
Synthesis of New Tricyclic 1,2-Thiazine Derivatives with Anti-Inflammatory Activity

 ,
,  ,
,  and
and 
Abstract
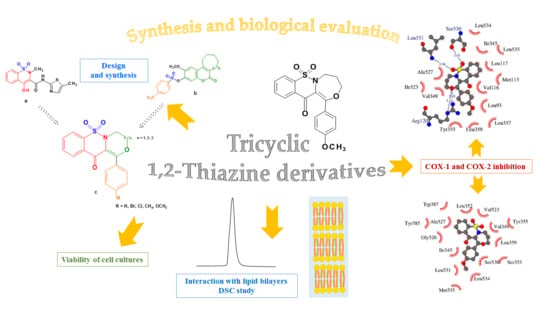
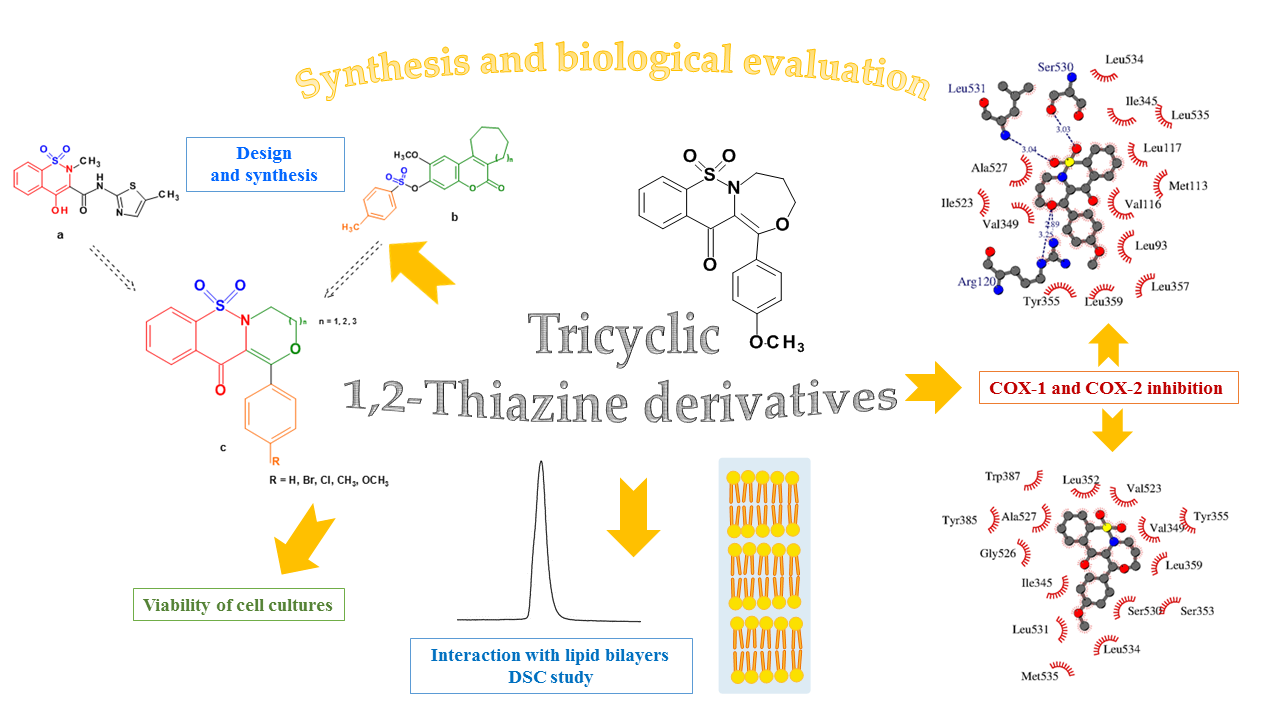
1. Introduction
2. Results and Discussion
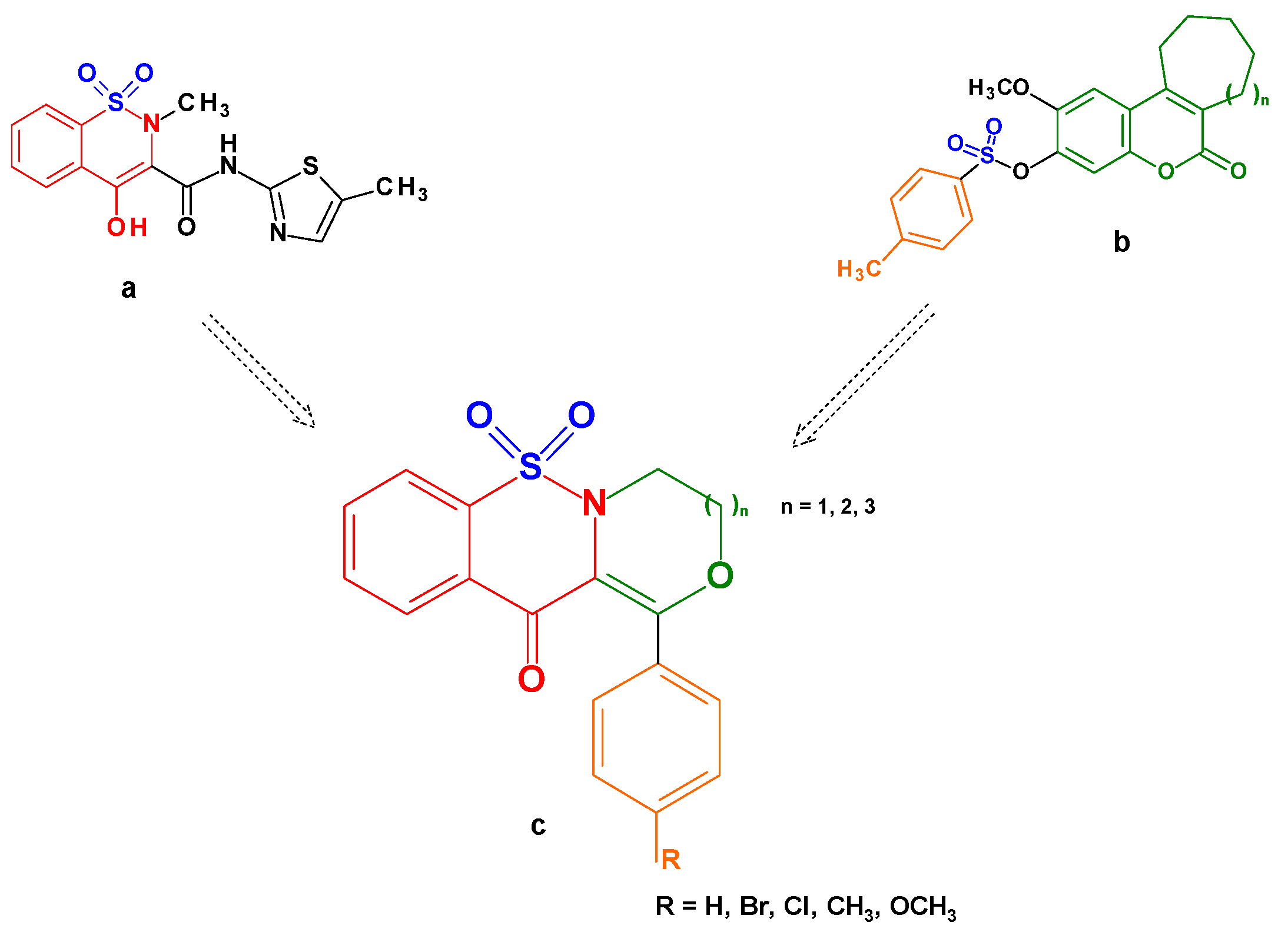
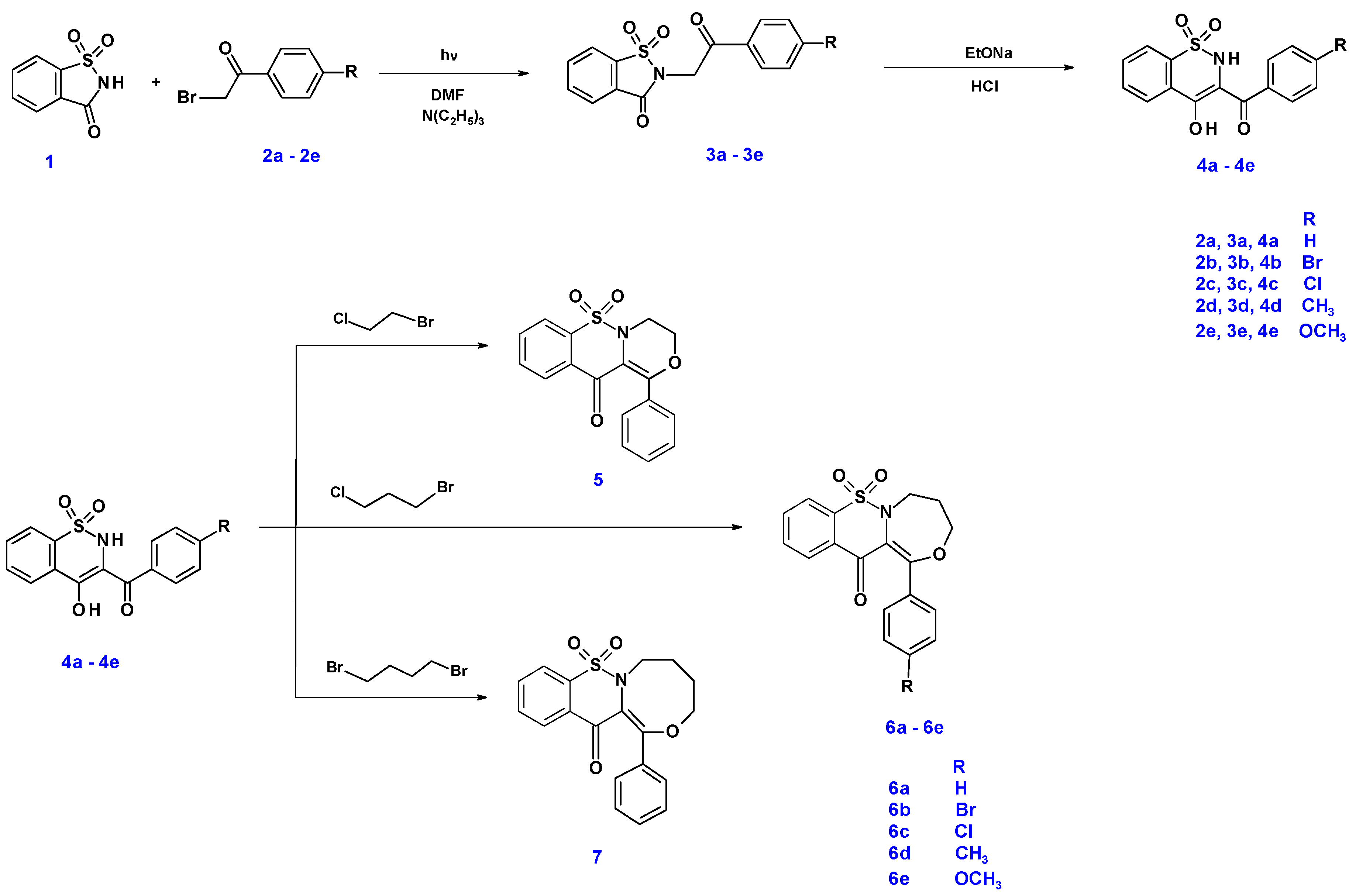
2.1. Chemistry
2.2. QSAR Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Symbol | Chemical Structure | LogP 1 |
|---|---|---|
| 4a |  | 3.43 |
| 4c |  | 3.96 |
| 4d |  | 3.81 |
| 5 |  | 3.53 |
