
Roles of PRR-Mediated Signaling Pathways in the Regulation of Oxidative Stress and Inflammatory Diseases



Abstract
:1. Introduction
2. Oxidative Stress Mediated by PRR-Dependent Pathways
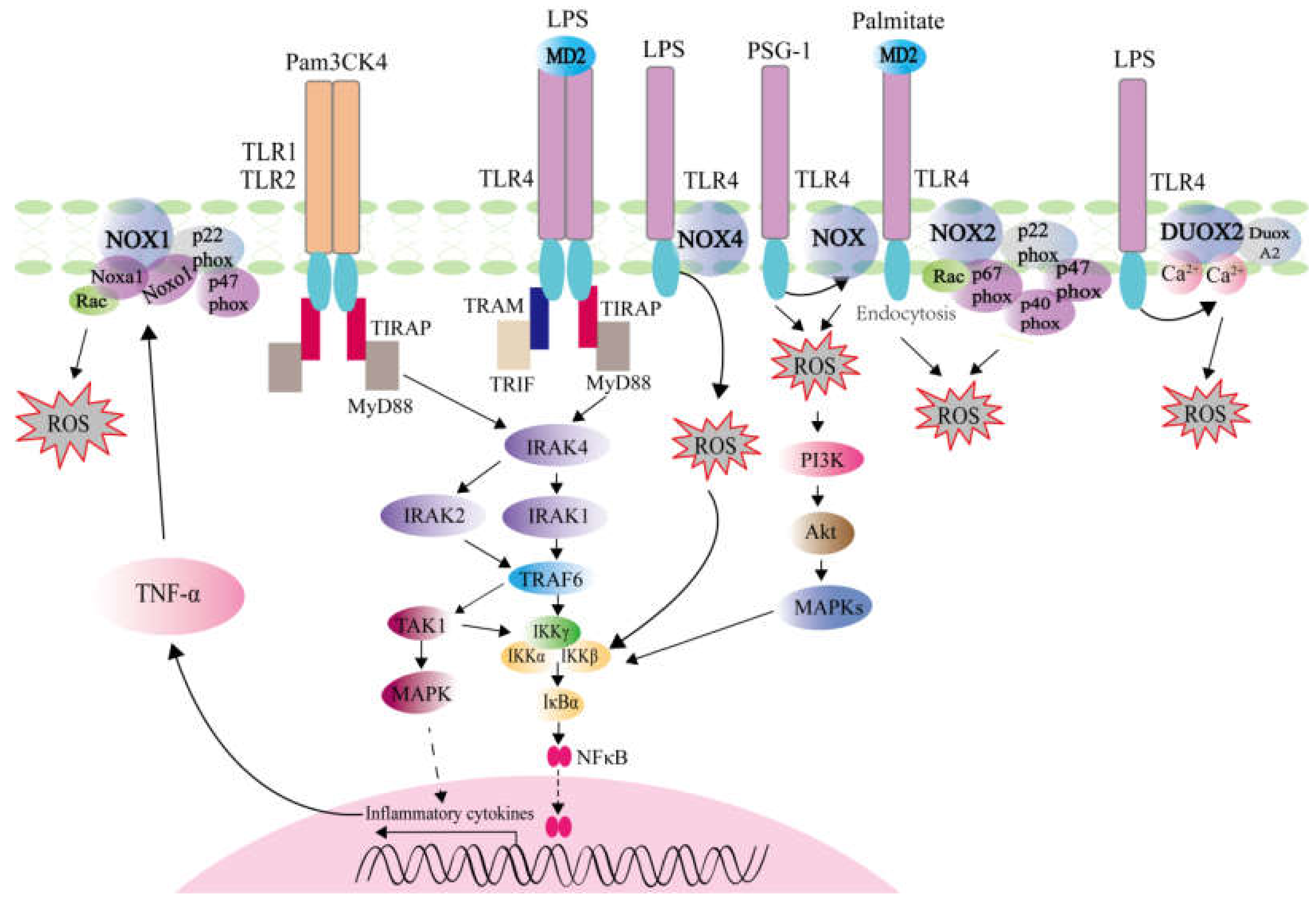
