
A Fork Trap in the Chromosomal Termination Area Is Highly Conserved across All Escherichia coli Phylogenetic Groups




Abstract
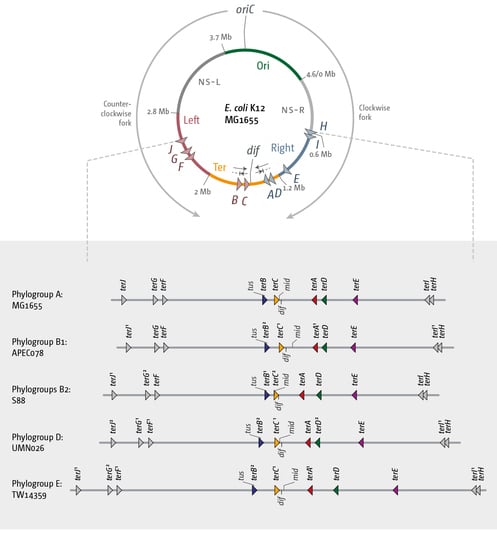
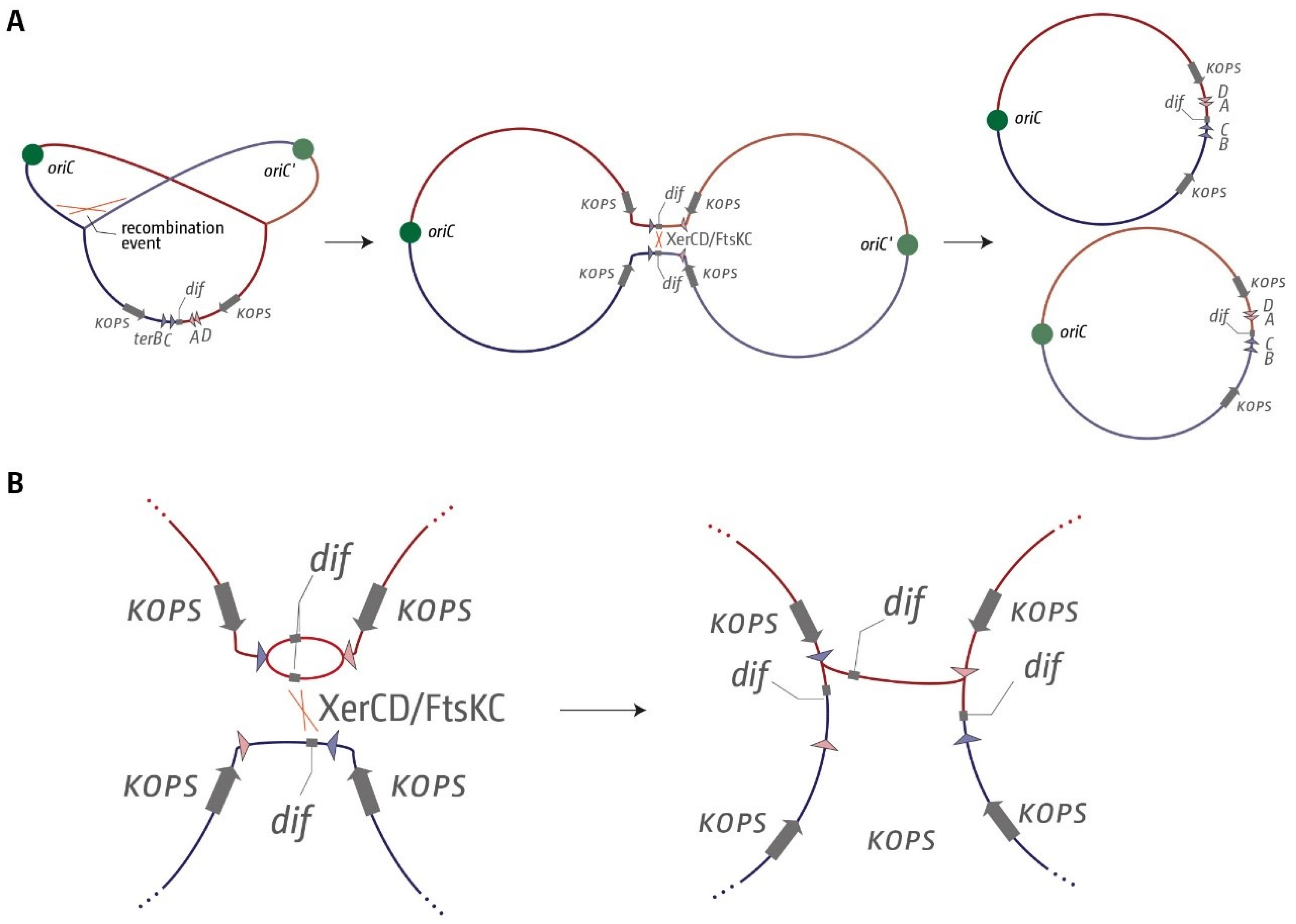
1. Introduction
2. Material and Methods
2.1. Strains, Media and General Methods
2.2. Growth Curves
2.3. Genome Sequences
2.4. Alignment of ter Sequences
2.5. Architecture of the Termination Area across Phylogroups
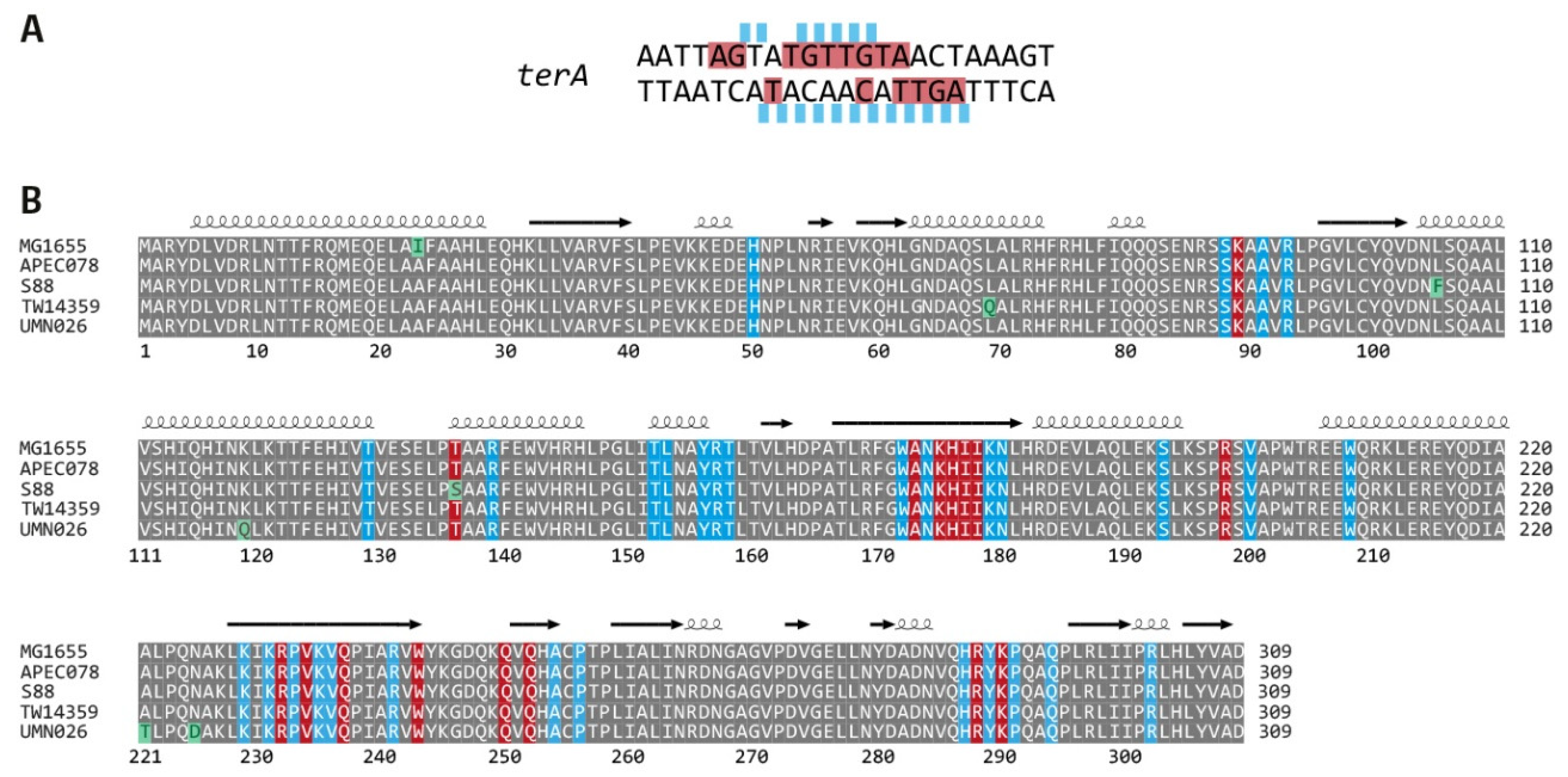
2.6. Sequence Alignment of Tus Proteins from All Phylogroups
2.7. Detection of ter Sites within Coding Sequences
2.8. Availability of Scripts Used in This Study
3. Results
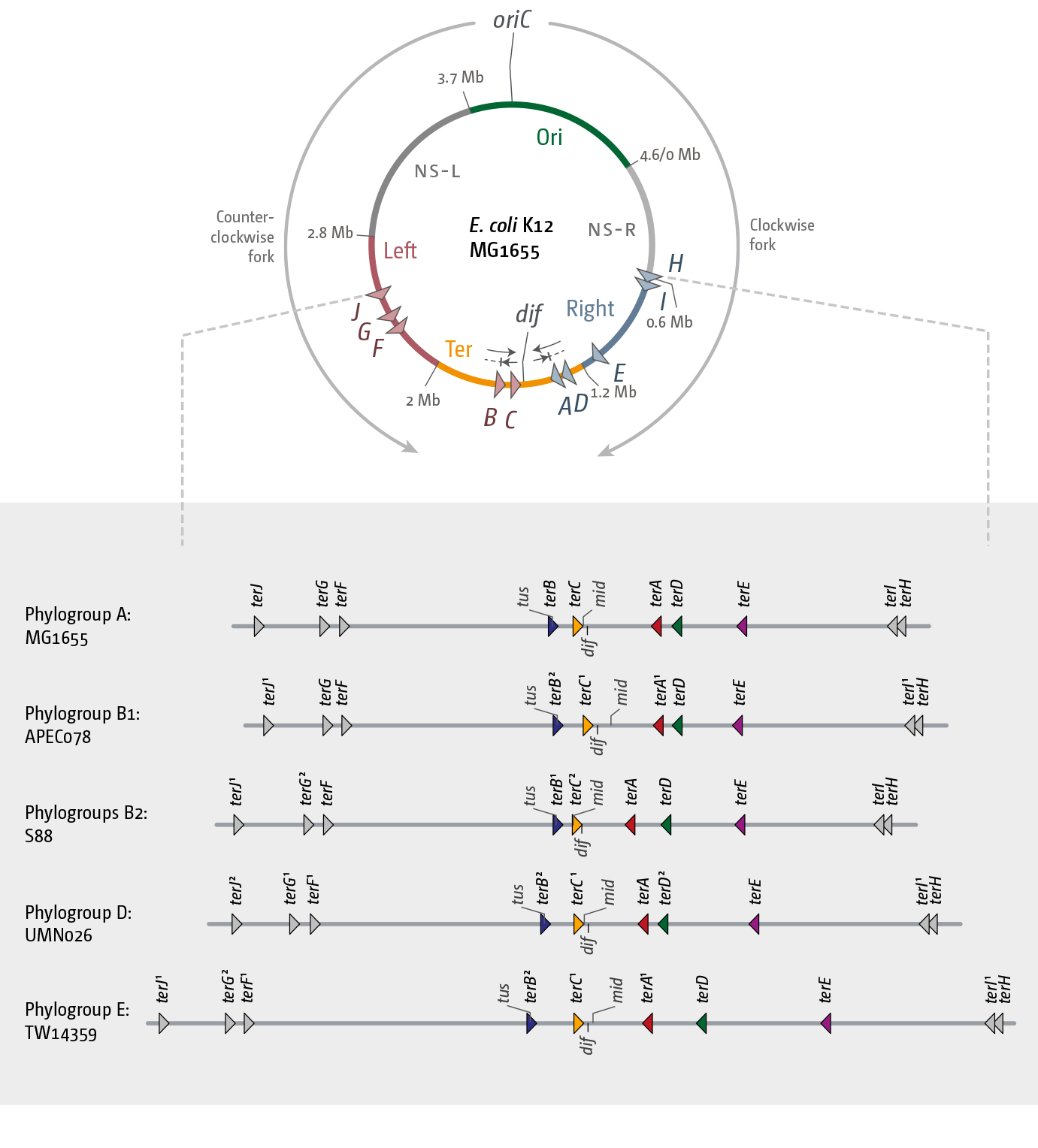
3.1. A Functional Replication Fork Trap Might Provide a Growth Advantage in E. coli
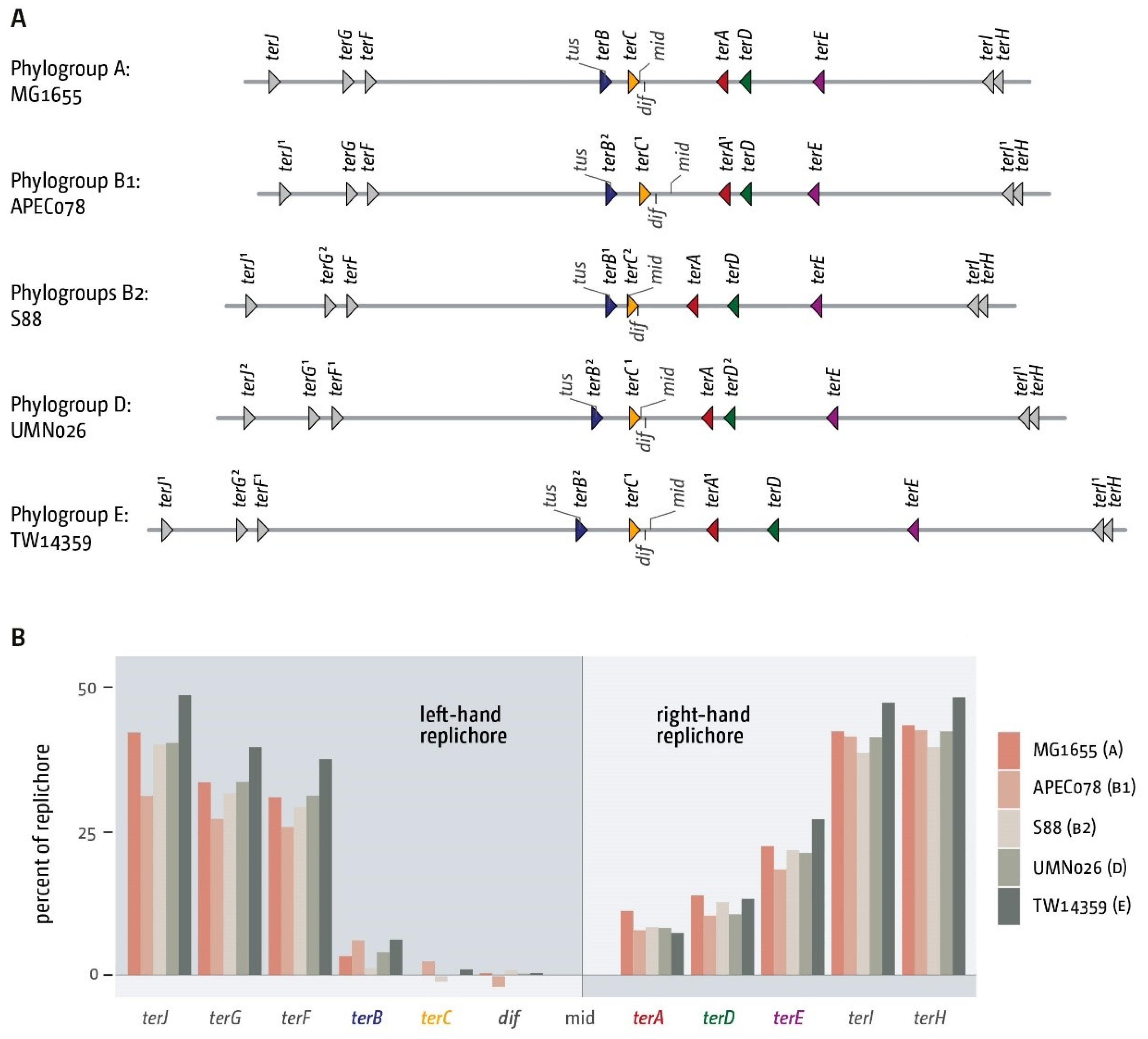
3.2. The Number and Distribution of ter Sites Is Similar in All E. coli Phylogroups
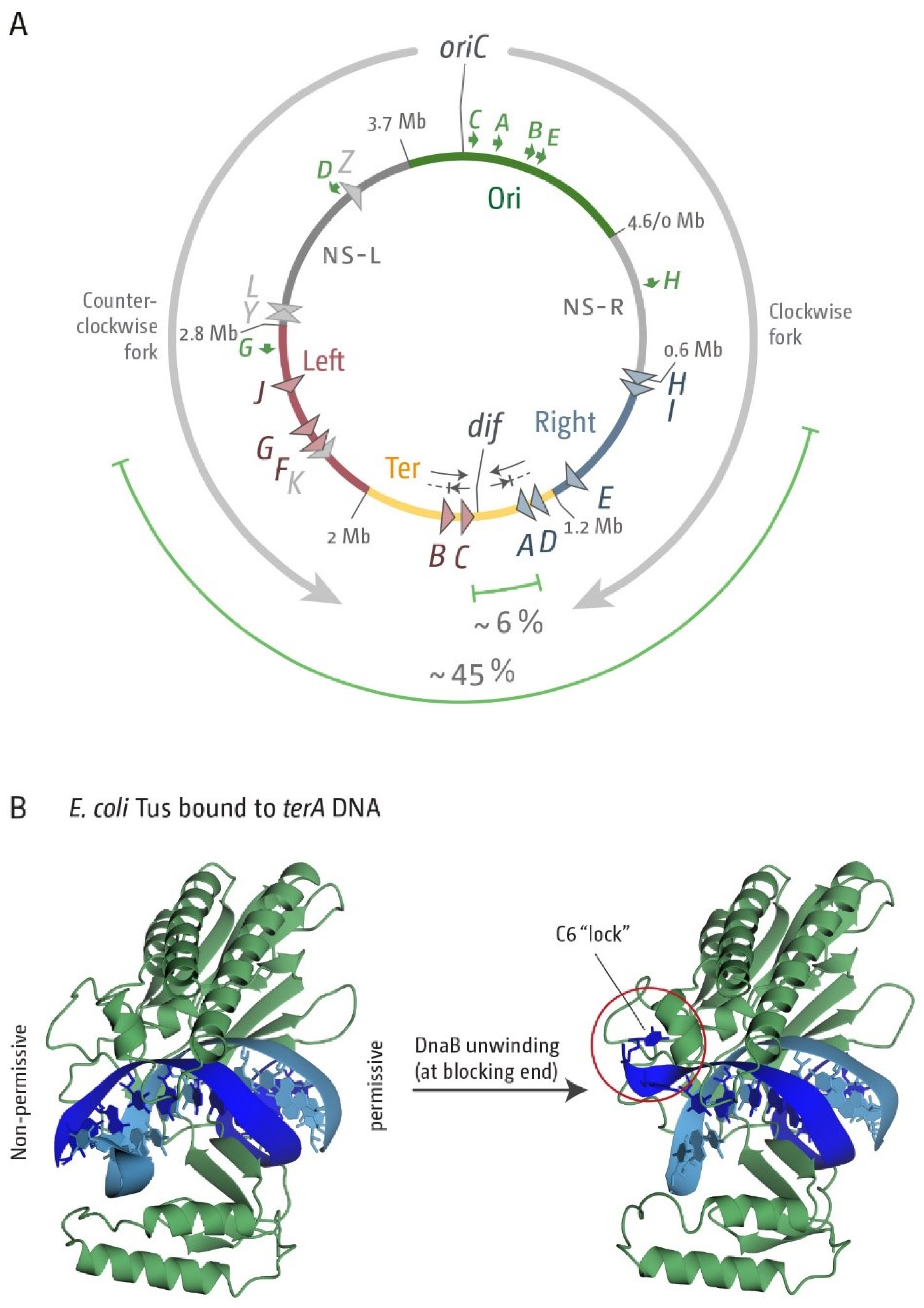
3.3. Functionality of the Fork Trap in the Phylogroups
3.4. Analysis of Secondary ter Sites
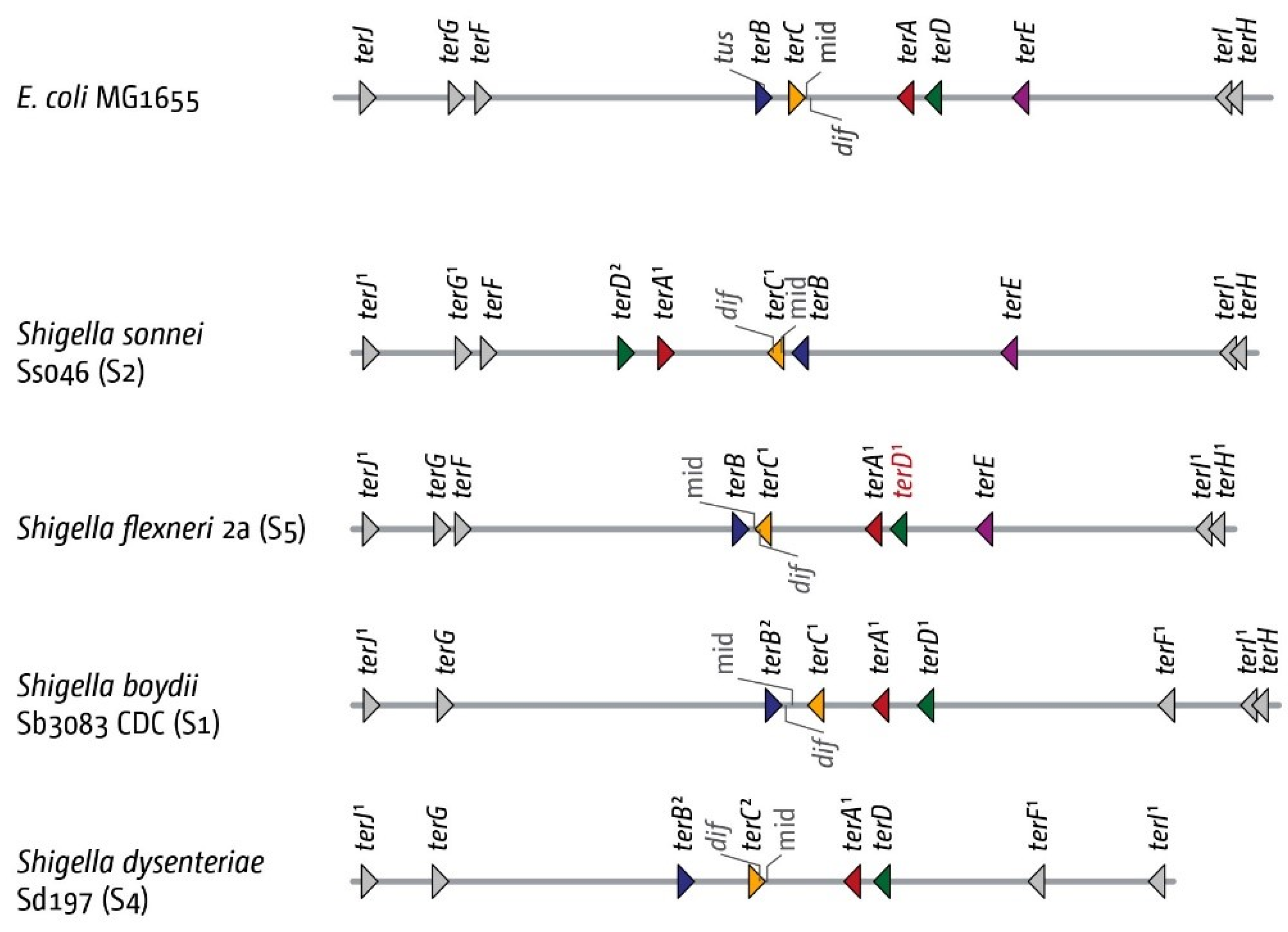
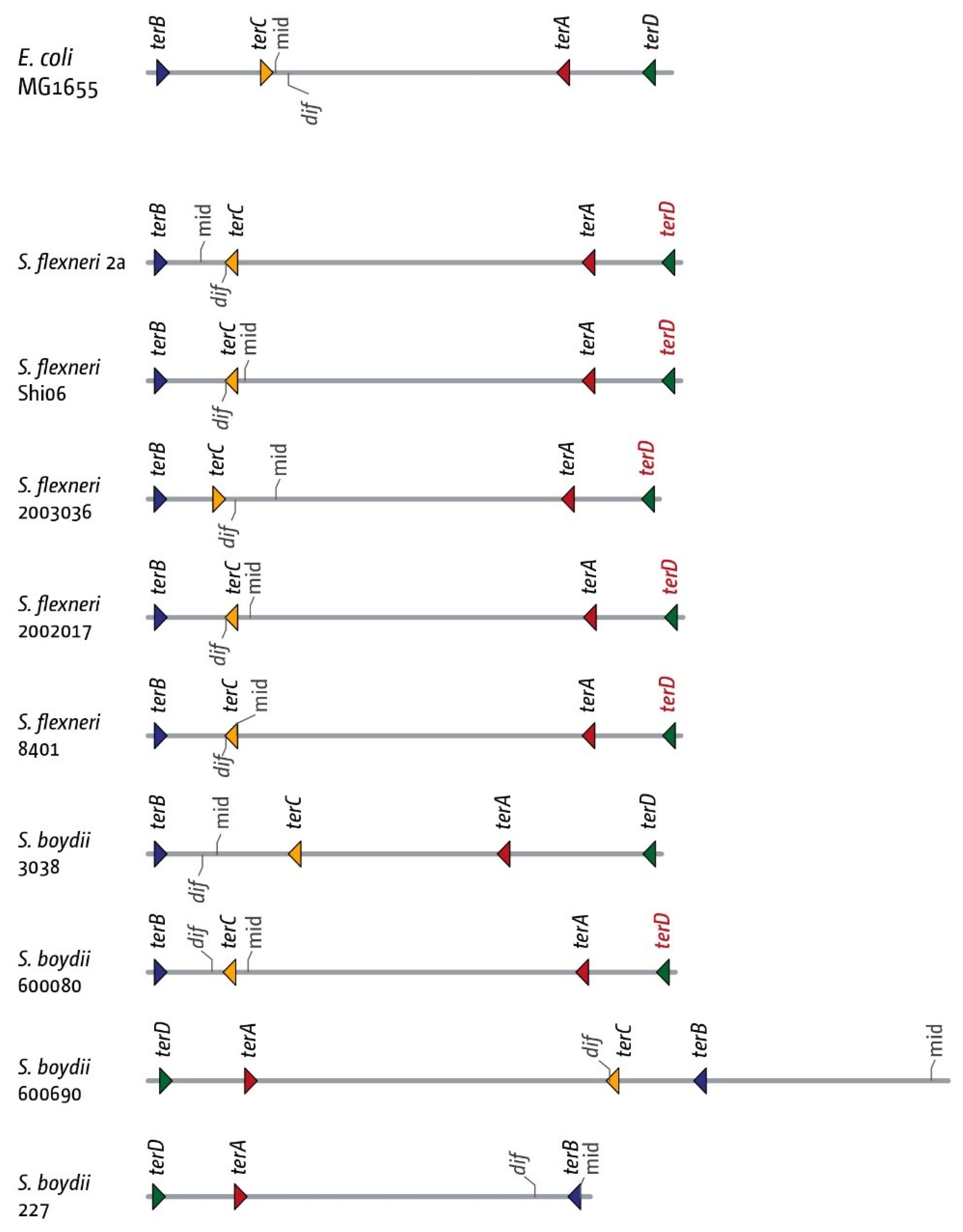
3.5. The Replication Fork Trap in Shigellae
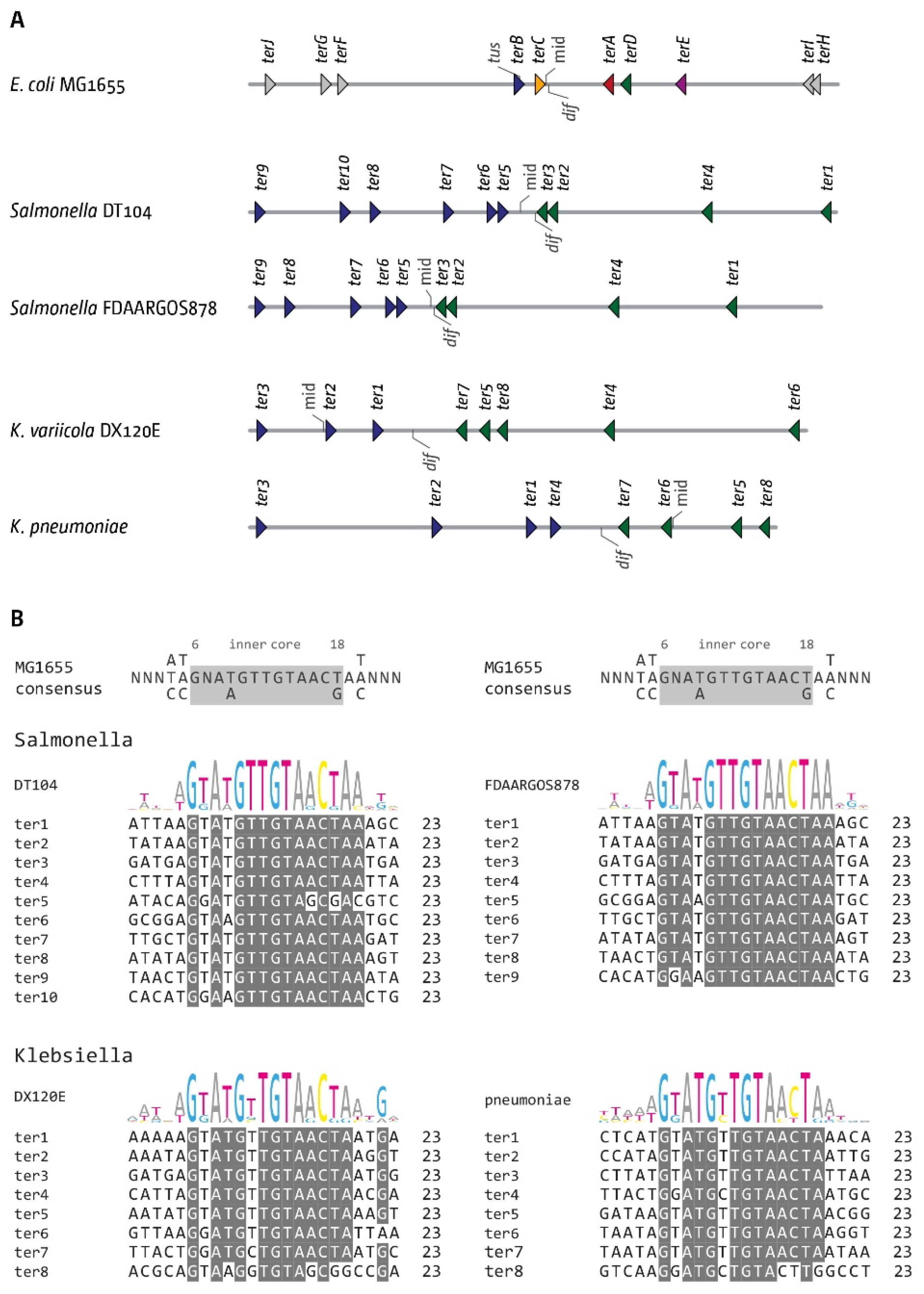
3.6. The Replication Fork Trap in Salmonella and Klebsiella
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mott, M.L.; Berger, J.M. DNA Replication Initiation: Mechanisms and Regulation in Bacteria. Nat. Rev. Microbiol. 2007, 5, 343–354. [Google Scholar] [CrossRef]
- Chodavarapu, S.; Kaguni, J.M. Replication Initiation in Bacteria. Enzymes 2016, 39, 1–30. [Google Scholar] [CrossRef]
- Syeda, A.H.; Dimude, J.U.; Skovgaard, O.; Rudolph, C.J. Too Much of a Good Thing: How Ectopic DNA Replication Affects Bacterial Replication Dynamics. Front. Microbiol. 2020, 11, 534. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Dimude, J.U.; Midgley-Smith, S.L.; Stein, M.; Rudolph, C.J. Replication Termination: Containing Fork Fusion-Mediated Pathologies in Escherichia coli. Genes 2016, 7, 40. [Google Scholar] [CrossRef]