| 6a |  | 3.64 |
| 6b |  | 2.64 |
| 6c |  | 2.76 |
| 6d |  | 2.64 |
| 6e |  | 1.48 |
| 7 |  | 3.54 |
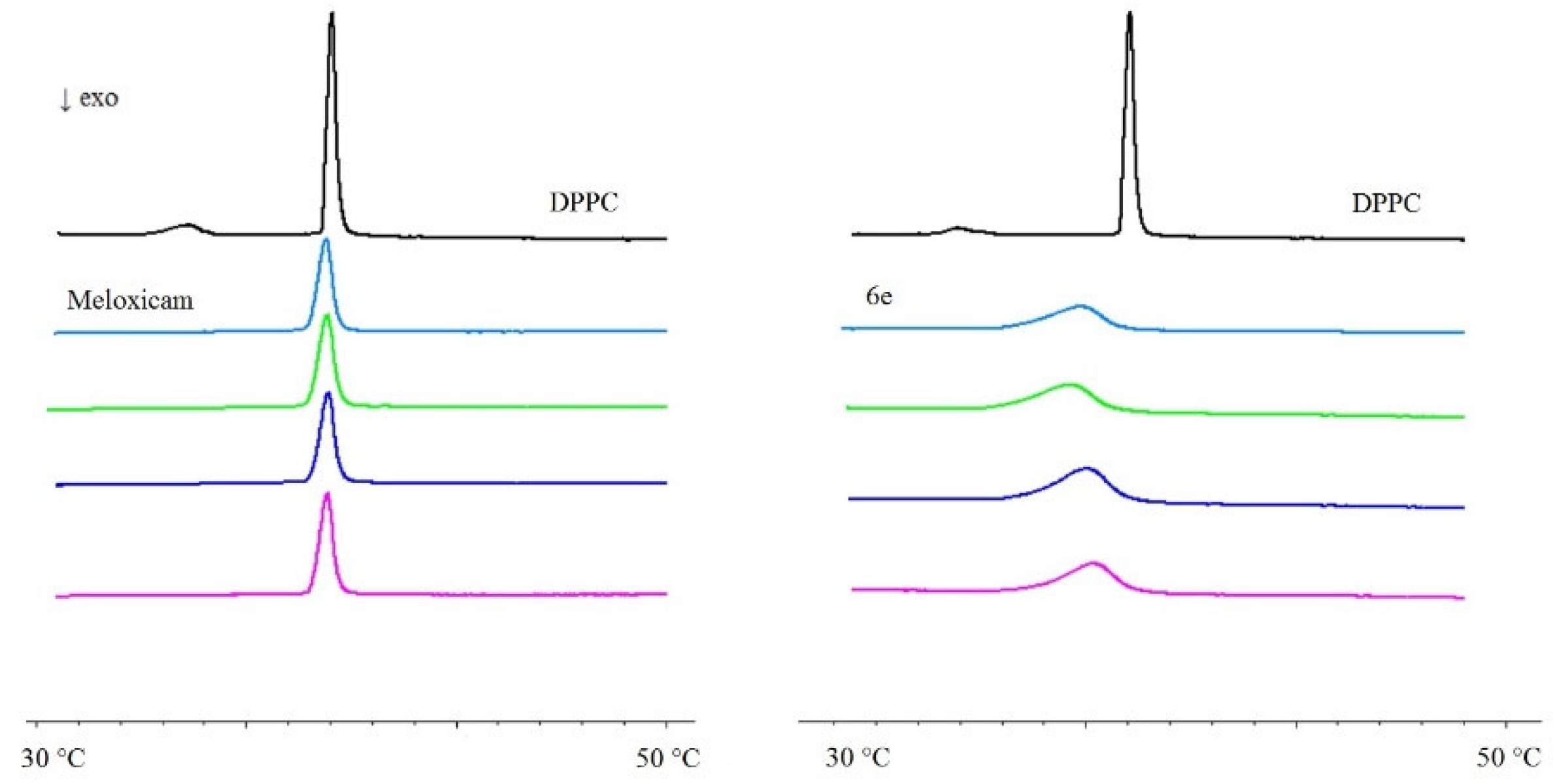
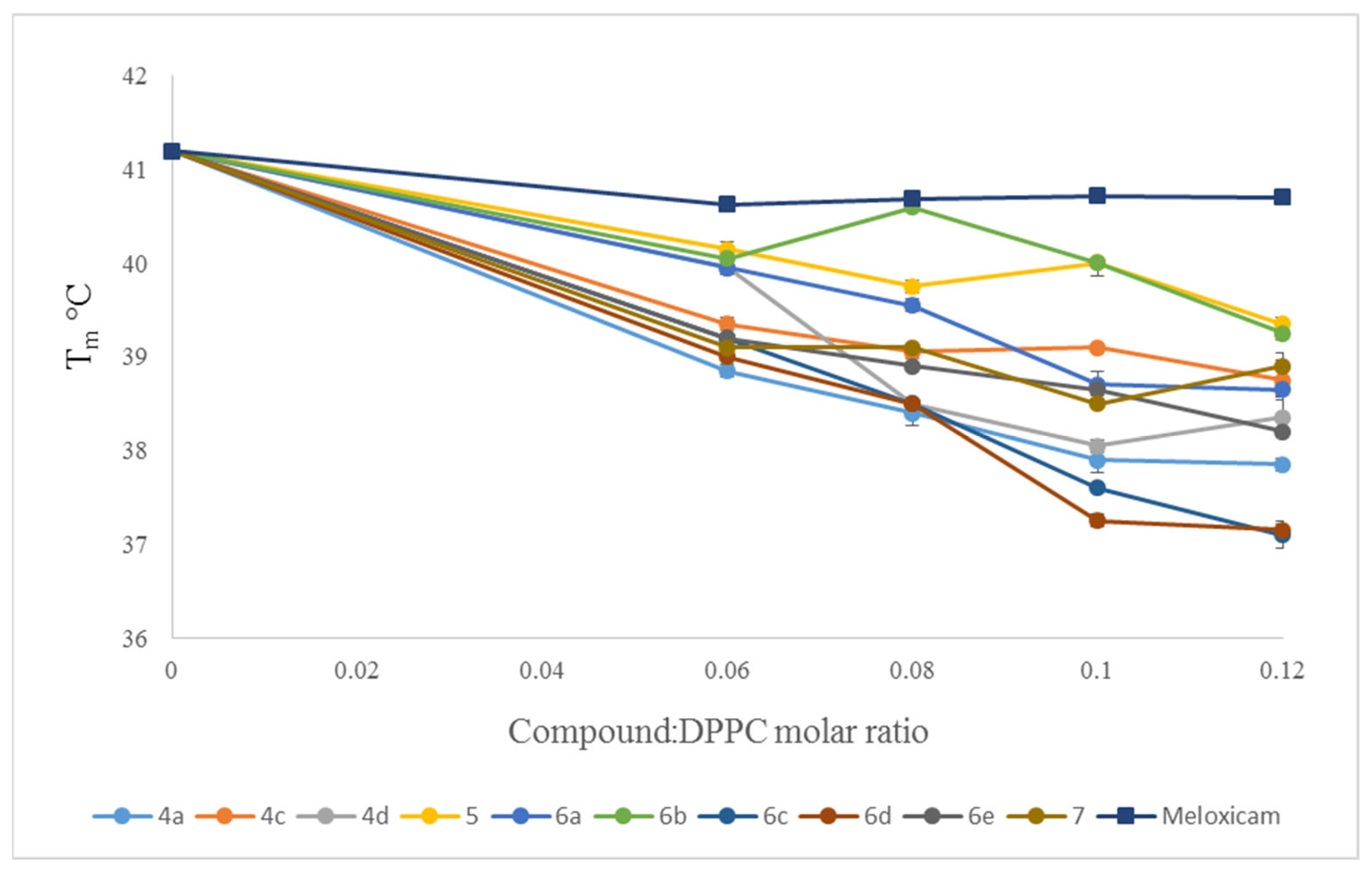
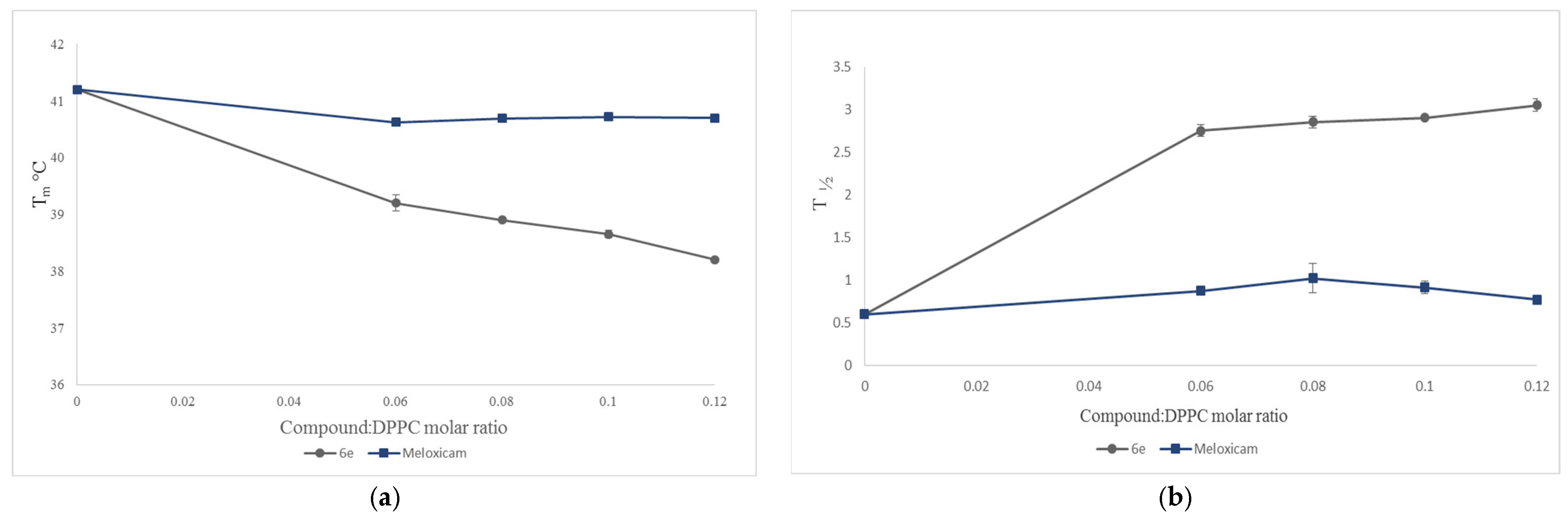
2.3. Interaction with Model Membranes
2.4. Biological Tests
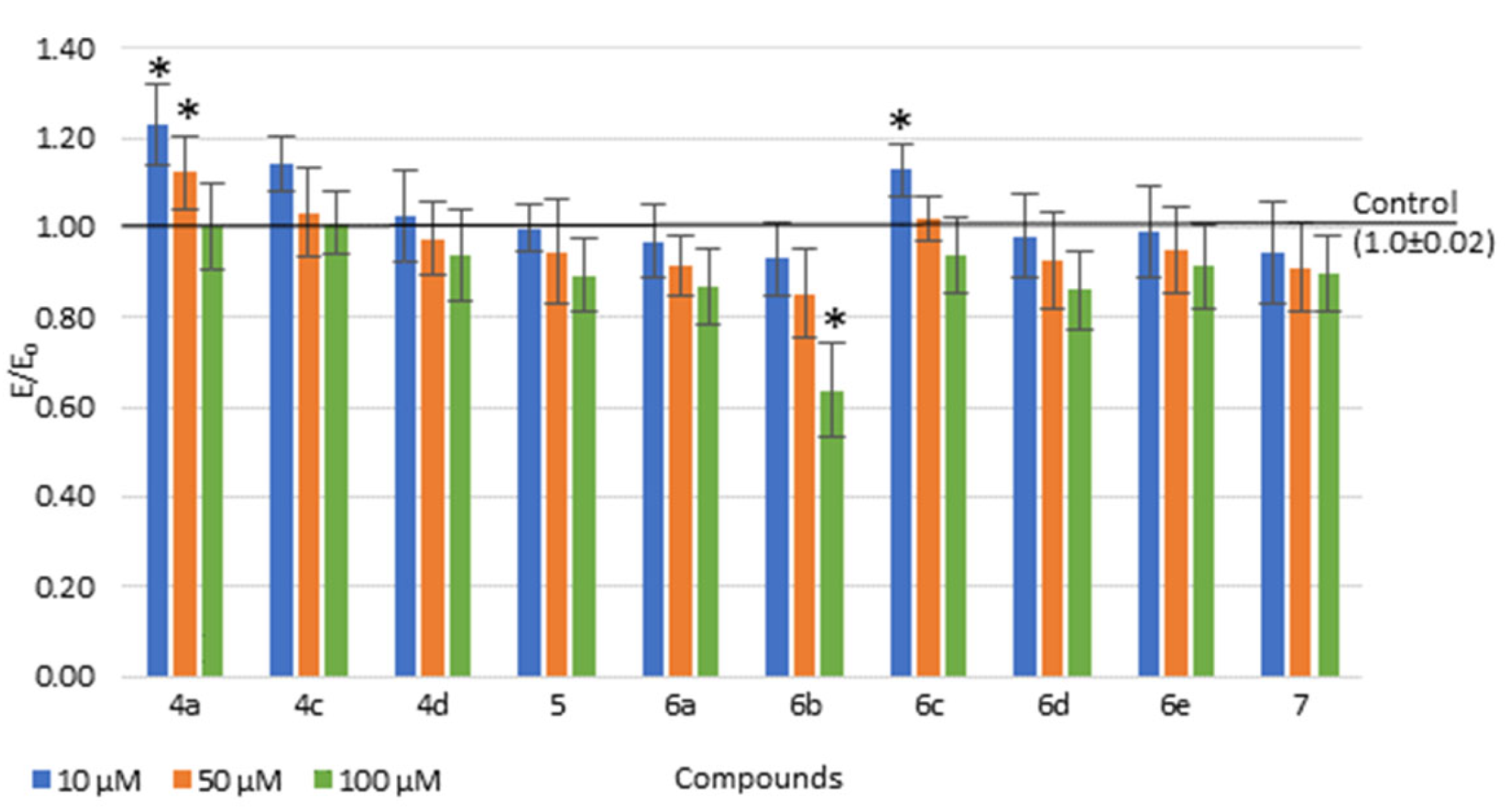
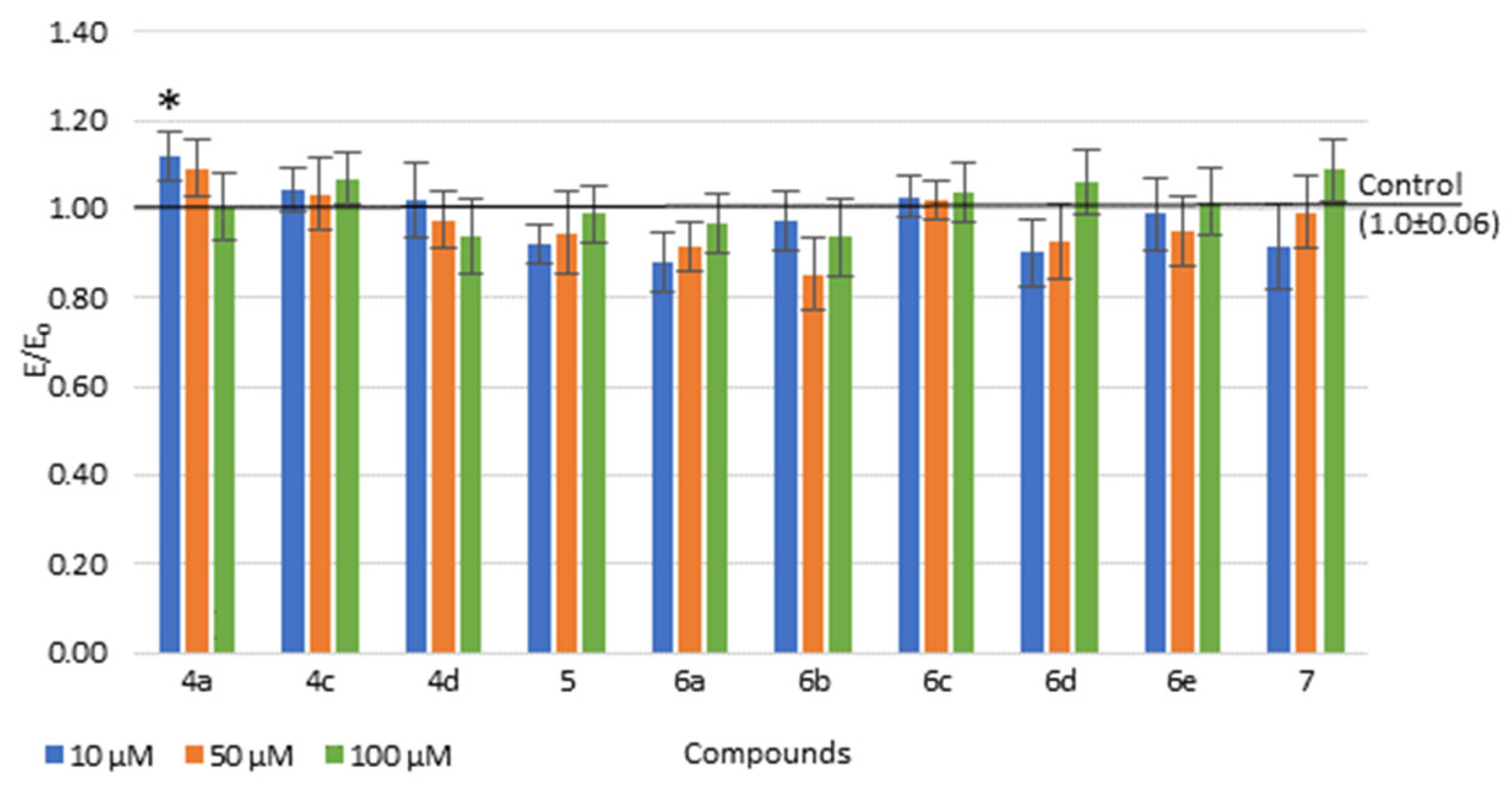
2.4.1. Viability of Cell Cultures
2.4.2. The COX Colorimetric Inhibitor Screening Assay
