
Reference Gene Selection for Gene Expression Analyses in Mouse Models of Acute Lung Injury

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Selection of Candidate Reference Genes
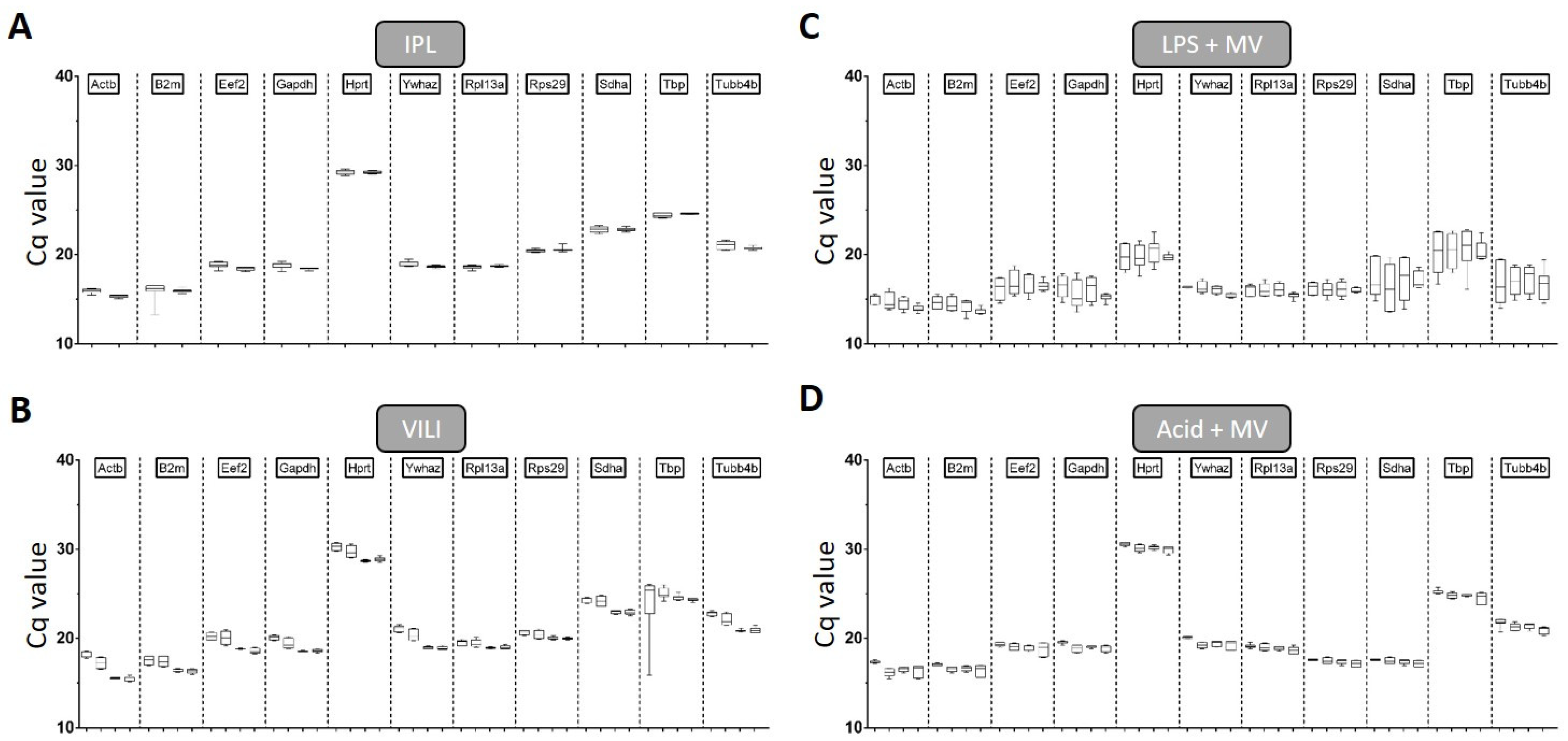
2.2. Amplification Efficiency and General qPCR Parameters
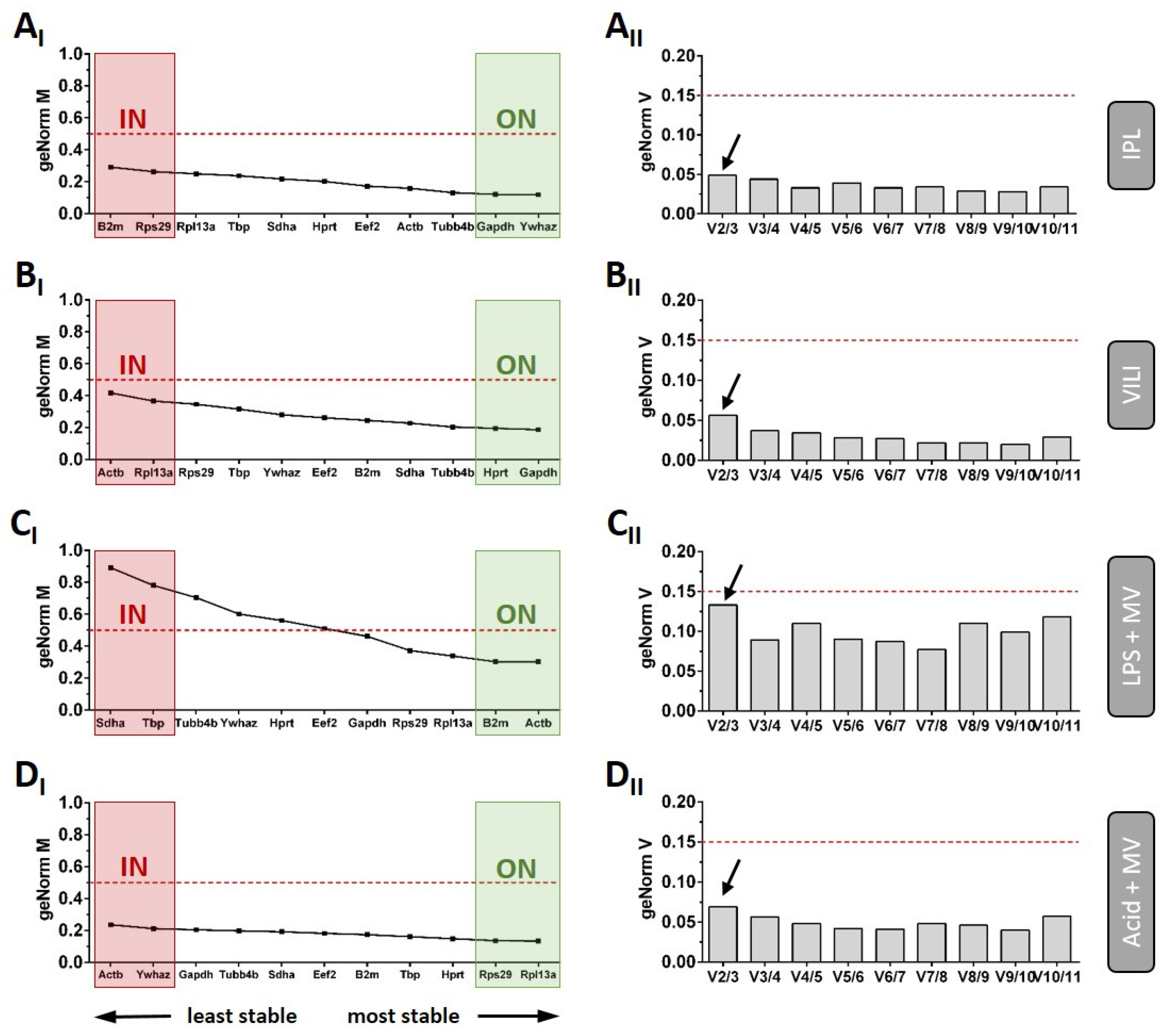
2.3. Normalisation Strategies
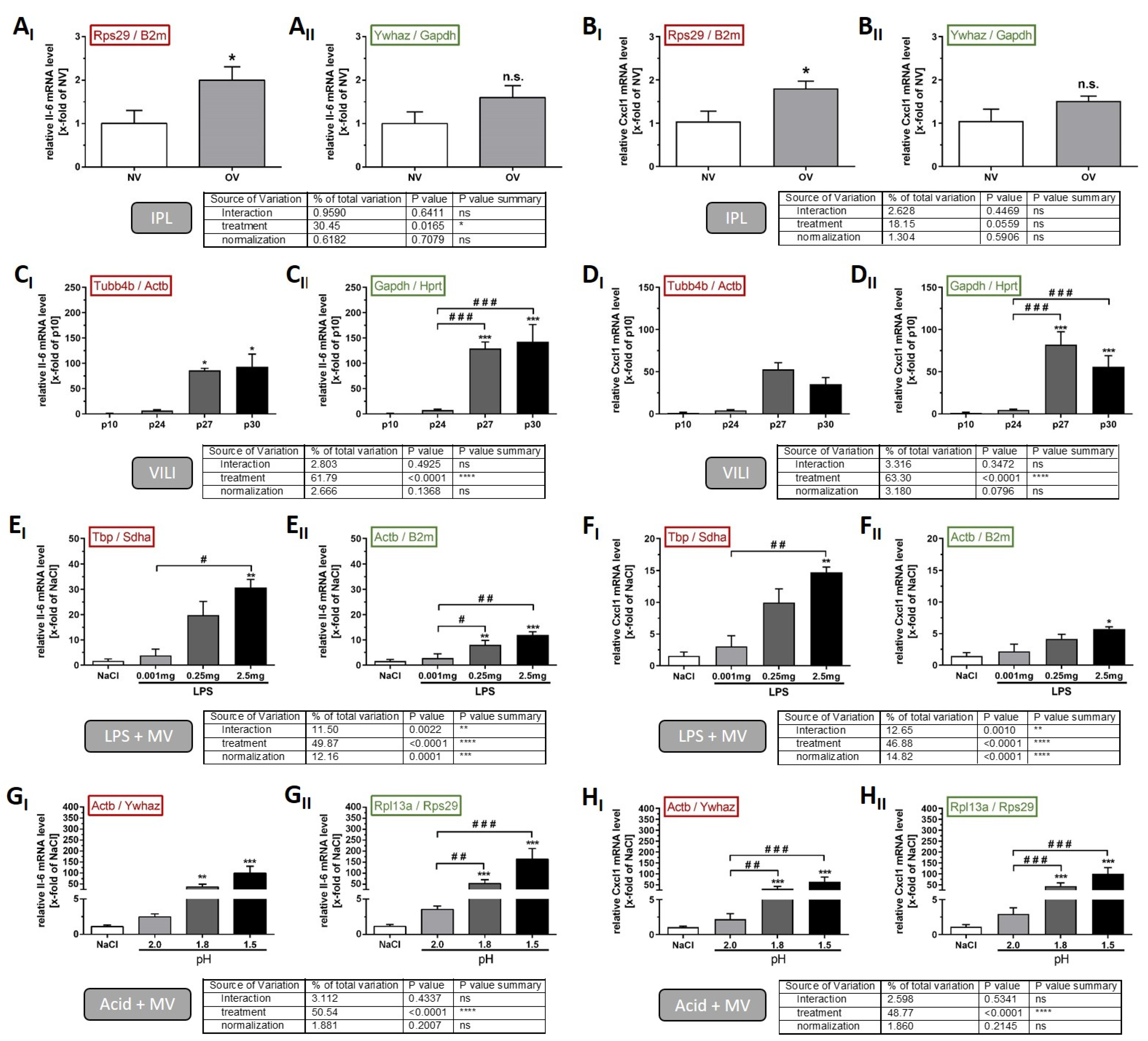
2.4. Comparison of IL-6 and Cxcl1 Gene Expression Using an Optimised vs. Inappropriate Normalisation Strategy
3. Discussion
4. Materials and Methods
4.1. Animals and Sample Collection
4.2. RNA Isolation, cDNA Synthesis, and qPCR
4.3. Primer Design
4.4. Data Analysis
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Actb | beta-actin |
| ALI | acute lung injury |
| ARDS | acute respiratory distress syndrome |
| B2m | beta-2 microglobulin |
| Cq | cycle of quantification |
| Cxcl1 | C-X-C motif ligand 1 |
| Eef2 | eukaryotic translation elongation factor 2 |
| Gapdh | glyceraldehyde-3-phosphate dehydrogenase |
| HCl | hydrochloric acid |
| Hprt | hypoxanthine guanine phosphoribosyl transferase |
| Il-6 | Interleukin 6 |
| Il-8 | Interleukin 8 |
| IN | inappropriate normalisation |
| IPL | isolated perfused lung |
| NV | normal ventilation |
| MV | mechanical ventilation |
| ON | optimized ventilation |
| OV | overventilation |
| RGI | reference gene index |
| RIN | RNA integrity number |
| Rpl13a | ribosomal protein L13A |
| Rps29 | ribosomal protein S29 |
| Sdha | succinate dehydrogenase complex, subunit A flavoprotein |
| Tbp | TATA box binding protein |
| Tubb4b | tubulin, beta 4B class IVB |
| VILI | ventilator-induced lung injury |
| Ywhaz | tyrosine 3-monooxygenase/tryptophan 5 monooxygenase activation protein zeta |
References
- McNicholas, B.A.; Rooney, G.M.; Laffey, J.G. Lessons to learn from epidemiologic studies in ards. Curr. Opin. Crit. Care 2018, 24, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Grieco, D.L.; Bongiovanni, F.; Chen, L.; Menga, L.S.; Cutuli, S.L.; Pintaudi, G.; Carelli, S.; Michi, T.; Torrini, F.; Lombardi, G.; et al. Respiratory physiology of covid-19-induced respiratory failure compared to ards of other etiologies. Crit. Care 2020, 24, 529. [Google Scholar] [CrossRef]