- Liu, X.; Wang, X.; Reyes-Lamothe, R.; Sherratt, D. Replication-Directed Sister Chromosome Alignment in Escherichia coli. Mol. Microbiol. 2010, 75, 1090–1097. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duigou, S.; Boccard, F. Long Range Chromosome Organization in Escherichia coli: The Position of the Replication Origin Defines the Non-Structured Regions and the Right and Left Macrodomains. PLoS Genet. 2017, 13, e1006758. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Qian, Z.; Adhya, S.L. Architecture of the Escherichia coli Nucleoid. PLoS Genet. 2019, 15, e1008456. [Google Scholar] [CrossRef]
- Mercier, R.; Petit, M.-A.; Schbath, S.; Robin, S.; El Karoui, M.; Boccard, F.; Espéli, O. The MatP/MatS Site-Specific System Organizes the Terminus Region of the E. coli Chromosome into a Macrodomain. Cell 2008, 135, 475–485. [Google Scholar] [CrossRef] [PubMed]
- den Blaauwen, T. Prokaryotic Cell Division: Flexible and Diverse. Curr. Opin. Microbiol. 2013, 16, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Barre, F.-X. FtsK and SpoIIIE: The Tale of the Conserved Tails. Mol. Microbiol. 2007, 66, 1051–1055. [Google Scholar] [CrossRef]
- Sherratt, D.J.; Arciszewska, L.K.; Crozat, E.; Graham, J.E.; Grainge, I. The Escherichia coli DNA Translocase FtsK. Biochem. Soc. Trans. 2010, 38, 395–398. [Google Scholar] [CrossRef]
- Bigot, S.; Saleh, O.A.; Lesterlin, C.; Pages, C.; El Karoui, M.; Dennis, C.; Grigoriev, M.; Allemand, J.-F.; Barre, F.-X.; Cornet, F. KOPS: DNA Motifs That Control, E. coli Chromosome Segregation by Orienting the FtsK Translocase. EMBO J. 2005, 24, 3770–3780. [Google Scholar] [CrossRef]
- Levy, O.; Ptacin, J.L.; Pease, P.J.; Gore, J.; Eisen, M.B.; Bustamante, C.; Cozzarelli, N.R. Identification of Oligonucleotide Sequences That Direct the Movement of the Escherichia coli FtsK Translocase. Proc. Natl. Acad. Sci. USA 2005, 102, 17618–17623. [Google Scholar] [CrossRef]
- Duggin, I.G.; Bell, S.D. Termination Structures in the Escherichia coli Chromosome Replication Fork Trap. J. Mol. Biol. 2009, 387, 532–539. [Google Scholar] [CrossRef]
- Neylon, C.; Kralicek, A.V.; Hill, T.M.; Dixon, N.E. Replication Termination in Escherichia coli: Structure and Antihelicase Activity of the Tus-Ter Complex. Microbiol. Mol. Biol. Rev. MMBR 2005, 69, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Duggin, I.G.; Wake, R.G.; Bell, S.D.; Hill, T.M. The Replication Fork Trap and Termination of Chromosome Replication. Mol. Microbiol. 2008, 70, 1323–1333. [Google Scholar] [CrossRef]