| Compound | IC50 [µM] | Ratio: COX-2/COX-1 | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| 4a | 91.2 (0.18) | 54.6 (0.06) | 0.6 |
| 4c | 89.0 (0.16) | 55.9 (0.08) | 0.63 |
| 4d | 89.3 (0.09) | 55.3 (0.10) | 0.62 |
| 5 | 66.1 (0.08) | 56.9 (0.13) | 0.86 |
| 6a | 105.5 (0.15) | 89.9 (0.14) | 0.85 |
| 6b | 86.9 (0.15) | 95.1(0.10) | 1.09 |
| 6c | 89.8 (0.14) | 94.1 (0.12) | 1.05 |
| 6d | 125.7 (0.07) | 92.9 (0.09) | 0.74 |
| 6e | 115.3 (0.11) | 56.9 (0.06) | 0.49 |
| 7 | 86.1 (0.08) | 54.0 (0.09) | 0.63 |
| meloxicam | 83.7 (0.10) | 59.2 (0.12) | 0.71 |
2.4.3. Reactive Oxygen Species (ROS) and Nitric Oxide (NO)
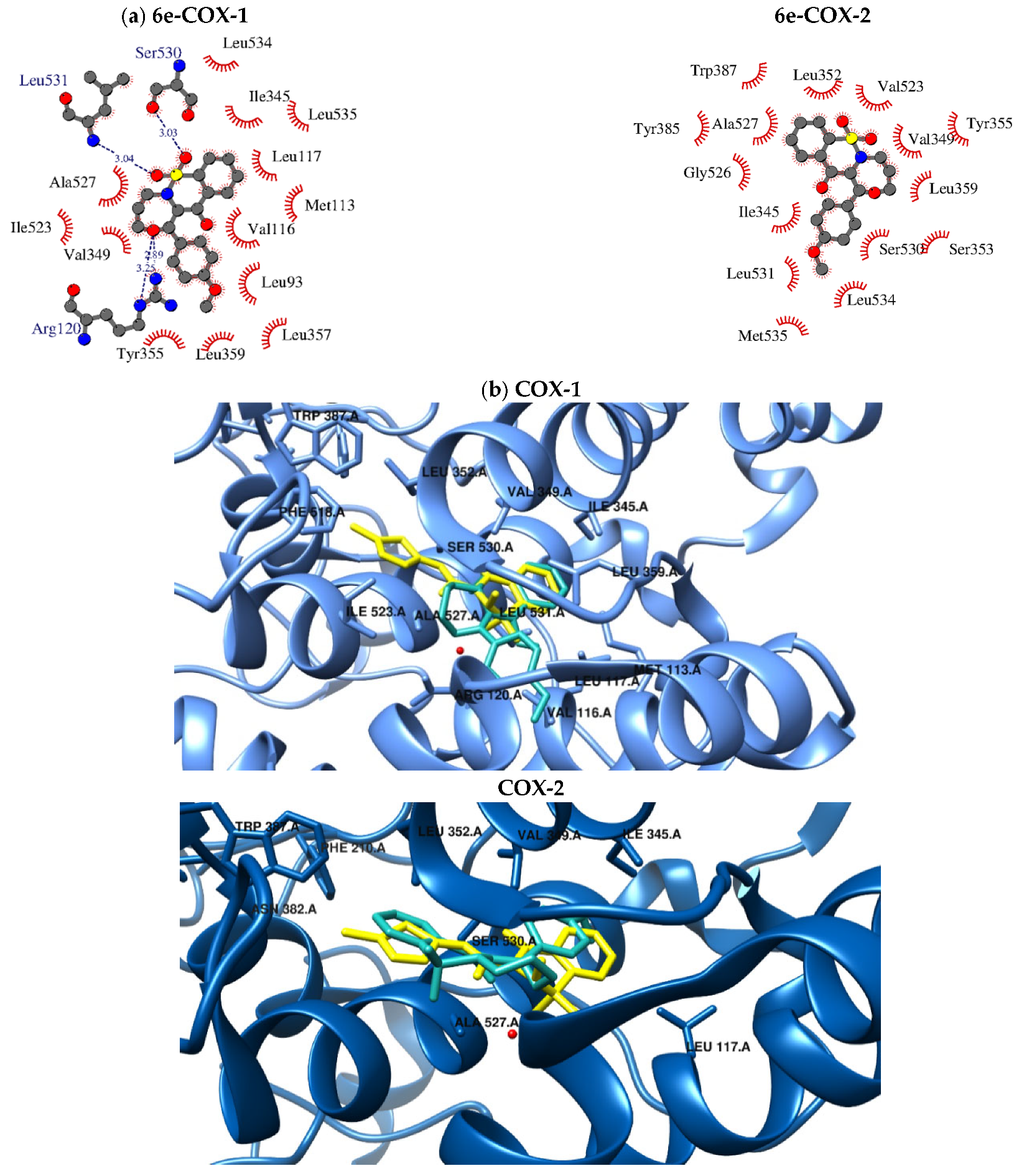
2.5. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. The Microwave Synthesis of Compounds 3a–3e
3.1.2. Synthesis and Experimental Data of Tricyclic 1,2-Benzothiazine Derivatives (5, 6a–6e and 7)
6,6-dioxo-1-phenyl-3,4-dihydro-[1,4] oxazino [4,3-b][1,2]benzothiazin-11-one (5)
7,7-dioxo-1-phenyl-4,5-dihydro-3H-[1,4]oxazepino[4,3-b][1,2]benzothiazin-12-one (6a)
1-(4-bromophenyl)-7,7-dioxo-4,5-dihydro-3H-[1,4]oxazepino[4,3-b][1,2]benzothiazin-12-one (6b)
