
Simulations of Promising Indolizidine—α6-β2 Nicotinic Acetylcholine Receptor Complexes

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Homology Model of 62 Nicotinic Acetylcholine Receptor
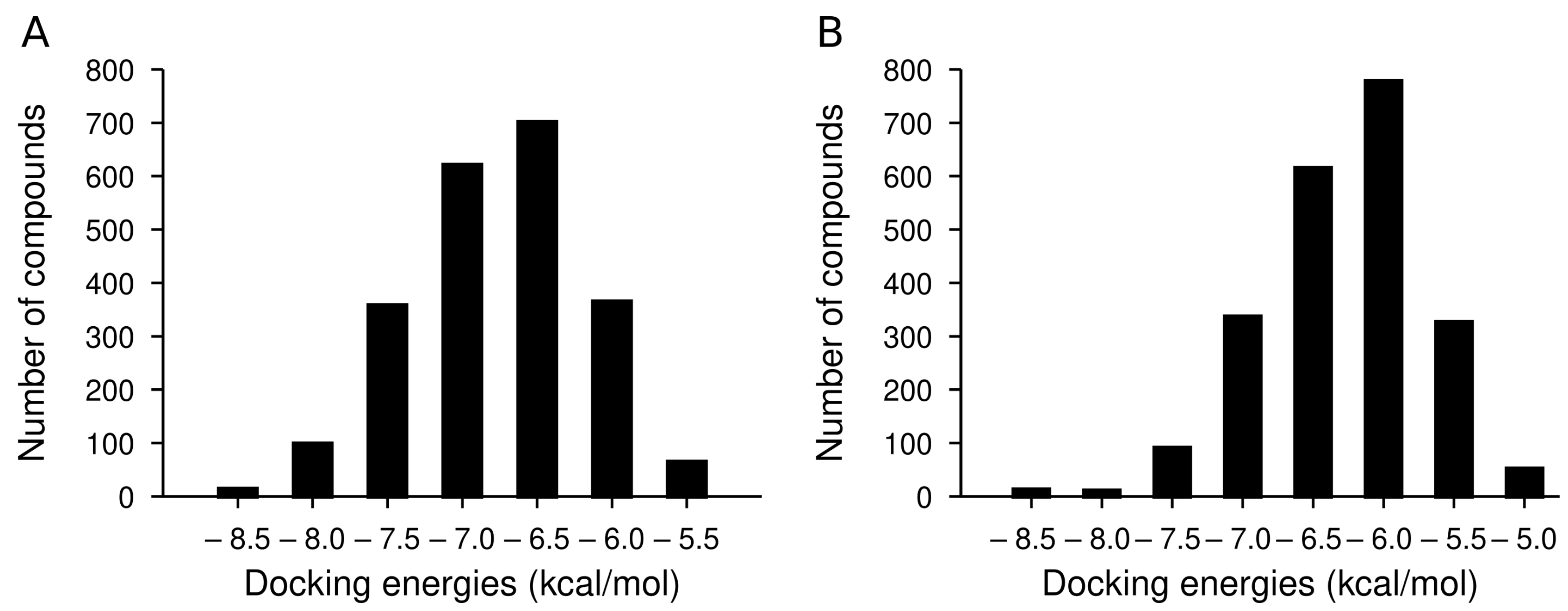
2.2. Library of Indolizidine Analogs
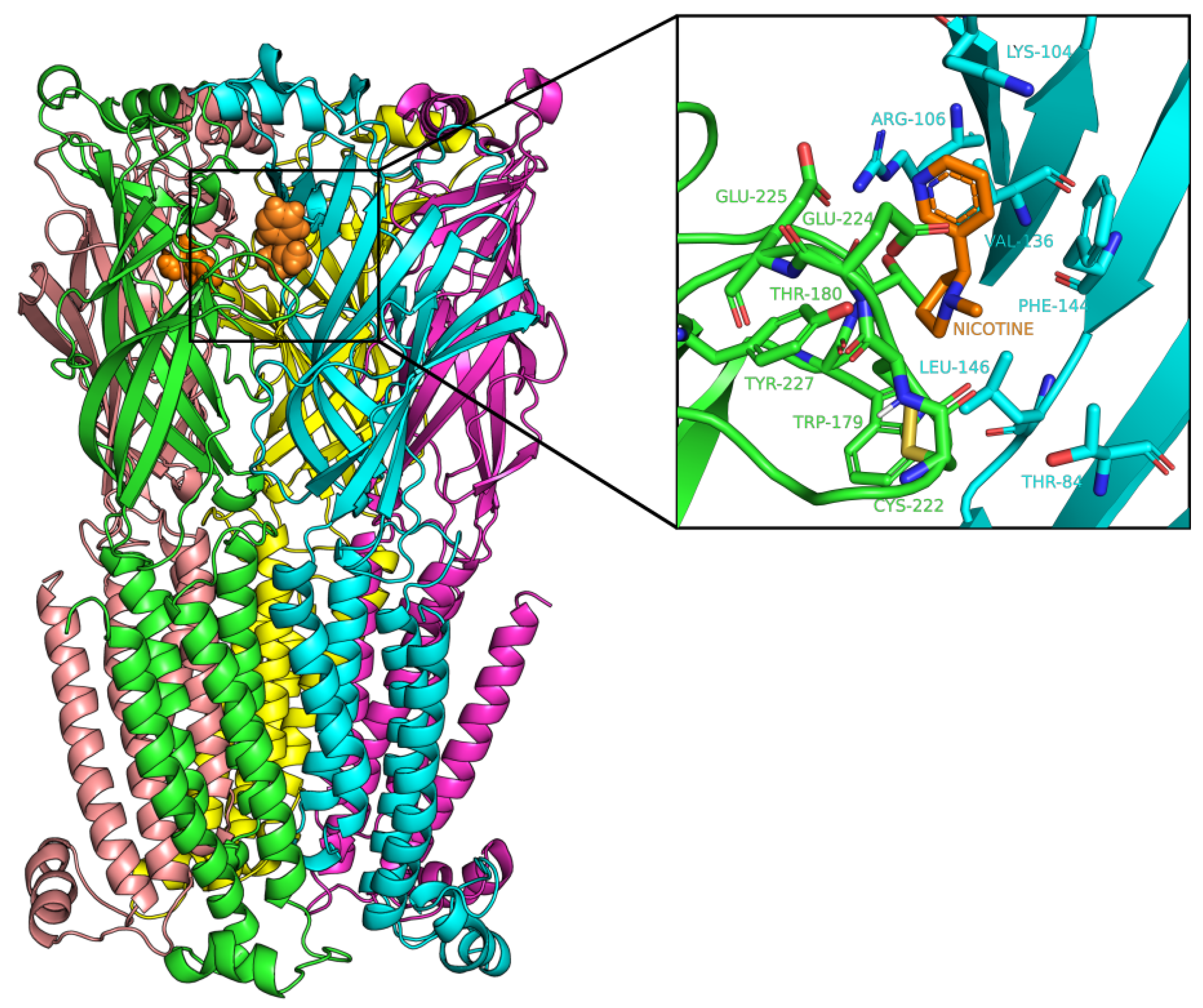
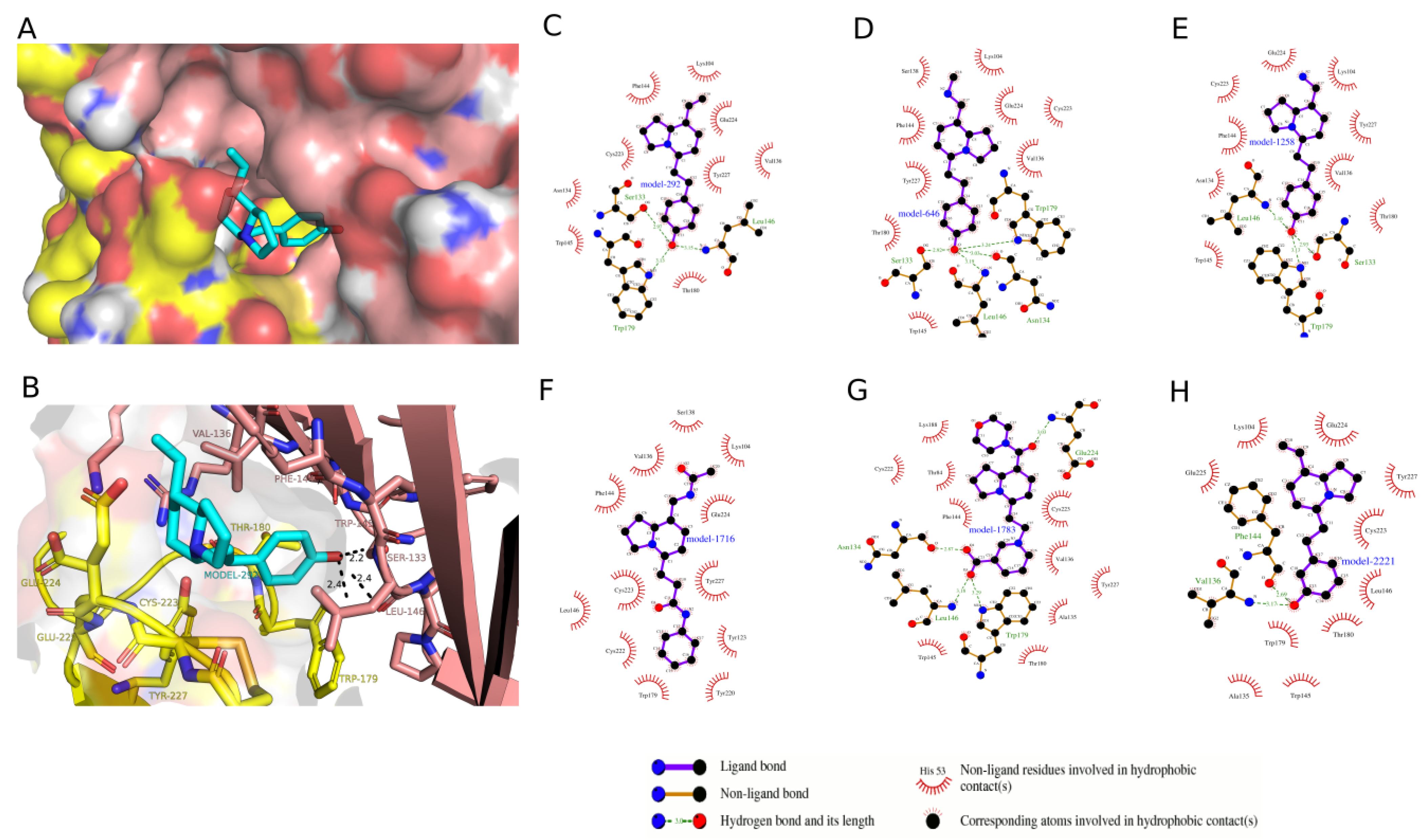
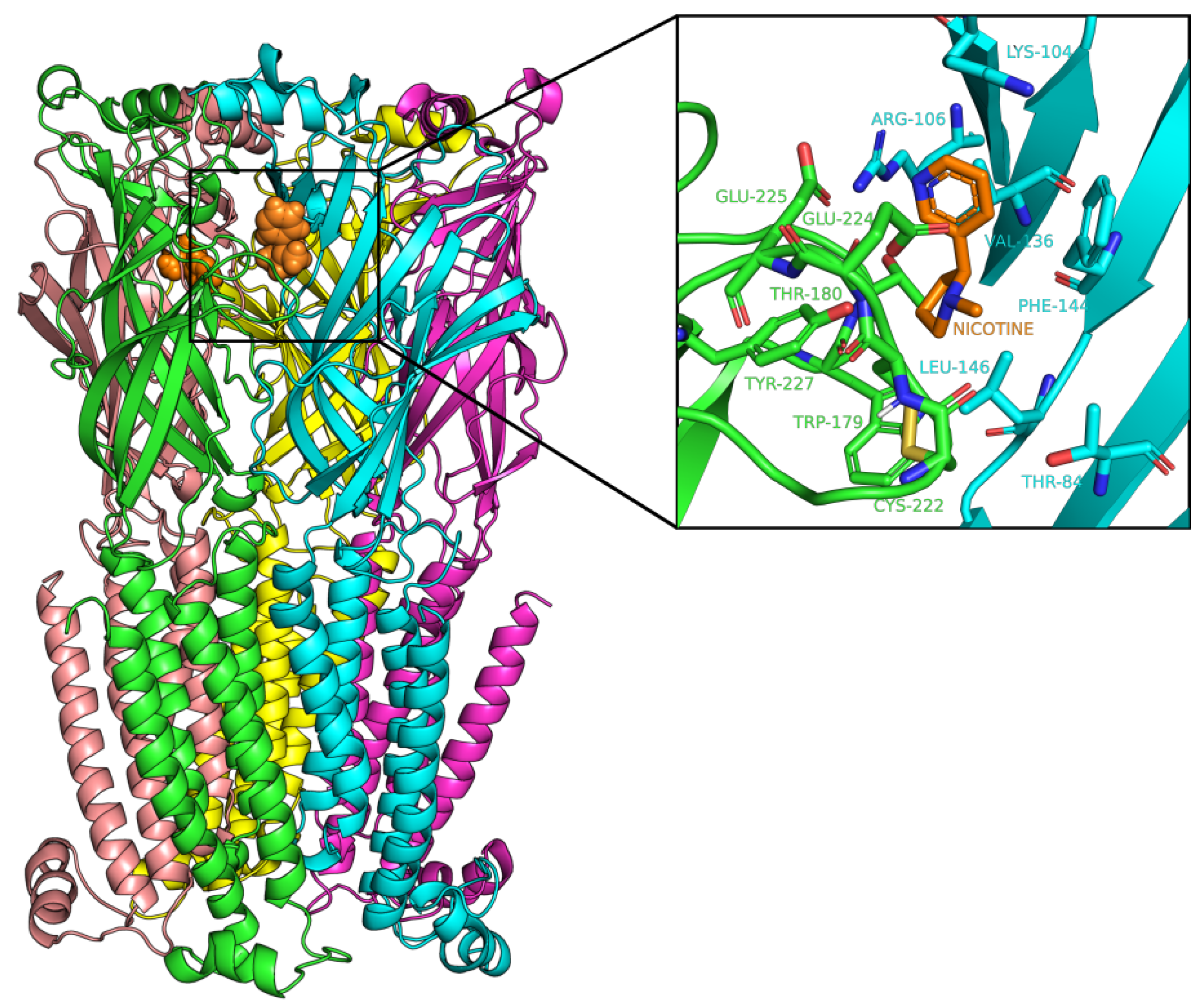
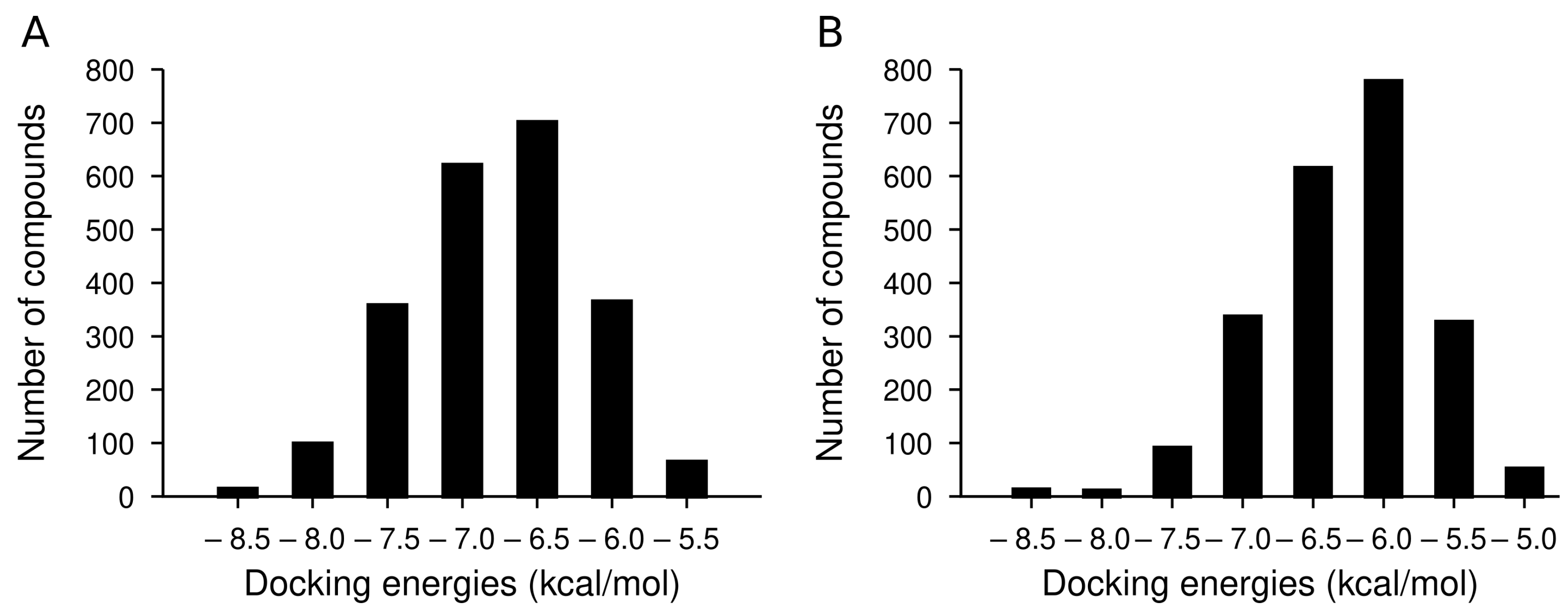
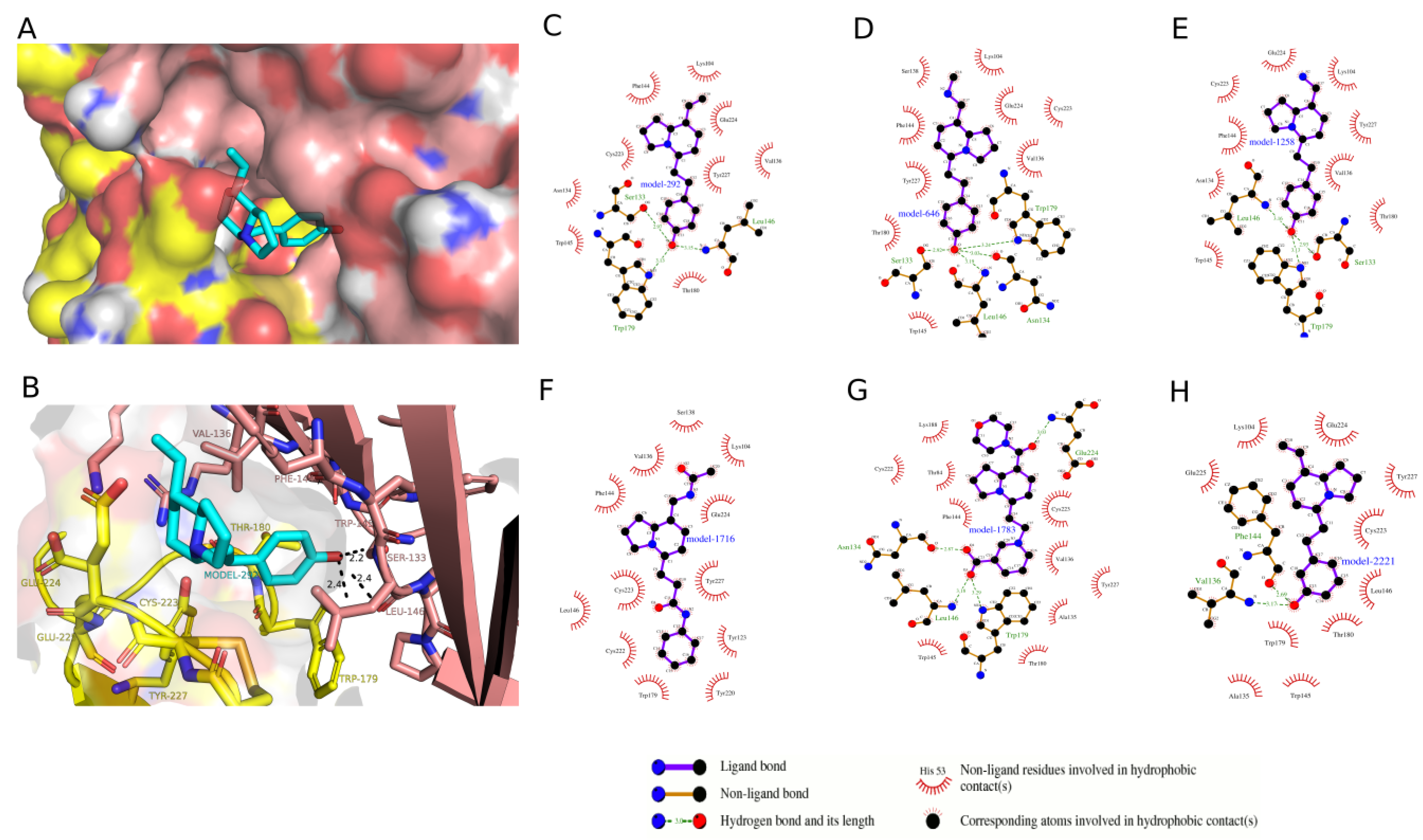
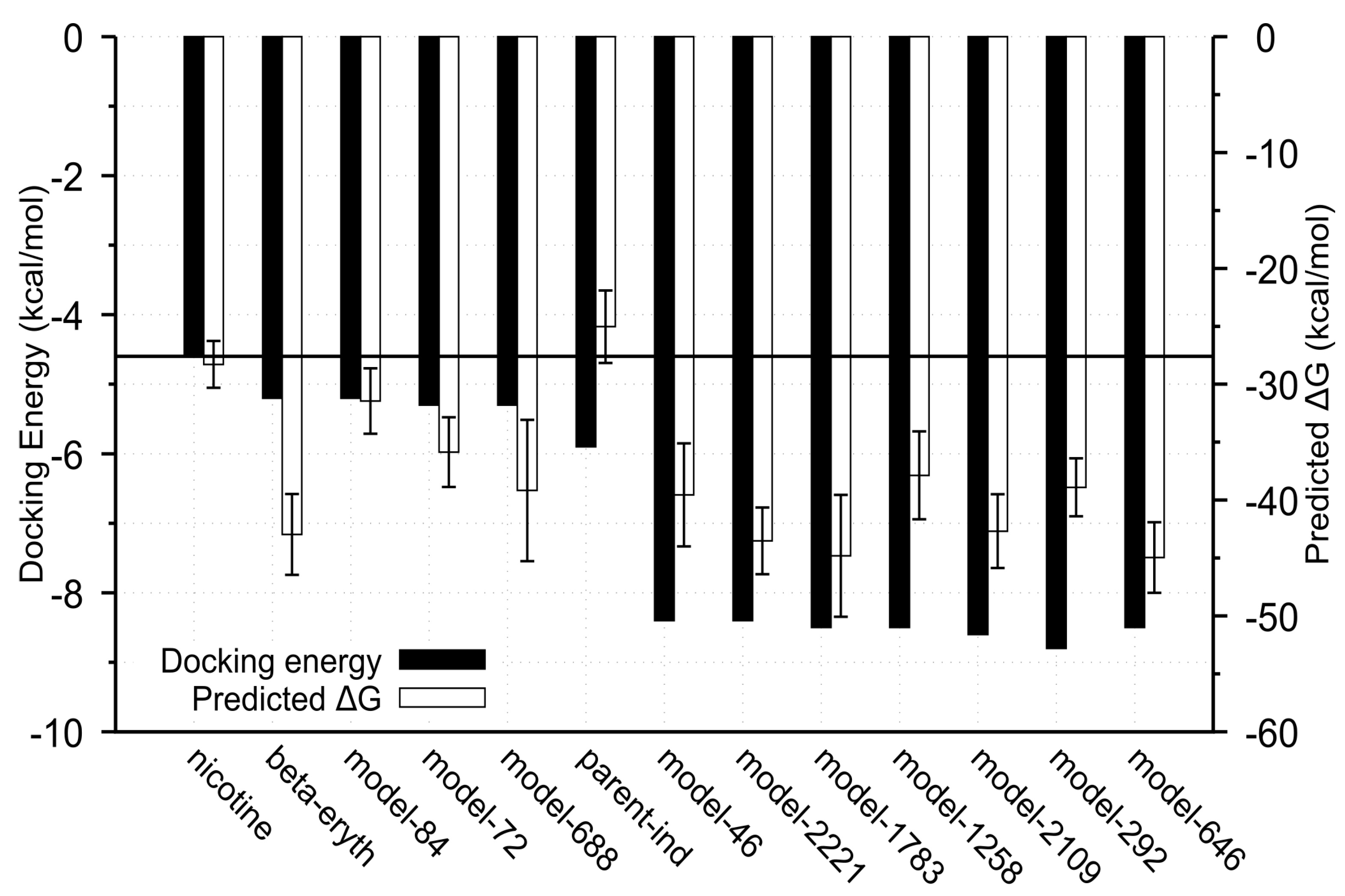
2.3. Docking Analysis of IND (-)-237D against 62 nAChR
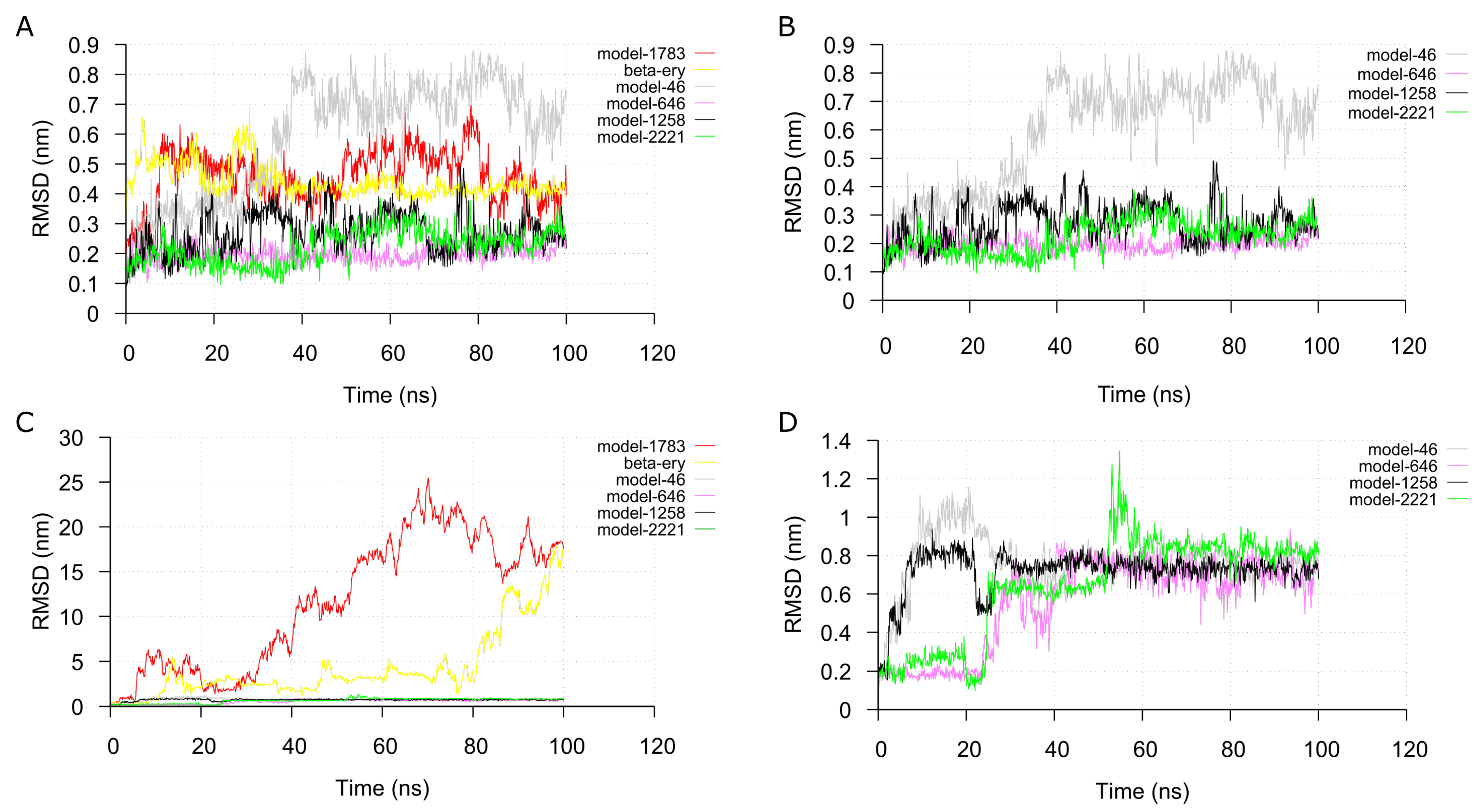
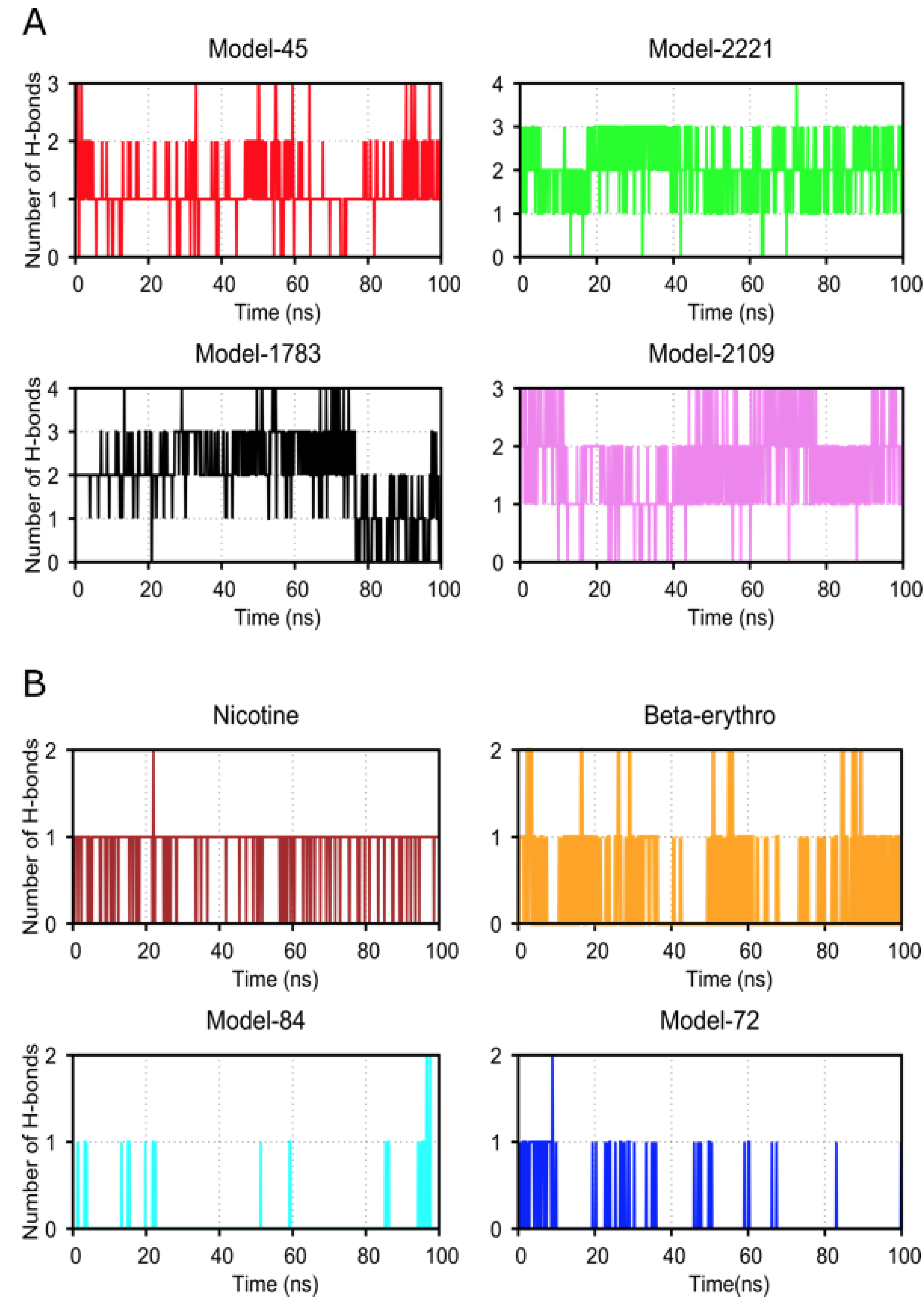
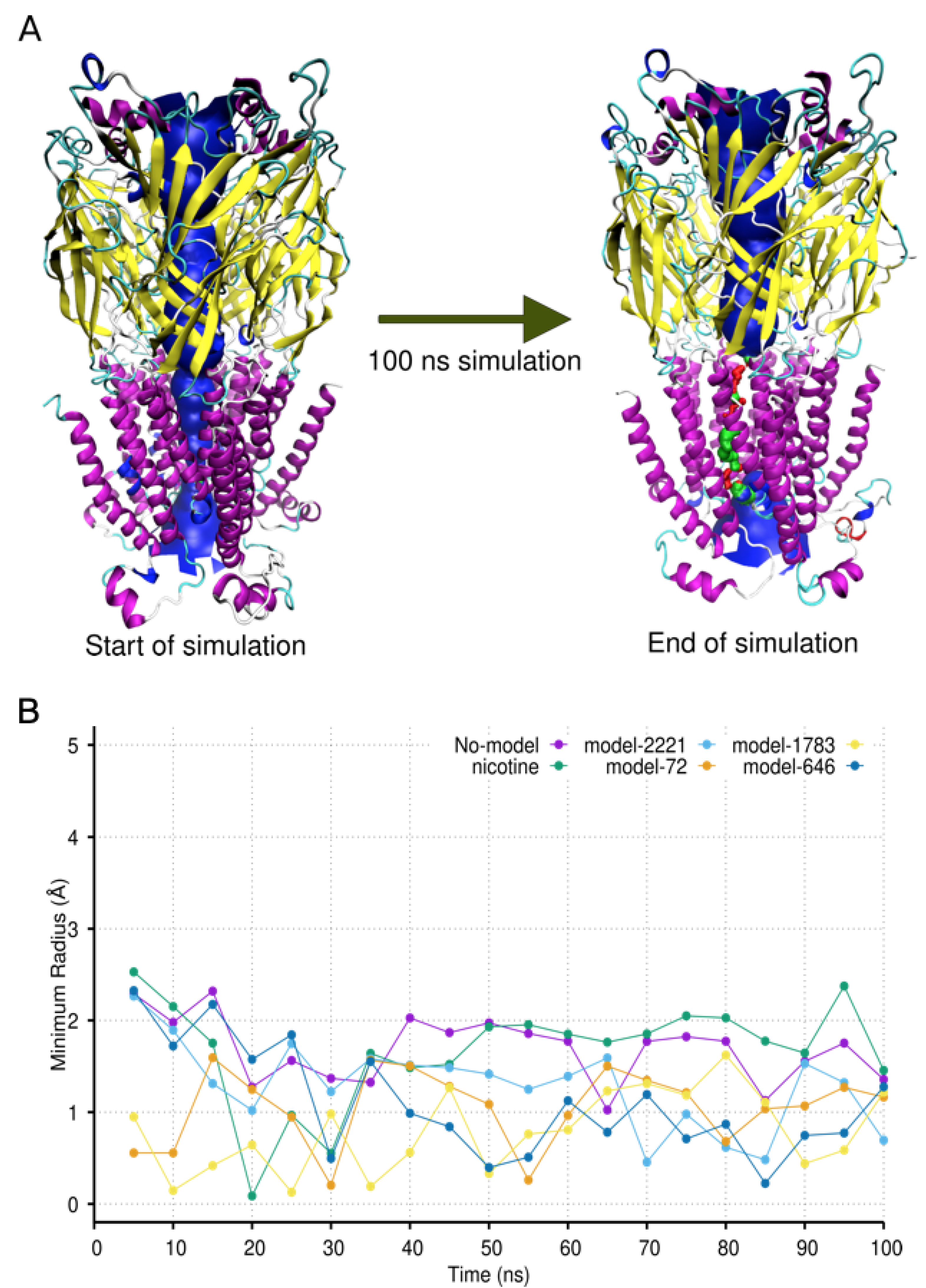
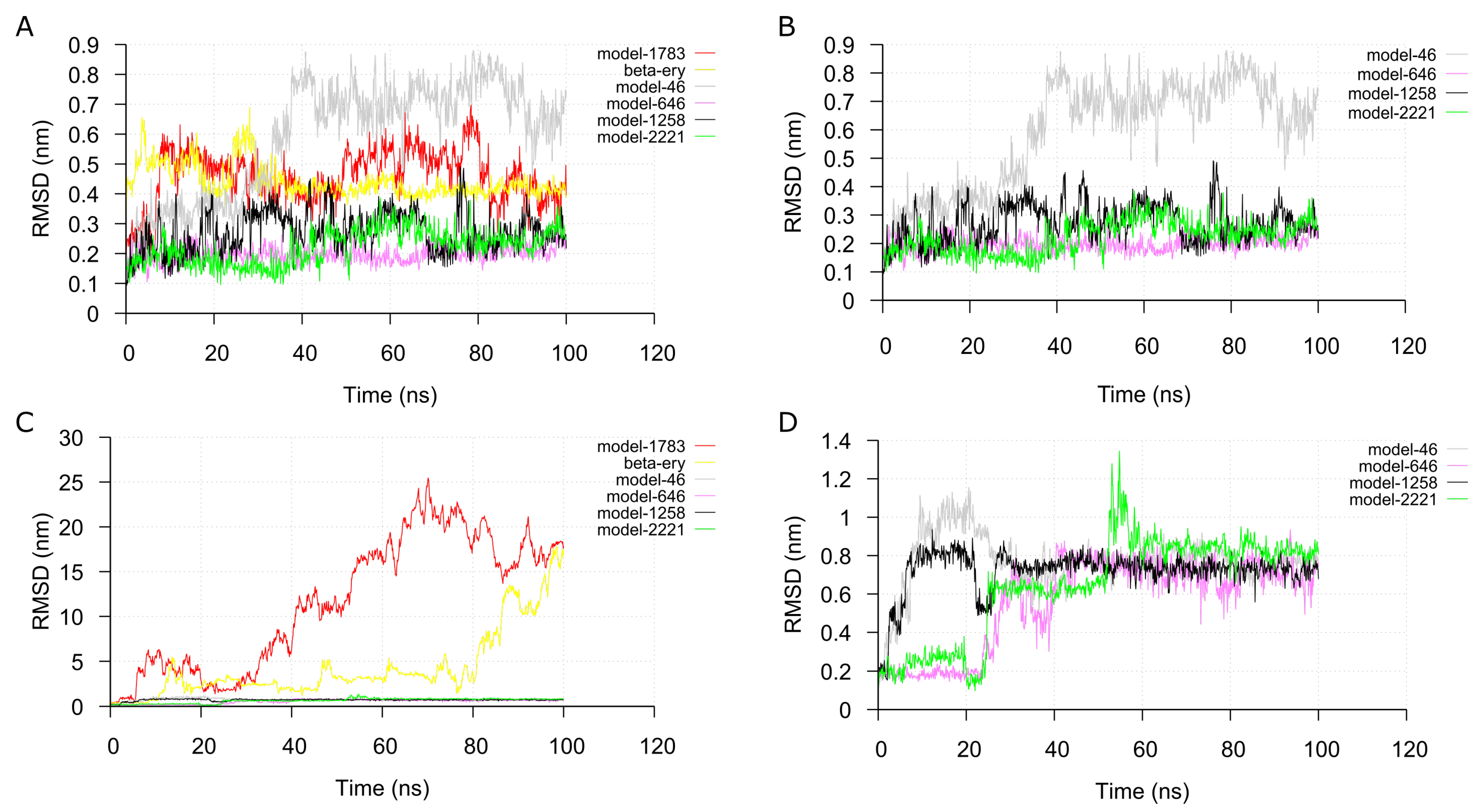
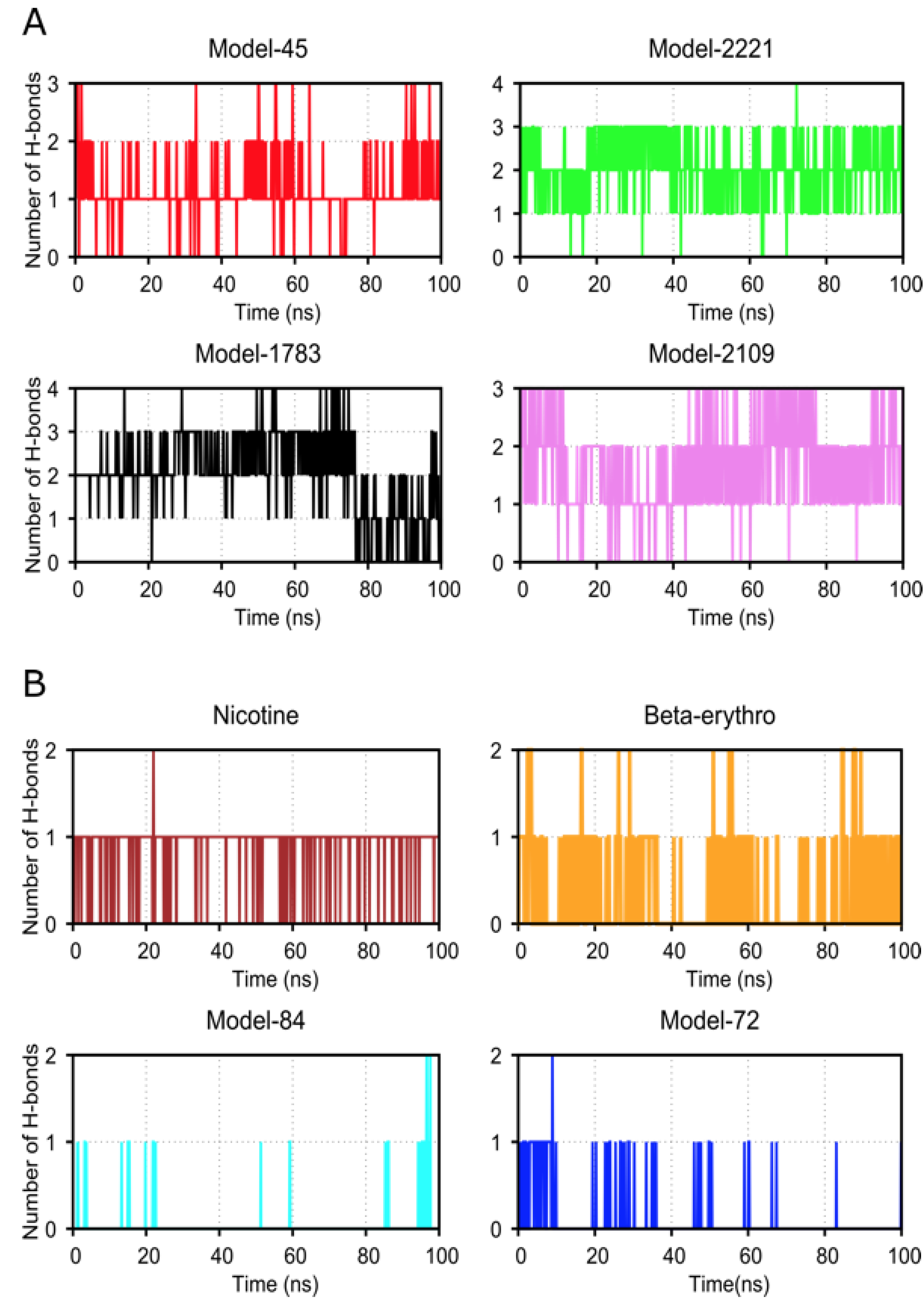
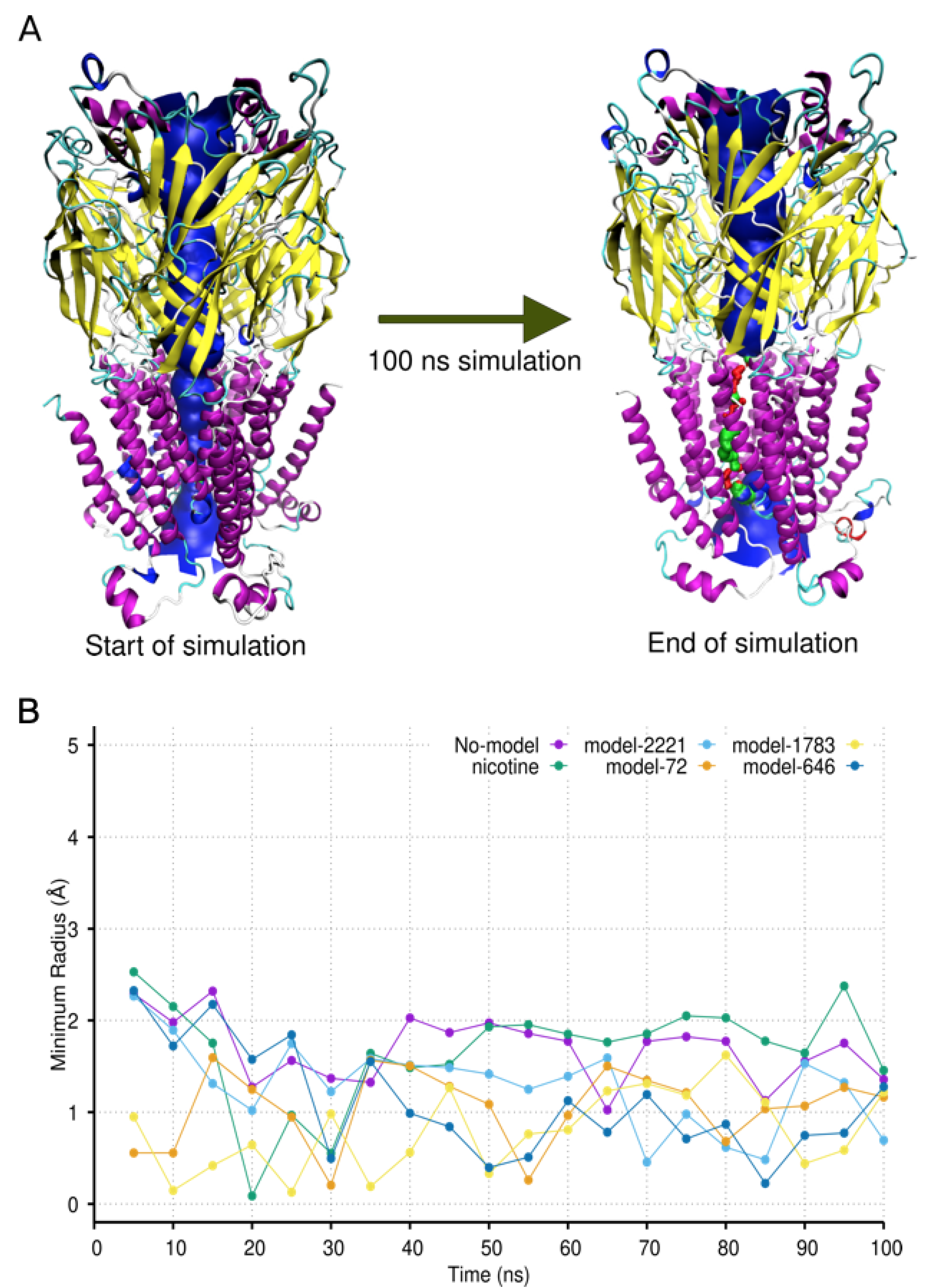
2.4. Molecular Dynamic Simulation and Analysis
3. Discussion
4. Materials and Methods
4.1. Homology Modeling of 62 Nicotinic Acetylcholine Receptor
4.2. Structure Assessment, Validation, Refinement of the 62 nAChR Homology Model
4.3. Docking of Indolizidne (-)-237D Derivatives
4.3.1. Modeling of Indolizidne (-)-237D Derivatives
4.3.2. Protein Preparation and Molecular Docking
4.4. Molecular Dynamic Simulation
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMET | absorption, distribution, metabolism, excretion, toxicity |
| ADT | AutoDock Tools |
| CNS MPO | Central Nervous System Multiparameter Optimization |