- Bidnenko, V.; Ehrlich, S.D.; Michel, B. Replication Fork Collapse at Replication Terminator Sequences. EMBO J. 2002, 21, 3898–3907. [Google Scholar] [CrossRef] [PubMed]
- Moolman, M.C.; Tiruvadi Krishnan, S.; Kerssemakers, J.W.J.; de Leeuw, R.; Lorent, V.; Sherratt, D.J.; Dekker, N.H. The Progression of Replication Forks at Natural Replication Barriers in Live Bacteria. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.; Taylor, T.; Smith, S.L.; Dimude, J.U.; Upton, A.L.; Mehrjouy, M.M.; Skovgaard, O.; Sherratt, D.J.; Retkute, R.; Rudolph, C.J. Shaping the Landscape of the Escherichia coli Chromosome: Replication-Transcription Encounters in Cells with an Ectopic Replication Origin. Nucleic Acids Res. 2015, 43, 7865–7877. [Google Scholar] [CrossRef] [PubMed]
- Labib, K.; Hodgson, B. Replication Fork Barriers: Pausing for a Break or Stalling for Time? EMBO Rep. 2007, 8, 346–353. [Google Scholar] [CrossRef]
- Bouché, J.P.; Gélugne, J.P.; Louarn, J.; Louarn, J.M.; Kaiser, K. Relationships between the Physical and Genetic Maps of a 470 × 10(3) Base-Pair Region around the Terminus of Escherichia coli K12 DNA Replication. J. Mol. Biol. 1982, 154, 21–32. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Stockum, A.; Nieduszynski, C.A.; Lloyd, R.G. Avoiding Chromosome Pathology When Replication Forks Collide. Nature 2013, 500, 608–611. [Google Scholar] [CrossRef]
- Galli, E.; Ferat, J.-L.; Desfontaines, J.-M.; Val, M.-E.; Skovgaard, O.; Barre, F.-X.; Possoz, C. Replication Termination without a Replication Fork Trap. Sci. Rep. 2019, 9, 8315. [Google Scholar] [CrossRef] [PubMed]
- Roecklein, B.; Pelletier, A.; Kuempel, P. The tus Gene of Escherichia coli: Autoregulation, Analysis of Flanking Sequences and Identification of a Complementary System in Salmonella Typhimurium. Res. Microbiol. 1991, 142, 169–175. [Google Scholar] [CrossRef]
- Hill, T.M. Arrest of Bacterial DNA Replication. Annu. Rev. Microbiol. 1992, 46, 603–633. [Google Scholar] [CrossRef] [PubMed]
- Pupo, G.M.; Lan, R.; Reeves, P.R. Multiple Independent Origins of Shigella Clones of Escherichia coli and Convergent Evolution of Many of Their Characteristics. Proc. Natl. Acad. Sci. USA 2000, 97, 10567–10572. [Google Scholar] [CrossRef]