1-(4-chlorophenyl)-7,7-dioxo-4,5-dihydro-3H-[1,4]oxazepino[4,3-b][1,2]benzothiazin-12-one (6c)
7,7-dioxo-1-(p-tolyl)-4,5-dihydro-3H-[1,4]oxazepino[4,3-b][1,2]benzothiazin-12-one (6d)
1-(4-methoxyphenyl)-7,7-dioxo-4,5-dihydro-3H-[1,4]oxazepino[4,3-b][1,2]benzothiazin-12-one (6e)
8,8-dioxo-1-phenyl-3,4,5,6-tetrahydro-[1,4]oxazocino[4,3-b][1,2]benzothiazin-13-one (7)
3.2. Interaction with Model Membranes
3.2.1. Chemicals
3.2.2. Differential Scanning Calorimetry (DSC)
3.3. Biological Assay
3.3.1. Cell Line
Cell Culture Media
Tested Compounds for Viability and ROS/NO Studies
3.3.2. Viability of Cell Cultures
3.3.3. Reactive Oxygen Species (ROS) and Nitric Oxide (NO)
3.3.4. The COX Colorimetric Inhibitor Screening Assay
3.3.5. Statistical Analysis
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Pahwa, R.; Goyal, A.; Bansal, P.; Jialal, I. Chronic Inflammation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: www.ncbi.nlm.nih.gov/books/NBK493173/ (accessed on 7 June 2021).