- Matthay, M.A.; Leligdowicz, A.; Liu, K.D. Biological mechanisms of covid-19 acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 1489–1491. [Google Scholar] [CrossRef]
- Reiss, L.K.; Schuppert, A.; Uhlig, S. Inflammatory processes during acute respiratory distress syndrome: A complex system. Curr. Opin. Crit. Care 2018, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef] [Green Version]
- Reiss, L.K.; Kowallik, A.; Uhlig, S. Recurrent recruitment manoeuvres improve lung mechanics and minimize lung injury during mechanical ventilation of healthy mice. PLoS ONE 2011, 6, e24527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, L.K.; Uhlig, U.; Uhlig, S. Models and mechanisms of acute lung injury caused by direct insults. Eur. J. Cell Biol. 2012, 91, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Lex, D.; Uhlig, S. One-hit models of ventilator-induced lung injury: Benign inflammation versus inflammation as a by-product. Anesthesiology 2017, 126, 909–922. [Google Scholar] [CrossRef]
- Grune, J.; Tabuchi, A.; Kuebler, W.M. Alveolar dynamics during mechanical ventilation in the healthy and injured lung. Intensive Care Med. Exp. 2019, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, S.; Wollin, L. An improved setup for the isolated perfused rat lung. J. Pharmacol. Toxicol. Methods 1994, 31, 85–94. [Google Scholar] [CrossRef]
- von Bethmann, A.N.; Brasch, F.; Nusing, R.; Vogt, K.; Volk, H.D.; Muller, K.M.; Wendel, A.; Uhlig, S. Hyperventilation induces release of cytokines from perfused mouse lung. Am. J. Respir. Crit. Care Med. 1998, 157, 263–272. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; O’Neill, L.A. Signal transduction by the lipopolysaccharide receptor, toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef]