| COBRE | Center of Biomedical Research Excellence |
| GPU | Graphical Computing Unit |
| IND | indolizidines |
| MD | molecular dynamics |
| nAChRs | nicotinic acetylcholine receptors |
| OCAST | Oklahoma Center for the Advancement of Science and Technology |
| OSCER | Oklahoma Center for Supercomputing in Education and Research |
| PAINS | Pan-Assay Interference Compounds |
| PDB | Protein Data Bank |
| QMEAN | Qualitative Model Energy Analysis |
| RMSD | root mean square deviation |
| SDF | Structure Data File |
References
- WHO. WHO Report on the Global Tobacco Epidemic 2019: Offer Help to Quit Tobacco Use; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Benowitz, N.L. Nicotine addiction. N. Engl. J. Med. 2010, 362, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Devi, R.E.; Barman, D.; Sinha, S.; Hazarika, S.J.; Das, S. Nicotine replacement therapy: A friend or foe. J. Fam. Med. Prim. Care 2020, 9, 2615–2620. [Google Scholar] [CrossRef]
- Mersha, A.G.; Eftekhari, P.; Bovill, M.; Tollosa, D.N.; Gould, G.S. Evaluating level of adherence to nicotine replacement therapy and its impact on smoking cessation: A protocol for systematic review and meta-analysis. BMJ Open 2020, 10, e039775. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [Green Version]
