsay, Y.-G.; Yu, J.-S.; Liao, P.-C. Combining Alkaline Phosphatase Treatment and Hybrid Linear Ion Trap/Orbitrap High Mass Accuracy Liquid Chromatography−Mass Spectrometry Data for the Efficient and Confident Identification of Protein Phosphorylation. Anal. Chem. 2009, 81, 7778–7787. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, D.K.; Rigas, J.R.; Gerber, S.A. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J. Proteom. 2013, 91, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Klammer, M.; Kaminski, M.; Zedler, A.; Oppermann, F.; Blencke, S.; Marx, S.; Müller, S.; Tebbe, A.; Godl, K.; Schaab, C. Phosphosignature Predicts Dasatinib Response in Non-small Cell Lung Cancer. Mol. Cell. Proteom. 2012, 11, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-F.; Wang, Y.-T.; Yen, H.-Y.; Tsou, C.-C.; Ku, W.-C.; Lin, P.-Y.; Chen, H.-Y.; Nesvizhskii, A.; Ishihama, Y.; Chen, Y.-J. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef]
- Casado, P.; Alcolea, M.P.; Iorio, F.; Rodríguez-Prados, J.-C.; Vanhaesebroeck, B.; Saez-Rodriguez, J.; Joel, S.; Cutillas, P.R. Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol. 2013, 14, R37. [Google Scholar] [CrossRef]
- Olsen, J.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.; et al. Quantitative Phosphoproteomics Reveals Widespread Full Phosphorylation Site Occupancy During Mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.; Ahn, N.G. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol. Cell. Proteom. 2015, 14, 1599–1615. [Google Scholar] [CrossRef]
- Weimann, M.; Grossmann, A.; Woodsmith, J.; Özkan, Z.; Birth, P.; Meierhofer, D.; Benlasfer, N.; Valovka, T.; Timmermann, B.; Wanker, E.; et al. A Y2H-seq approach defines the human protein methyltransferase interactome. Nat. Methods 2013, 10, 339–342. [Google Scholar] [CrossRef]
- Kimura, K.; Tanaka, N.; Nakamura, N.; Takano, S.; Ohkuma, S. Knockdown of Mitochondrial Heat Shock Protein 70 Promotes Progeria-like Phenotypes in Caenorhabditis elegans. J. Biol. Chem. 2007, 282, 5910–5918. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.C.; Yaguchi, T.; Taira, K.; Reddel, R.R.; Wadhwa, R. Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp. Cell Res. 2003, 286, 96–101. [Google Scholar] [CrossRef]
- Yokoyama, K.; Fukumoto, K.; Murakami, T.; Harada, S.-I.; Hosono, R.; Wadhwa, R.; Mitsui, Y.; Ohkuma, S. Extended longevity ofCaenorhabditis elegansby knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 2002, 516, 53–57. [Google Scholar] [CrossRef]
- Meriin, A.B.; Sherman, M.Y. Role of molecular chaperones in neurodegenerative disorders. Int. J. Hyperth. 2005, 21, 403–419. [Google Scholar] [CrossRef]
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive Oxygen Species: Stuck in the Middle of Neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S357–S367. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain Regional Correspondence Between Alzheimer’s Disease Histopathology and Biomarkers of Protein Oxidation. J. Neurochem. 2002, 65, 2146–2156. [Google Scholar] [CrossRef]
- Renkawek, K.; Bosman, G.I.C.G.M.; De Jong, W.W. Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol. 1994, 87, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-Q.; Wang, X.-L.; Cao, X.-H.; Ye, Z.; Li, L.; Cai, W.-Q. Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res. 2013, 1519, 105–111. [Google Scholar] [CrossRef]
- Osorio, C.; Sullivan, P.M.; He, N.N.; Mace, B.E.; Ervin, J.F.; Strittmatter, W.J.; Alzate, O. Mortalin is regulated by APOE in hippocampus of AD patients and by human APOE in TR mice. Neurobiol. Aging 2007, 28, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, L. A Comprehensive Review of Alzheimer’s Association with Related Proteins: Pathological Role and Therapeutic Significance. Curr. Neuropharmacol. 2020, 18, 674–695. [Google Scholar] [CrossRef]
- DeKroon, R.M.; Osorio, C.; Robinette, J.B.; Mocanu, M.; Winnik, W.M.; Alzate, O. Simultaneous Detection of Changes in Protein Expression and Oxidative Modification as a Function of Age andAPOEGenotype. J. Proteome Res. 2011, 10, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Hulette, C.; Wang, Y.; Zhang, T.; Pan, C.; Wadhwa, R.; Zhang, J. Proteomic Identification of a Stress Protein, Mortalin/mthsp70/GRP75. Mol. Cell. Proteom. 2006, 5, 1193–1204. [Google Scholar] [CrossRef]
- Shi, M.; Jin, J.; Wang, Y.; Beyer, R.P.; Kitsou, E.; Albin, R.L.; Gearing, M.; Pan, C.; Zhang, J. Mortalin: A Protein Associated With Progression of Parkinson Disease? J. Neuropathol. Exp. Neurol. 2008, 67, 117–124. [Google Scholar] [CrossRef]