2.1. ROS Generation Mediated by TLRs Activation
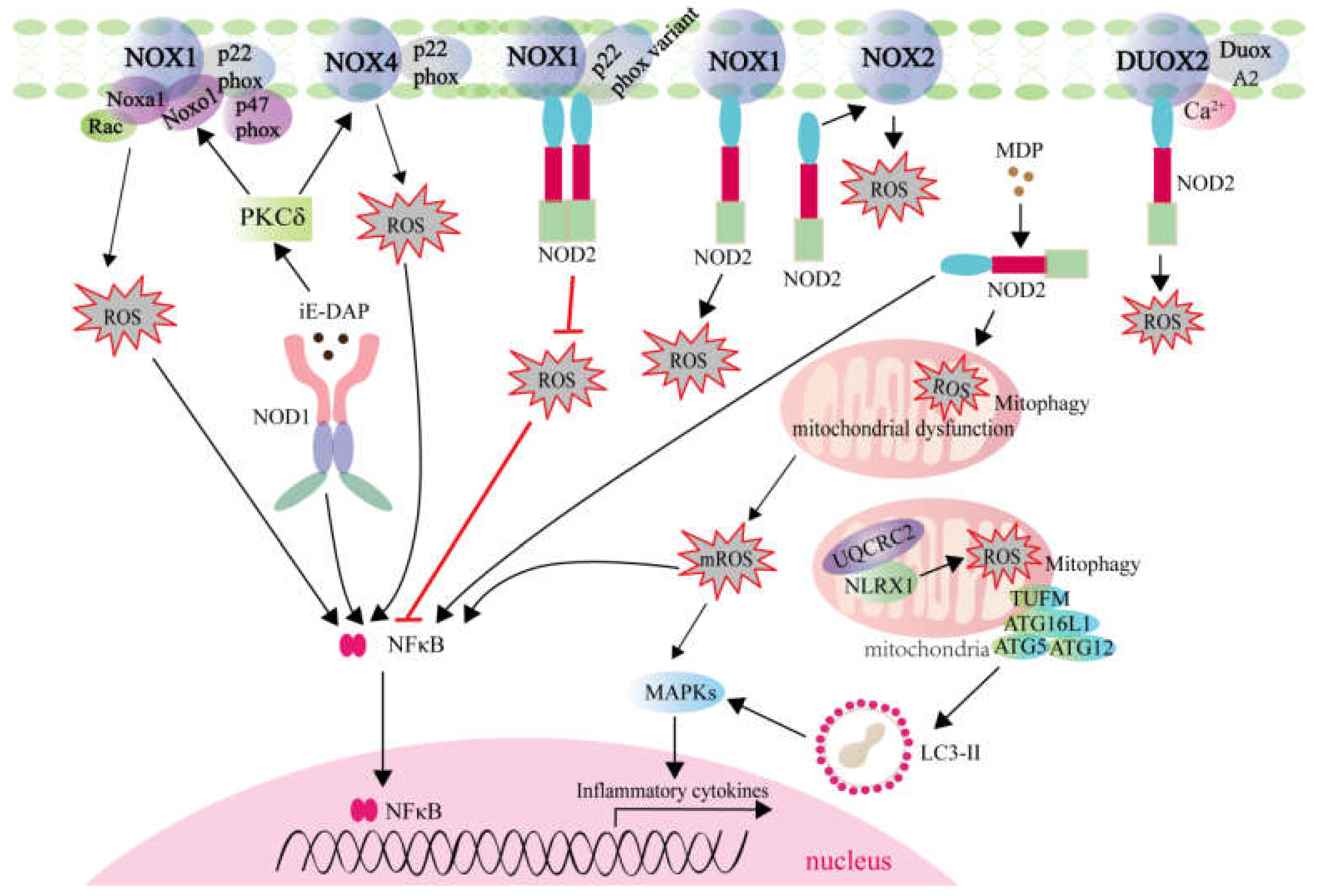
2.2. ROS Generation Mediated by NLRs Activation
2.2.1. ROS Generation Mediated by NLRs Involving in Signaling Transduction and Autophagy