- Vidal, C. Nicotinic receptors in the brain. Molecular biology, function, and therapeutics. Mol. Chem. Neuropathol. 1996, 28, 3–11. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Caldarone, B.J.; King, S.L.; Zachariou, V. Nicotinic receptors in the brain. Links between molecular biology and behavior. Neuropsychopharmacology 2000, 22, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Dani, J.A. Overview of nicotinic receptors and their roles in the central nervous system. Biol. Psychiatry 2001, 49, 166–174. [Google Scholar] [CrossRef]
- Dani, J.A. Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int. Rev. Neurobiol. 2015, 124, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Picciotto, M.R.; Kenny, P.J. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb. Perspect. Med. 2013, 3, a012112. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P. Nicotine addiction and nicotinic receptors: Lessons from genetically modified mice. Nat. Rev. Neurosci. 2010, 11, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Dwoskin, L.P.; Smith, A.M.; Wooters, T.E.; Zhang, Z.; Crooks, P.A.; Bardo, M.T. Nicotinic receptor-based therapeutics and candidates for smoking cessation. Biochem. Pharmacol. 2009, 78, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Gotti, C.; Guiducci, S.; Tedesco, V.; Corbioli, S.; Zanetti, L.; Moretti, M.; Zanardi, A.; Rimondini, R.; Mugnaini, M.; Clementi, F.; et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: Primary role of ventral tegmental area α6-β2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J. Neurosci. 2010, 30, 5311–5325. [Google Scholar] [CrossRef] [Green Version]