- Sahl, J.W.; Morris, C.R.; Emberger, J.; Fraser, C.M.; Ochieng, J.B.; Juma, J.; Fields, B.; Breiman, R.F.; Gilmour, M.; Nataro, J.P.; et al. Defining the Phylogenomics of Shigella Species: A Pathway to Diagnostics. J. Clin. Microbiol. 2015, 53, 951–960. [Google Scholar] [CrossRef]
- Thomason, L.C.; Costantino, N.; Court, D.L. E. coli Genome Manipulation by P1 Transduction. Curr. Protoc. Mol. Biol. 2007. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-Step Inactivation of Chromosomal Genes in Escherichia coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Luria, S.E.; Burrous, J.W. Hybridization between Escherichia coli and Shigella. J. Bacteriol. 1957, 74, 461–476. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinforma. Oxf. Engl. 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Markovitz, A. A New in Vivo Termination Function for DNA Polymerase I of Escherichia coli K12. Mol. Microbiol. 2005, 55, 1867–1882. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.J.; Upton, A.L.; Lloyd, R.G. Replication Fork Collisions Cause Pathological Chromosomal Amplification in Cells Lacking RecG DNA Translocase. Mol. Microbiol. 2009, 74, 940–955. [Google Scholar] [CrossRef]
- Wendel, B.M.; Courcelle, C.T.; Courcelle, J. Completion of DNA Replication in Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, 16454–16459. [Google Scholar] [CrossRef]
- Dimude, J.U.; Stockum, A.; Midgley-Smith, S.L.; Upton, A.L.; Foster, H.A.; Khan, A.; Saunders, N.J.; Retkute, R.; Rudolph, C.J. The Consequences of Replicating in the Wrong Orientation: Bacterial Chromosome Duplication without an Active Replication Origin. mBio 2015, 6. [Google Scholar] [CrossRef]
- Midgley-Smith, S.L.; Dimude, J.U.; Taylor, T.; Forrester, N.M.; Upton, A.L.; Lloyd, R.G.; Rudolph, C.J. Chromosomal Over-Replication in Escherichia coli recG Cells Is Triggered by Replication Fork Fusion and Amplified If Replichore Symmetry Is Disturbed. Nucleic Acids Res. 2018, 46, 7701–7715. [Google Scholar] [CrossRef]
- Dimude, J.U.; Stein, M.; Andrzejewska, E.E.; Khalifa, M.S.; Gajdosova, A.; Retkute, R.; Skovgaard, O.; Rudolph, C.J. Origins Left, Right, and Centre: Increasing the Number of Initiation Sites in the Escherichia coli Chromosome. Genes 2018, 9, 376. [Google Scholar] [CrossRef]
- Esnault, E.; Valens, M.; Espéli, O.; Boccard, F. Chromosome Structuring Limits Genome Plasticity in Escherichia coli. PLoS Genet. 2007, 3, e226. [Google Scholar] [CrossRef] [PubMed]