- Liu, B.; Qu, L.; Tao, H. Cyclo-oxygenase 2 up-regulates the effect of multidrug resistance. Cell Biol. Int. 2010, 34, 21–25. [Google Scholar]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 1–26. [Google Scholar] [CrossRef]
- Mims, J.W. Asthma: Definitions and pathophysiology. Int. Forum Allergy Rhinol. 2015, 5, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- DeMizio, D.J.; Geraldino-Pardilla, L.B. Autoimmunity and Inflammation Link to Cardiovascular Disease Risk in Rheumatoid Arthritis. Rheumatol. Ther. 2020, 7, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Huh, Y.; Ji, R.R. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J. Anesth. 2019, 33, 131–139. [Google Scholar] [CrossRef]
- Piotrowski, I.; Kulcenty, K.; Suchorska, W. Interplay between inflammation and cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Khatami, M. Chronic inflammation: Synergistic interactions of recruiting macrophages (TAMs) and eosinophils (EoS) with host mast cells (MCs) and tumorigenesis in CALTs. M-CSF, suitable biomarker for cancer diagnosis. Cancers 2014, 6, 297–322. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 809–824. [Google Scholar]
- Ricciotti, E.; Fitzgerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef]
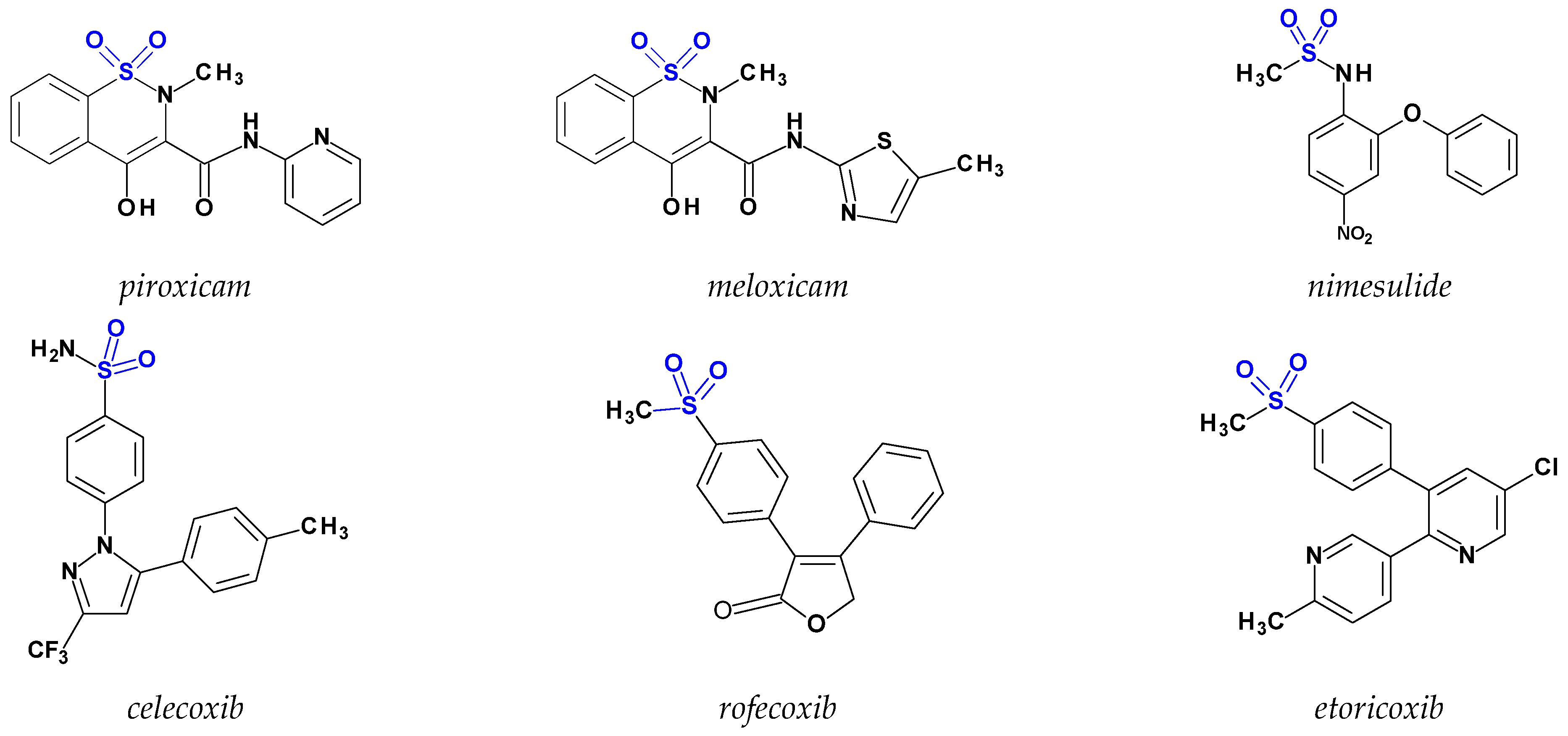
- Dhingra, A.K.; Chopra, B.; Dass, R.; Mittal, S.K. A review on COX and their inhibitors: Present and future. Innov. Pharm. Pharmacother. 2014, 2, 470–485. [Google Scholar]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.T.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, S.M.I.M.; Zarghi, A. Selective COX-2 inhibitors as anticancer agents: A patent review (2014–2018). Expert Opin. Ther. Pat. 2019, 29, 407–427. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.; Gębczak, K.; Gębarowski, T.; Maniewska, J. Synthesis, COX-1/2 inhibition and antioxidant activities of new oxicam analogues designed as potential chemopreventive agents. Acta Biochim. Pol. 2018, 65, 199–207. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Lee, W.S.; Shin, J.S.; Oh, C.H.; Lee, K.T.; Choi, J.; Myoung, N.; Baek, D. Synthesis of New Tricyclic and Tetracyclic Fused Coumarin Sulfonate Derivatives and Their Inhibitory Effects on LPS-Induced Nitric Oxide and PGE2 Productions in RAW 264.7 Macrophages: Part 2. Arch. Pharm. 2016, 349, 853–863. [Google Scholar] [CrossRef] [PubMed]
- El-Kenawi, A.; Ruffell, B. Inflammation, ROS, and Mutagenesis. Cancer Cell 2017, 32, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Cuzzocrea, S. Superoxide, NO, peroxynitrite and PARP in circulatory shock and inflammation. Front. Biosci. 2009, 14, 263–296. [Google Scholar] [CrossRef] [PubMed]
- Krzyżak, E.; Szczęśniak-Siȩga, B.; Malinka, W. Synthesis and thermal behaviour of new benzo-1,2-thiazine long-chain aryl-piperazine derivatives. J. Therm. Anal. Calorim. 2014, 115, 793–802. [Google Scholar] [CrossRef][Green Version]
- Yoshimura, A.; Koski, S.R.; Fuchs, J.M.; Saito, A.; Nemykin, V.N.; Zhdankin, V.V. Saccharin-based μ-oxo imidoiodane: A readily available and highly reactive reagent for electrophilic amination. Chem. A Eur. J. 2015, 21, 5328–5331. [Google Scholar] [CrossRef] [PubMed]