- Exley, R.; Clements, M.A.; Hartung, H.; McIntosh, J.M.; Cragg, S.J. α 6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology 2008, 33, 2158–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuneki, H.; You, Y.; Toyooka, N.; Kagawa, S.; Kobayashi, S.; Sasaoka, T.; Nemoto, H.; Kimura, I.; Dani, J.A. Alkaloids indolizidine 235B’, quinolizidine 1-epi-207I, and the tricyclic 205B are potent and selective noncompetitive inhibitors of nicotinic acetylcholine receptors. Mol. Pharmacol. 2004, 66, 1061–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pivavarchyk, M.; Smith, A.M.; Zhang, Z.; Zhou, D.; Wang, X.; Toyooka, N.; Tsuneki, H.; Sasaoka, T.; McIntosh, J.M.; Crooks, P.A.; et al. Indolizidine (-)-235B’ and related structural analogs: Discovery of nicotinic receptor antagonists that inhibit nicotine-evoked [3H]dopamine release. Eur. J. Pharmacol. 2011, 658, 132–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunzell, D.H. Preclinical evidence that activation of mesolimbic alpha 6 subunit containing nicotinic acetylcholine receptors supports nicotine addiction phenotype. Nicotine Tob. Res. 2012, 14, 1258–1269. [Google Scholar] [CrossRef] [Green Version]