- Goswami, A.V.; Samaddar, M.; Sinha, D.; Purushotham, J.; D’Silva, P. Enhanced J-protein interaction and compromised protein stability of mtHsp70 variants lead to mitochondrial dysfunction in Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3317–3332. [Google Scholar] [CrossRef]
- Takano, S.; Wadhwa, R.; Yoshii, Y.; Nose, T.; Kaul, S.C.; Mitsui, Y. Elevated Levels of Mortalin Expression in Human Brain Tumors. Exp. Cell Res. 1997, 237, 38–45. [Google Scholar] [CrossRef]
- Bini, L.; Magi, B.; Marzocchi, B.; Arcuri, F.; Tripodi, S.; Cintorino, M.; Sanchez, J.-C.; Frutiger, S.; Hughes, G.; Pallini, V.; et al. Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis 1997, 18, 2832–2841. [Google Scholar] [CrossRef]
- Cho, W.; Jin, X.; Pang, J.; Wang, Y.; Mivechi, N.F.; Moskophidis, D. The Molecular Chaperone Heat Shock Protein 70 Controls Liver Cancer Initiation and Progression by Regulating Adaptive DNA Damage and Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Signaling Pathways. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef]
- Starenki, D.; Sosonkina, N.; Hong, S.-K.; Lloyd, R.V.; Park, J.-I. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2069. [Google Scholar] [CrossRef]
- Dundas, S.R.; Lawrie, L.C.; Rooney, P.H.; I Murray, G. Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J. Pathol. 2005, 205, 74–81. [Google Scholar] [CrossRef]
- Cook, T.J.; Hoekstra, J.G.; Eaton, D.L.; Zhang, J. Mortalin is Expressed by Astrocytes and Decreased in the Midbrain of Parkinson’s Disease Patients. Brain Pathol. 2015, 26, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Fitzgerald, J.C.; Stegen, K.; Westermeier, J.; Thost, A.-K.; Kato, H.; Mokranjac, D.; Sauerwald, J.; Martins, L.M.; Woitalla, D.; et al. Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis. 2014, 5, e1180. [Google Scholar] [CrossRef]
- Ma, Z.; Izumi, H.; Kanai, M.; Kabuyama, Y.; Ahn, N.G.; Fukasawa, K. Mortalin controls centrosome duplication via modulating centrosomal localization of p53. Oncogene 2006, 25, 5377–5390. [Google Scholar] [CrossRef]
- Lu, W.-J.; Lee, N.P.; Kaul, S.; Lan, F.; Poon, R.T.P.; Wadhwa, R.; Luk, J.M. Mortalin–p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef]
- Rendón, O.Z.; Shoubridge, E.A. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Savel’Ev, A.S.; Novikova, L.A.; Kovaleva, I.E.; Luzikov, V.N.; Neupert, W.; Langer, T. ATP-dependent Proteolysis in Mitochondria. J. Biol. Chem. 1998, 273, 20596–20602. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a Putative Mitochondrial Iron Transporter Gene (ABC7) in X-Linked Sideroblastic Anemia and Ataxia (XLSA/A). Hum. Mol. Genet. 1999, 8, 743–749. [Google Scholar] [CrossRef]
- Liu, G.; Guo, S.; Anderson, G.; Camaschella, C.; Han, B.; Nie, G. Heterozygous missense mutations in the GLRX5 gene cause sideroblastic anemia in a Chinese patient. Blood 2014, 124, 2750–2751. [Google Scholar] [CrossRef] [PubMed]
- Crispin, A.; Guo, C.; Chen, C.; Campagna, D.R.; Schmidt, P.J.; Lichtenstein, D.A.; Cao, C.; Sendamarai, A.K.; Hildick-Smith, G.J.; Huston, N.C.; et al. Mutations in the iron-sulfur cluster biogenesis protein HSCB cause congenital sideroblastic anemia. J. Clin. Investig. 2020, 130, 5245–5256. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Havalová, H.; Ondrovičová, G.; Keresztesová, B.; Bauer, J.A.; Pevala, V.; Kutejová, E.; Kunová, N. Mitochondrial HSP70 Chaperone System—The Influence of Post-Translational Modifications and Involvement in Human Diseases. Int. J. Mol. Sci. 2021, 22, 8077. https://doi.org/10.3390/ijms22158077
Havalová H, Ondrovičová G, Keresztesová B, Bauer JA, Pevala V, Kutejová E, Kunová N. Mitochondrial HSP70 Chaperone System—The Influence of Post-Translational Modifications and Involvement in Human Diseases. International Journal of Molecular Sciences. 2021; 22(15):8077. https://doi.org/10.3390/ijms22158077
Chicago/Turabian StyleHavalová, Henrieta, Gabriela Ondrovičová, Barbora Keresztesová, Jacob A. Bauer, Vladimír Pevala, Eva Kutejová, and Nina Kunová. 2021. "Mitochondrial HSP70 Chaperone System—The Influence of Post-Translational Modifications and Involvement in Human Diseases" International Journal of Molecular Sciences 22, no. 15: 8077. https://doi.org/10.3390/ijms22158077
APA StyleHavalová, H., Ondrovičová, G., Keresztesová, B., Bauer, J. A., Pevala, V., Kutejová, E., & Kunová, N. (2021). Mitochondrial HSP70 Chaperone System—The Influence of Post-Translational Modifications and Involvement in Human Diseases. International Journal of Molecular Sciences, 22(15), 8077. https://doi.org/10.3390/ijms22158077