- Maniewska, J.; Szczęśniak-Sięga, B.; Poła, A.; Środa-Pomianek, K.; Malinka, W.; Michalak, K. The interaction of new piroxicam analogues with lipid bilayers—A calorimetric and fluorescence spectroscopic study. Acta Pol. Pharm. Drug Res. 2014, 71, 1004–1012. [Google Scholar]
- Chmiel, T.; Mieszkowska, A.; Kempińska-Kupczyk, D.; Kot-Wasik, A.; Namieśnik, J.; Mazerska, Z. The impact of lipophilicity on environmental processes, drug delivery and bioavailability of food components. Microchem. J. 2019, 146, 393–406. [Google Scholar] [CrossRef]
- Potemkin, V.; Galimova, O.; Grishina, M. Cinderella’s Shoe for virtual drug discovery screening and design. Drug Future 2010, 35, 14–15. [Google Scholar]
- Kyrikou, I.; Hadjikakou, S.K.; Kovala-Demertzi, D.; Viras, K.; Mavromoustakos, T. Effects of non-steroid anti-inflammatory drugs in membrane bilayers. Chem. Phys. Lipids 2004, 132, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Maniewska, J.; Gąsiorowska, J.; Szczęśniak-Sięga, B.; Michalak, K. The interaction of new oxicam derivatives with lipid bilayers as measured by calorimetry and fluorescence spectroscopy. Acta Biochim. Pol. 2018, 65, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Szczęśniak-Sięga, B.M.; Wiatrak, B.; Czyżnikowska, Ż.; Janczak, J.; Wiglusz, R.J.; Maniewska, J. Synthesis and biological evaluation as well as in silico studies of arylpiperazine-1,2-benzothiazine derivatives as novel anti-inflammatory agents. Bioorganic Chem. 2021, 106, 104476. [Google Scholar] [CrossRef]
- Jain, M.K.; Wu, N.M. Effect of small molecules on the dipalmitoyl lecithin liposomal bilayer: III. Phase transition in lipid bilayer. J. Membr. Biol. 1977, 34, 157–201. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to Isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.02 2016; Gaussian, Inc.: Wallingford CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated h-bonding network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maniewska, J.; Wiatrak, B.; Czyżnikowska, Ż.; Szczęśniak-Sięga, B.M. Synthesis of New Tricyclic 1,2-Thiazine Derivatives with Anti-Inflammatory Activity. Int. J. Mol. Sci. 2021, 22, 7818. https://doi.org/10.3390/ijms22157818
Maniewska J, Wiatrak B, Czyżnikowska Ż, Szczęśniak-Sięga BM. Synthesis of New Tricyclic 1,2-Thiazine Derivatives with Anti-Inflammatory Activity. International Journal of Molecular Sciences. 2021; 22(15):7818. https://doi.org/10.3390/ijms22157818
Chicago/Turabian StyleManiewska, Jadwiga, Benita Wiatrak, Żaneta Czyżnikowska, and Berenika M. Szczęśniak-Sięga. 2021. "Synthesis of New Tricyclic 1,2-Thiazine Derivatives with Anti-Inflammatory Activity" International Journal of Molecular Sciences 22, no. 15: 7818. https://doi.org/10.3390/ijms22157818
APA StyleManiewska, J., Wiatrak, B., Czyżnikowska, Ż., & Szczęśniak-Sięga, B. M. (2021). Synthesis of New Tricyclic 1,2-Thiazine Derivatives with Anti-Inflammatory Activity. International Journal of Molecular Sciences, 22(15), 7818. https://doi.org/10.3390/ijms22157818