- Quik, M.; Perez, X.A.; Grady, S.R. Role of α6 nicotinic receptors in CNS dopaminergic function: Relevance to addiction and neurological disorders. Biochem. Pharmacol. 2011, 82, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyooka, N.; Kobayashi, S.; Zhou, D.; Tsuneki, H.; Wada, T.; Sakai, H.; Nemoto, H.; Sasaoka, T.; Garraffo, H.M.; Spande, T.F.; et al. Synthesis of poison-frog alkaloids 233A, 235U, and 251AA and their inhibitory effects on neuronal nicotinic acetylcholine receptors. Bioorg. Med. Chem. Lett. 2007, 17, 5872–5875. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Miller III, B.R.; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Valdes-Tresanco, M.; Valdes-Tresanco, M.; Valiente, P.; Moreno, E. gmx_MMPBSA: A New Tool Aid to Perform End-State Free Energy Calculations with GROMACS Files. 2021. Available online: https://doi.org/10.5281/zenodo.4569307 (accessed on 11 June 2021).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lage, O.M.; Ramos, M.C.; Calisto, R.; Almeida, E.; Vasconcelos, V.; Vicente, F. Current screening methodologies in drug discovery for selected human diseases. Mar. Drugs 2018, 16, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Yu, R.; Tae, H.S.; Xu, Q.; Craik, D.J.; Adams, D.J.; Jiang, T.; Kaas, Q. Molecular dynamics simulations of dihydro-β-erythroidine bound to the human α4β2 nicotinic acetylcholine receptor. Br. J. Pharmacol. 2019, 176, 2750–2763. [Google Scholar] [CrossRef] [PubMed]