- Kaku, S.; Nguyen, C.D.; Htet, N.N.; Tutera, D.; Barr, J.; Paintal, H.S.; Kuschner, W.G. Acute respiratory distress syndrome: Etiology, pathogenesis, and summary on management. J. Intensive Care Med. 2019, 8, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.P.; Johnson, K.J.; Kunkel, R.G.; Ward, P.A.; Knight, P.R.; Finch, J.S. Acute acid aspiration lung injury in the rat: Biphasic pathogenesis. Anesth. Analg. 1989, 69, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Reiss, L.K.; Raffetseder, U.; Gibbert, L.; Drescher, H.K.; Streetz, K.L.; Schwarz, A.; Martin, C.; Uhlig, S.; Adam, D. Reevaluation of lung injury in tnf-induced shock: The role of the acid sphingomyelinase. Mediat. Inflamm. 2020, 2020, 3650508. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The miqe guidelines: Minimum information for publication of quantitative real-time pcr experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Fantini, P.M.; Rajapaksa, A.E.; Oakley, R.; Tingay, D.G. Selection of reference genes for gene expression studies related to lung injury in a preterm lamb model. Scient. Rep. 2016, 6, 26476. [Google Scholar] [CrossRef] [Green Version]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time rt-pcr normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative rt-pcr data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Hildyard, J.C.W.; Finch, A.M.; Wells, D.J. Identification of qpcr reference genes suitable for normalizing gene expression in the mdx mouse model of duchenne muscular dystrophy. PLoS ONE 2019, 14, e0211384. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: Bestkeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-pcr data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, S.; Khawaja, A.; Wieruszewski, P.M.; Gajic, O.; Odeyemi, Y. Diagnosis and treatment of acute pulmonary inflammation in critically ill patients: The role of inflammatory biomarkers. World J. Crit. Care Med. 2019, 8, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Tanaka, T. Il-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef]
- Chapman, J.R.; Waldenstrom, J. With reference to reference genes: A systematic review of endogenous controls in gene expression studies. PLoS ONE 2015, 10, e0141853. [Google Scholar]