- De Castro Stoppe, N.; Silva, J.S.; Carlos, C.; Sato, M.I.Z.; Saraiva, A.M.; Ottoboni, L.M.M.; Torres, T.T. Worldwide Phylogenetic Group Patterns of Escherichia coli from Commensal Human and Wastewater Treatment Plant Isolates. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.A.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The Pangenome Structure of Escherichia coli: Comparative Genomic Analysis of E. coli Commensal and Pathogenic Isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef]
- Abram, K.; Udaondo, Z.; Bleker, C.; Wanchai, V.; Wassenaar, T.M.; Robeson, M.S.; Ussery, D.W. Mash-Based Analyses of Escherichia coli Genomes Reveal 14 Distinct Phylogroups. Commun. Biol. 2021, 4, 117. [Google Scholar] [CrossRef]
- Herzer, P.J.; Inouye, S.; Inouye, M.; Whittam, T.S. Phylogenetic Distribution of Branched RNA-Linked Multicopy Single-Stranded DNA among Natural Isolates of Escherichia coli. J. Bacteriol. 1990, 172, 6175–6181. [Google Scholar] [CrossRef]
- Clermont, O.; Bonacorsi, S.; Bingen, E. Rapid and Simple Determination of the Escherichia coli Phylogenetic Group. Appl. Environ. Microbiol. 2000, 66, 4555–4558. [Google Scholar] [CrossRef]
- Picard, B.; Garcia, J.S.; Gouriou, S.; Duriez, P.; Brahimi, N.; Bingen, E.; Elion, J.; Denamur, E. The Link between Phylogeny and Virulence in Escherichia coli Extraintestinal Infection. Infect. Immun. 1999, 67, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Sims, G.E.; Kim, S.-H. Whole-Genome Phylogeny of Escherichia coli/Shigella Group by Feature Frequency Profiles (FFPs). Proc. Natl. Acad. Sci. USA 2011, 108, 8329–8334. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.M.; Weier, H.-U.G.; Cozzarelli, N.R. Independence of Replisomes in Escherichia coli Chromosomal Replication. Proc. Natl. Acad. Sci. USA 2005, 102, 3942–3947. [Google Scholar] [CrossRef]
- Castillo, F.; Benmohamed, A.; Szatmari, G. Xer Site Specific Recombination: Double and Single Recombinase Systems. Front. Microbiol. 2017, 8, 453. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Horiuchi, T.; Ohsumi, K.; Shimamoto, N.; Morikawa, K. Structure of a Replication-Terminator Protein Complexed with DNA. Nature 1996, 383, 598–603. [Google Scholar] [CrossRef]