- Post, M.R.; Limapichat, W.; Lester, H.A.; Dougherty, D.A. Heterologous expression and nonsense suppression provide insights into agonist behavior at α6β2 nicotinic acetylcholine receptors. Neuropharmacology 2015, 97, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Wall, T.R.; Henderson, B.J.; Voren, G.; Wageman, C.R.; Deshpande, P.; Cohen, B.N.; Grady, S.R.; Marks, M.J.; Yohannes, D.; Kenny, P.J.; et al. TC299423, a novel agonist for nicotinic acetylcholine receptors. Front. Pharmacol. 2017, 8, 641. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human α4β2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Tosatto, S.C.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins: Struct. Funct. Bioinform. 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. 9), 114–122. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of Pan Assay Interference Compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Amani, P.; Sneyd, T.; Preston, S.; Young, N.D.; Mason, L.; Bailey, U.M.; Baell, J.; Camp, D.; Gasser, R.B.; Gorse, A.D.; et al. A practical Java tool for small-molecule compound appraisal. J. Cheminform. 2015, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, R.F.; Watson, I.A. Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond Rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, LLC; Delano, W. The PyMOL Molecular Graphics System, Version 2.5.1. Available online: http://www.pymol.org/pymol (accessed on 1 July 2021).
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Verlet, L. Computer “Experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Results (Initial Model) | Results (Refined Model) |
|---|---|---|
| Molprobity score | 2.49 | 1.29 |
| Clashscore | 5.69 | 0.16 |
| Ramachandran Favoured | 87.36% | 92.33% |
| Ramachandran Outliers | 3.04% | 1.23% |
| Docking Energy (kcal/mol) | ||||
|---|---|---|---|---|
| Analog # | R1 | R2 | 62 | 42 |
| Model-292 | =CH2 | 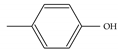 | −8.8 | −7.0 |
| Model-2109 | –CH3 | 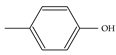 | −8.6 | −6.9 |
| Model-646 | 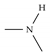 |  | −8.5 | −6.9 |
| Model-1258 | –NH2 |  | −8.5 | −6.8 |
| Model-1716 | 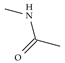 | 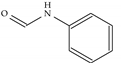 | −8.5 | −7.2 |
| Model-1783 | 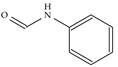 | 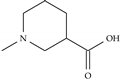 | −8.5 | −6.3 |
| Model-2221 | =CH2 | 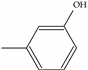 | −8.4 | −6.7 |
| Model-46 | =CH2 | 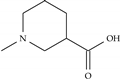 | −8.4 | −7.1 |
| Indolizidine (-)-237D | –H | –H | −5.9 | −5.3 |
| Nicotine | - | - | −4.6 | −5.4 |
| Dihydro-beta-erythroidine | - | - | −5.2 | −6.5 |
| Analog | Average Minimum Radius of Pore (Å) |
|---|---|
| No-model | 1.61 ± 0.3 |
| Nicotine | 1.88 ± 0.2 |
| Model-688 | 1.79 ± 0.2 |
| Parent-IND | 1.57 ± 0.2 |
| Model-2109 | 1.56 ± 0.5 |