- Della Latta, V.; Cabiati, M.; Burchielli, S.; Frenzilli, G.; Bernardeschi, M.; Cecchettini, A.; Viglione, F.; Morales, M.A.; Del Ry, S. Lung inflammation after bleomycin treatment in mice: Selection of an accurate normalization strategy for gene expression analysis in an ex-vivo and in-vitro model. Int. J. Biochem. Cell Biol. 2017, 88, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Griessl, M.; Gutknecht, M.; Cook, C.H. Determination of suitable reference genes for rt-qpcr analysis of murine cytomegalovirus in vivo and in vitro. J. Virol. Methods 2017, 248, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Dobersch, S.; Dammann, R.H.; Bellusci, S.; Ilinskaya, O.N.; Braun, T.; Barreto, G. Validation of tuba1a as appropriate internal control for normalization of gene expression analysis during mouse lung development. Int. J. Mol. Sci. 2015, 16, 4492–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, T.H.; Koeppen, K.; Bashor, L.; Stanton, B.A. Selection of reference genes for quantitative pcr: Identifying reference genes for airway epithelial cells exposed to pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L256–L265. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.R.; Jreij, P.; Fletcher, D.A. Mechanotransduction by the actin cytoskeleton: Converting mechanical stimuli into biochemical signals. Ann. Rev. Biophys. 2018, 47, 617–631. [Google Scholar] [CrossRef]
- Galván-Peña, S.; Carroll, R.G.; Newman, C.; Hinchy, E.C.; Palsson-McDermott, E.; Robinson, E.K.; Covarrubias, S. Malonylation of gapdh is an inflammatory signal in macrophages. Nat. Commun. 2019, 10, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, P.E.; Eisner, M.D.; Thompson, B.T.; Matthay, M.A.; Ancukiewicz, M.; Bernard, G.R.; Wheeler, A.P. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit. Care Med. 2005, 33, 1–6, discussion 230–232. [Google Scholar] [CrossRef]
- Meduri, G.U.; Kohler, G.; Headley, S.; Tolley, E.; Stentz, F.; Postlethwaite, A. Inflammatory cytokines in the bal of patients with ards. Persistent elevation over time predicts poor outcome. Chest 1995, 108, 1303–1314. [Google Scholar] [CrossRef]
- Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [PubMed]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An official american thoracic society workshop report: Features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Reiss, L.K.; Fragoulis, A.; Siegl, S.; Platen, C.; Kan, Y.W.; Nautiyal, J.; Parker, M.; Pufe, T.; Uhlig, U.; Martin, C.; et al. Interplay between nuclear factor erythroid 2-related factor 2 and amphiregulin during mechanical ventilation. Am. J. Respir. Cell Mol. Biol. 2014, 51, 668–677. [Google Scholar] [CrossRef]
- Müller, H.C.; Hellwig, K.; Rosseau, S.; Tschernig, T.; Schmiedl, A.; Gutbier, B.; Schmeck, B.; Hippenstiel, S.; Peters, H.; Morawietz, L.; et al. Simvastatin attenuates ventilator-induced lung injury in mice. Crit. Care 2010, 14, R143. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.E.; Chambers, R.C. The mercurial nature of neutrophils: Still an enigma in ards? Am. J. Phys. Lung Cell. Mol. Phys. 2014, 306, L217–L230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, L.D.; Schouten, L.R.; van Vught, L.A.; Wiewel, M.A.; Ong, D.S.Y.; Cremer, O.; Artigas, A.; Martin-Loeches, I.; Hoogendijk, A.J.; van der Poll, T.; et al. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax 2017, 72, 876–883. [Google Scholar] [CrossRef]
- Calfee, C.S.; Delucchi, K.; Parsons, P.E.; Thompson, B.T.; Ware, L.B.; Matthay, M.A. Subphenotypes in acute respiratory distress syndrome: Latent class analysis of data from two randomised controlled trials. Lancet Respir. Med. 2014, 2, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.G.; Simpson, L.J.; Ferreira, A.M.; Rustagi, A.; Roque, J.; Asuni, A.; Ranganath, T.; Grant, P.M.; Subramanian, A.; Rosenberg-Hasson, Y.; et al. Cytokine profile in plasma of severe covid-19 does not differ from ards and sepsis. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mrna and mrna-protein correlations in a xenograft model system. Scient. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegl, S.; Uhlig, S. Using the one-lung method to link p38 to pro-inflammatory gene expression during overventilation in c57bl/6 and balb/c mice. PLoS ONE 2012, 7, e41464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, L.K. A Mouse Intensive Care Unit to Study Tnf-Induced Sepsis and Acid-Induced Lung Injury. Ph.D. Thesis, RWTH Aachen University, Aachen, Germany, 2012. [Google Scholar]