- Coskun-Ari, F.F.; Hill, T.M. Sequence-Specific Interactions in the Tus-Ter Complex and the Effect of Base Pair Substitutions on Arrest of DNA Replication in Escherichia coli. J. Biol. Chem. 1997, 272, 26448–26456. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Hoede, C.; Tenaillon, O.; Barbe, V.; Baeriswyl, S.; Bidet, P.; Bingen, E.; Bonacorsi, S.; Bouchier, C.; Bouvet, O.; et al. Organised Genome Dynamics in the Escherichia coli Species Results in Highly Diverse Adaptive Paths. PLOS Genet. 2009, 5, e1000344. [Google Scholar] [CrossRef]
- Leimbach, A.; Poehlein, A.; Vollmers, J.; Görlich, D.; Daniel, R.; Dobrindt, U. No Evidence for a Bovine Mastitis Escherichia coli Pathotype. BMC Genom. 2017, 18, 359. [Google Scholar] [CrossRef]
- Mulcair, M.D.; Schaeffer, P.M.; Oakley, A.J.; Cross, H.F.; Neylon, C.; Hill, T.M.; Dixon, N.E. A Molecular Mousetrap Determines Polarity of Termination of DNA Replication in E. coli. Cell 2006, 125, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Ramisetty, B.C.M.; Sudhakari, P.A. Bacterial ‘Grounded’ Prophages: Hotspots for Genetic Renovation and Innovation. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and Bacterial Genomics: What Have We Learned so Far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.G.; Hendrix, R.W.; Casjens, S. Where Are the Pseudogenes in Bacterial Genomes? Trends Microbiol. 2001, 9, 535–540. [Google Scholar] [CrossRef]
- Niyogi, S.K. Shigellosis. J. Microbiol. Seoul Korea 2005, 43, 133–143. [Google Scholar]
- Ochman, H.; Whittam, T.S.; Caugant, D.A.; Selander, R.K. Enzyme Polymorphism and Genetic Population Structure in Escherichia coli and Shigella. J. Gen. Microbiol. 1983, 129, 2715–2726. [Google Scholar] [CrossRef]
- Hendrickson, H.; Lawrence, J.G. Mutational Bias Suggests That Replication Termination Occurs near the dif Site, Not at Ter Sites. Mol. Microbiol. 2007, 64, 42–56. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K.; Tomita, M. Comprehensive Prediction of Chromosome Dimer Resolution Sites in Bacterial Genomes. BMC Genom. 2011, 12, 19. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K.; Tomita, M. Validation of Bacterial Replication Termination Models Using Simulation of Genomic Mutations. PLoS ONE 2012, 7, e34526. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.A.; Nilles, A.F.; Valjavec-Gratian, M.; Hill, T.M. Site-Directed Mutagenesis and Phylogenetic Comparisons of the Escherichia coli Tus Protein: DNA-Protein Interactions Alone Can Not Account for Tus Activity. Mol. Genet. Genomics MGG 2001, 265, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Brewer, B.J. When Polymerases Collide: Replication and the Transcriptional Organization of the E. coli Chromosome. Cell 1988, 53, 679–686. [Google Scholar] [CrossRef]
- Horiuchi, T.; Fujimura, Y.; Nishitani, H.; Kobayashi, T.; Hidaka, M. The DNA Replication Fork Blocked at the Ter Site May Be an Entrance for the RecBCD Enzyme into Duplex DNA. J. Bacteriol. 1994, 176, 4656–4663. [Google Scholar] [CrossRef]
- Nordström, K. Plasmid R1--Replication and Its Control. Plasmid 2006, 55, 1–26. [Google Scholar] [CrossRef]
- Krabbe, M.; Zabielski, J.; Bernander, R.; Nordström, K. Inactivation of the Replication-Termination System Affects the Replication Mode and Causes Unstable Maintenance of Plasmid R1. Mol. Microbiol. 1997, 24, 723–735. [Google Scholar] [CrossRef]
- Hiasa, H.; Marians, K.J. Tus Prevents Overreplication of oriC Plasmid DNA. J. Biol. Chem. 1994, 269, 26959–26968. [Google Scholar] [CrossRef]
- Hasebe, T.; Narita, K.; Hidaka, S.; Su’etsugu, M. Efficient Arrangement of the Replication Fork Trap for In Vitro Propagation of Monomeric Circular DNA in the Chromosome-Replication Cycle Reaction. Life Basel Switz. 2018, 8, 43. [Google Scholar] [CrossRef]
- Médigue, C.; Krin, E.; Pascal, G.; Barbe, V.; Bernsel, A.; Bertin, P.N.; Cheung, F.; Cruveiller, S.; D’Amico, S.; Duilio, A.; et al. Coping with Cold: The Genome of the Versatile Marine Antarctica Bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res. 2005, 15, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.-B.; Rong, J.-C.; Tang, B.-L.; Wang, S.; Liu, G.; Qin, Q.-L.; Zhang, X.-Y.; Zhang, W.; She, Q.; Chen, Y.; et al. Evolutionary Trajectory of the Replication Mode of Bacterial Replicons. mBio 2021, 12. [Google Scholar] [CrossRef]
- Midgley-Smith, S.L.; Dimude, J.U.; Rudolph, C.J. A Role for 3’ Exonucleases at the Final Stages of Chromosome Duplication in Escherichia coli. Nucleic Acids Res. 2018, 47, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.J.; Upton, A.L.; Briggs, G.S.; Lloyd, R.G. Is RecG a General Guardian of the Bacterial Genome? DNA Repair 2010, 9, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.J.; Cagliero, C.; Zhou, Y.N. Growth Rate Regulation in Escherichia coli. FEMS Microbiol. Rev. 2012, 36, 269–287. [Google Scholar] [CrossRef] [PubMed]