| Model-46 | 1.3 ± 0.3 |
| Beta-ery | 1.30 ± 0.6 |
| Model-1258 | 1.26 ± 0.7 |
| Model-84 | 1.15 ± 0.3 |
| Model-45 | 1.25 ± 0.3 |
| Model-292 | 1.11 ± 0.2 |
| Model-2221 | 1.06 ± 0.4 |
| Model-72 | 1.05 ± 0.3 |
| Model-1783 | 0.96 ± 0.4 |
| Model-646 | 0.78 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acquah, F.A.; Paramel, M.; Kuta, A.; Hussaini, S.R.; Wallace, D.R.; Mooers, B.H.M. Simulations of Promising Indolizidine—α6-β2 Nicotinic Acetylcholine Receptor Complexes. Int. J. Mol. Sci. 2021, 22, 7934. https://doi.org/10.3390/ijms22157934
Acquah FA, Paramel M, Kuta A, Hussaini SR, Wallace DR, Mooers BHM. Simulations of Promising Indolizidine—α6-β2 Nicotinic Acetylcholine Receptor Complexes. International Journal of Molecular Sciences. 2021; 22(15):7934. https://doi.org/10.3390/ijms22157934
Chicago/Turabian StyleAcquah, Francis A., Matthew Paramel, Adama Kuta, Syed R. Hussaini, David R. Wallace, and Blaine H. M. Mooers. 2021. "Simulations of Promising Indolizidine—α6-β2 Nicotinic Acetylcholine Receptor Complexes" International Journal of Molecular Sciences 22, no. 15: 7934. https://doi.org/10.3390/ijms22157934
APA StyleAcquah, F. A., Paramel, M., Kuta, A., Hussaini, S. R., Wallace, D. R., & Mooers, B. H. M. (2021). Simulations of Promising Indolizidine—α6-β2 Nicotinic Acetylcholine Receptor Complexes. International Journal of Molecular Sciences, 22(15), 7934. https://doi.org/10.3390/ijms22157934