- Kanzler, S.S. A Murine Two-Hit Model to Investigate the Early Time-Course of Inflammation in Acute Respiratory Distress Syndrome. Ph.D. Thesis, RWTH Aachen University, Aachen, Germany, 2021. [Google Scholar]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.L.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (pcr) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Target Type | Gene | Accesion Number | Primer Sequence 5′′→3′′ | Annealing Temp [°C] | Amplicon Length [bp] | Tm [°C] |
|---|---|---|---|---|---|---|
| REF | Actb | NM_007393.5 | F: CAC TGT CGA GTC GCG TCC | 60 | 89 | 88.20 |
| R: TCA TCC ATG GCG AAC TGG TG | ||||||
| B2m | NM_009735.3 | F: TTC TGG TGC TTG TCT CAC TGA | 61 | 104 | 83.27 | |
| R: CAG TAT GTT CGG CTT CCC ATT C | ||||||
| Eef2 | NM_007907.2 | F: TCA CAA TCA AAT CCA CCG CC | 60 | 122 | 83.87 | |
| R: ATG GCC TGG AGA GTC GAT GA | ||||||
| Gapdh | NM_008084.3 | F: CAT GGC CTT CCG TGT TCC TA | 60 | 74 | 85.63 | |
| R: ACT TGG CAG GTT TCT CCA GG | ||||||
| Hprt | NM_013556.2 | F: TCA GTC AAC GGG GGA CAT AAA | 61 | 142 | 79.10 | |
| R: GGG GCT GTA CTG CTT AAC CAG | ||||||
| Rpl13a | NM_009438.5 | F: GCGGATGAATACCAACCCCT | 61 | 179 | 90.19 | |
| R: CCACCATCCGCTTTTTCTTGT | ||||||
| Rps29 | NM_009093.2 | F: CCTTTCTCCTCGTTGGGCG | 61 | 105 | 87.52 | |
| R: GAGCAGACGCGGCAAGAG | ||||||
| Sdha | NM_023281.1 | F: GGAACACTCCAAAAACAGACCT | 60 | 106 | 80.41 | |
| R: CCACCACTGGGTATTGAGTAGAA | ||||||
| Tbp | NM_013684.3 | F: ATCTACCGTGAATCTTGGCTGT | 61 | 183 | 82.21 | |
| R: GATTGTTCTTCACTCTTGGCTC | ||||||
| Tubb4b | NM_146116.2 | F: TCTTCTACAGCTGTTCCGCAG | 61 | 143 | 89.73 | |
| R: GTGGTAAGTGCCAGTGGGAT | ||||||
| Ywhaz | NM_011740.3 | F: GAAAAGTTCTTGATCCCCAATGC | 62 | 134 | 82.18 | |
| R: TGTGACTGGTCCACAATTCCTT | ||||||
| GOI | Cxcl1 | NM_008176.3 | F: CAAACCGAAGTCATAGCCAC | 60 | 106 | 83.10 |
| R: TGGGGACACCTTTTAGCATC | ||||||
| IL-6 | NM_031168.1 | F: TGCAAGAGACTTCCATCCAGTTGCC | 59 | 147 | 84.92 | |
| R: AAGCCTCCGACTTGTGAAGTGGT |
| Gene | Amplification Factor | Efficiency [%] | R2 | Slope | Y-Intercept |
|---|---|---|---|---|---|
| Actb | 2.010 | 101.01 | 0.994 | −3.298 | 21.475 |