- Lioy, V.S.; Cournac, A.; Marbouty, M.; Duigou, S.; Mozziconacci, J.; Espéli, O.; Boccard, F.; Koszul, R. Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell 2018, 172, 771–783.e18. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Background | Doubling Time (min) | SD | Reference |
|---|---|---|---|
| MG1655 | 19.1 a | ±1.3 | This study |
| ∆tus | 19.6 a | ±1.2 | This study |
| oriC+ oriZ+ | 20.6 | ±1.3 | [20] |
| oriC+ oriZ+ ∆tus | 21.5 | ±0.7 | [20] |
| oriC+ oriX+ | 22.3 | ±1.2 | [38] |
| oriC+ oriX+ ∆tus | 23.1 | ±0.7 | [38] |
| Background | Phylogroup | Genome Size (Mbp) | Inner Termination Area (kb) | % of Total Genome Size |
|---|---|---|---|---|
| MG1655 | A | 4.641 | 267 | 5.76 |
| APEC078 | B1 | 4.798 | 243 | 5.05 |
| S88 | B2 | 5.032 | 190 | 3.77 |
| UMN026 | D | 5.202 | 225 | 4.32 |
| TW14359 | E | 5.528 | 238 | 4.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goodall, D.J.; Jameson, K.H.; Hawkins, M.; Rudolph, C.J. A Fork Trap in the Chromosomal Termination Area Is Highly Conserved across All Escherichia coli Phylogenetic Groups. Int. J. Mol. Sci. 2021, 22, 7928. https://doi.org/10.3390/ijms22157928
Goodall DJ, Jameson KH, Hawkins M, Rudolph CJ. A Fork Trap in the Chromosomal Termination Area Is Highly Conserved across All Escherichia coli Phylogenetic Groups. International Journal of Molecular Sciences. 2021; 22(15):7928. https://doi.org/10.3390/ijms22157928
Chicago/Turabian StyleGoodall, Daniel J., Katie H. Jameson, Michelle Hawkins, and Christian J. Rudolph. 2021. "A Fork Trap in the Chromosomal Termination Area Is Highly Conserved across All Escherichia coli Phylogenetic Groups" International Journal of Molecular Sciences 22, no. 15: 7928. https://doi.org/10.3390/ijms22157928
APA StyleGoodall, D. J., Jameson, K. H., Hawkins, M., & Rudolph, C. J. (2021). A Fork Trap in the Chromosomal Termination Area Is Highly Conserved across All Escherichia coli Phylogenetic Groups. International Journal of Molecular Sciences, 22(15), 7928. https://doi.org/10.3390/ijms22157928