| B2m | 2.027 | 102.69 | 0.999 | −3.259 | 22.111 |
| Eef2 | 2.021 | 102.08 | 0.998 | −3.273 | 24.188 |
| Gapdh | 2.012 | 101.22 | 0.999 | −3.293 | 24.408 |
| Hprt | 1.850 | 85.03 | 0.990 | −3.742 | 36.555 |
| Rpl13a | 1.847 | 84.75 | 0.998 | −3.751 | 25.222 |
| Rps29 | 2.036 | 103.58 | 0.996 | −3.239 | 23.730 |
| Sdha | 1.958 | 95.83 | 0.992 | −3.426 | 28.847 |
| Tbp | 2.046 | 104.62 | 0.993 | −3.216 | 30.321 |
| Tubb4b | 1.923 | 92.28 | 0.995 | −3.522 | 27.734 |
| Ywhaz | 2.006 | 100.58 | 0.996 | −3.308 | 24.772 |
| IPL | VILI | LPS + MV | Acid + MV | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Mean Cq | SD | CV [%] | Mean Cq | SD | CV [%] | Mean Cq | SD | CV [%] | Mean Cq | SD | CV [%] |
| Actb | 15.61 | 0.35 | 2.24% | 16.46 | 1.21 | 7.35% | 14.53 | 0.74 | 5.09% | 16.94 | 0.75 | 4.43% |
| B2m | 15.97 | 0.45 | 2.82% | 16.89 | 0.64 | 3.79% | 14.19 | 0.72 | 5.07% | 17.05 | 0.65 | 3.81% |
| Eef2 | 18.60 | 0.32 | 1.72% | 19.37 | 0.84 | 4.34% | 16.61 | 1.03 | 6.20% | 19.52 | 0.66 | 3.38% |
| Gapdh | 18.62 | 0.29 | 1.56% | 19.11 | 0.68 | 3.56% | 15.78 | 0.96 | 6.08% | 19.29 | 0.51 | 2.64% |
| Hprt | 29.24 | 0.18 | 0.62% | 29.33 | 0.73 | 2.49% | 19.92 | 1.07 | 5.37% | 26.21 | 0.48 | 1.83% |
| Rpl13a | 18.67 | 0.13 | 0.70% | 19.20 | 0.35 | 1.82% | 15.91 | 0.62 | 3.90% | 19.09 | 0.41 | 2.15% |
| Rps29 | 20.50 | 0.16 | 0.78% | 20.25 | 0.38 | 1.88% | 16.10 | 0.63 | 3.91% | 18.55 | 0.33 | 1.78% |
| Sdha | 22.83 | 0.24 | 1.05% | 23.54 | 0.74 | 3.14% | 16.98 | 2.04 | 12.01% | 23.29 | 0.61 | 2.62% |
| Tbp | 24.50 | 0.19 | 0.78% | 24.85 | 0.54 | 2.17% | 20.39 | 1.73 | 8.48% | 24.89 | 0.56 | 2.25% |
| Tubb4b | 20.91 | 0.36 | 1.72% | 21.69 | 0.92 | 4.24% | 16.94 | 1.59 | 9.39% | 21.44 | 0.65 | 3.03% |
| Ywhaz | 18.86 | 0.30 | 1.59% | 19.76 | 0.98 | 4.96% | 15.80 | 1.24 | 7.85% | 20.10 | 0.65 | 3.23% |
| IPL | VILI | LPS + MV | Acid + MV | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Efficiency | R2 | Slope | Y-Intercept | Efficiency | R2 | Slope | Y-Intercept | Efficiency | R2 | Slope | Y-Intercept | Efficiency | R2 | Slope | Y-Intercept |
| Actb | 2.008 | 0.997 | −3.304 | 21.531 | 1.974 | 0.999 | −3.386 | 21.637 | 1.984 | 0.999 | −3.362 | 22.172 | ||||
| B2m | 2.027 | 0.999 | −3.259 | 22.111 | 1.982 | 0.998 | −3.367 | 21.278 | ||||||||
| Eef2 | 2.021 | 0.998 | −3.273 | 24.188 | ||||||||||||
| Gapdh | 2.012 | 0.999 | −3.293 | 24.408 | 2.039 | 0.996 | −3.232 | 24.428 | ||||||||
| Hprt | 1.918 | 0.994 | −3.535 | 35.044 | ||||||||||||
| Rpl13a | 1.847 | 0.998 | −3.751 | 25.222 | ||||||||||||
| Rps29 | 2.036 | 0.996 | −3.239 | 23.730 | ||||||||||||
| Sdha | 1.958 | 0.992 | −3.426 | 28.847 | 1.996 | 0.996 | −3.331 | 28.245 | ||||||||
| Tbp | 2.046 | 0.993 | −3.216 | 30.321 | ||||||||||||
| Tubb4b | 1.923 | 0.995 | −3.522 | 27.734 | ||||||||||||
| Ywhaz | 2.006 | 0.996 | −3.308 | 24.772 | ||||||||||||
| Cxcl1 | 1.948 | 0.992 | −3.452 | 26.908 | 1.943 | 0.996 | −3.466 | 27.398 | 1.887 | 0.998 | −3.627 | 25.379 | 1.961 | 0.997 | −3.418 | 27.915 |
| Il-6 | 1.965 | 0.999 | −3.409 | 24.709 | 1.965 | 0.996 | −3.410 | 25.633 | 1.922 | 0.996 | −3.523 | 25.332 | 2.005 | 0.998 | −3.310 | 28.257 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fragoulis, A.; Biller, K.; Fragoulis, S.; Lex, D.; Uhlig, S.; Reiss, L.K. Reference Gene Selection for Gene Expression Analyses in Mouse Models of Acute Lung Injury. Int. J. Mol. Sci. 2021, 22, 7853. https://doi.org/10.3390/ijms22157853
Fragoulis A, Biller K, Fragoulis S, Lex D, Uhlig S, Reiss LK. Reference Gene Selection for Gene Expression Analyses in Mouse Models of Acute Lung Injury. International Journal of Molecular Sciences. 2021; 22(15):7853. https://doi.org/10.3390/ijms22157853
Chicago/Turabian StyleFragoulis, Athanassios, Kristina Biller, Stephanie Fragoulis, Dennis Lex, Stefan Uhlig, and Lucy Kathleen Reiss. 2021. "Reference Gene Selection for Gene Expression Analyses in Mouse Models of Acute Lung Injury" International Journal of Molecular Sciences 22, no. 15: 7853. https://doi.org/10.3390/ijms22157853
APA StyleFragoulis, A., Biller, K., Fragoulis, S., Lex, D., Uhlig, S., & Reiss, L. K. (2021). Reference Gene Selection for Gene Expression Analyses in Mouse Models of Acute Lung Injury. International Journal of Molecular Sciences, 22(15), 7853. https://doi.org/10.3390/ijms22157853