2.2.2. ROS Generation Mediated by NLRs Involving in Transcription Activation
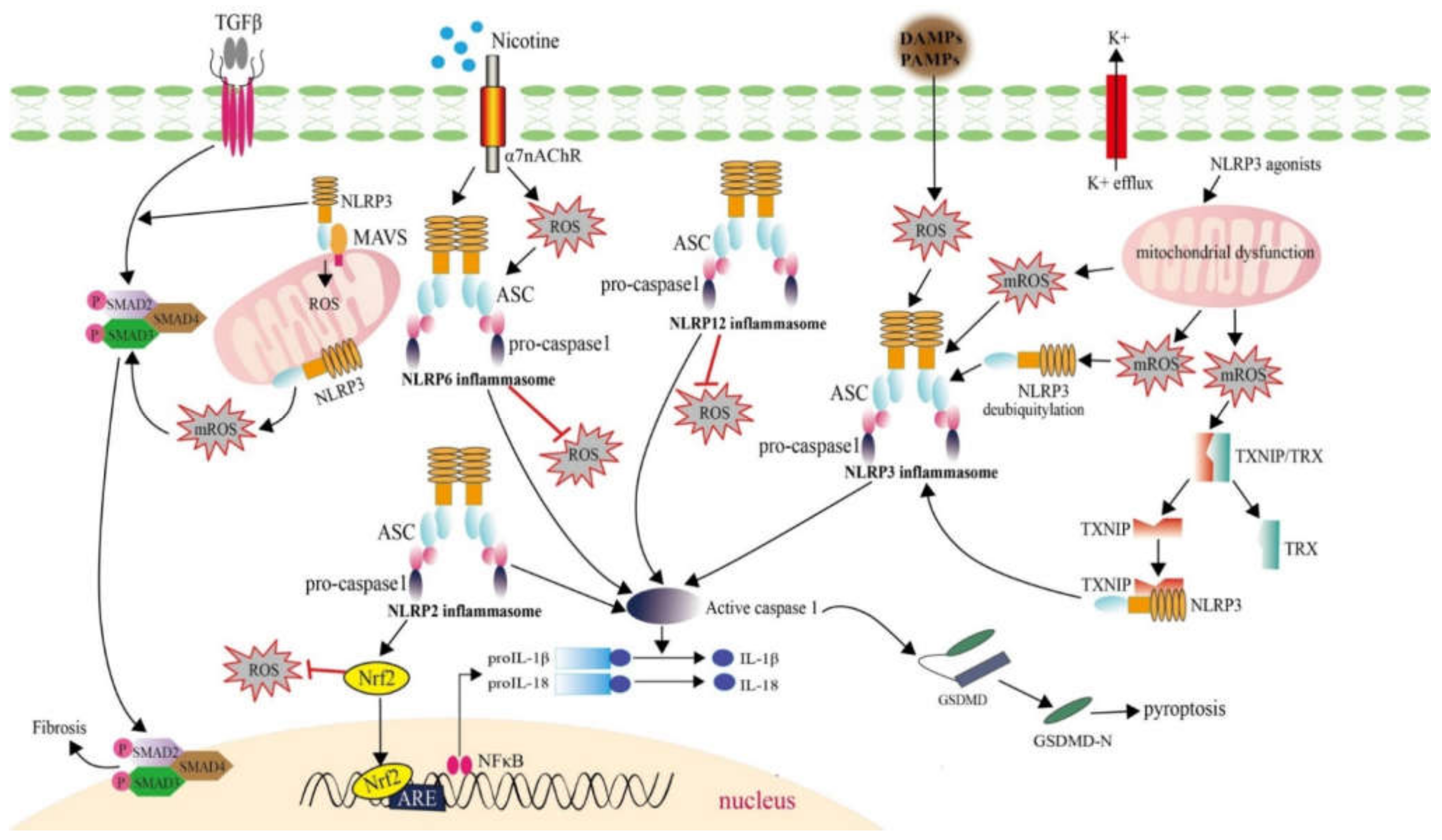
2.2.3. ROS Generation Mediated by NLRs Involving in Inflammasome Formation
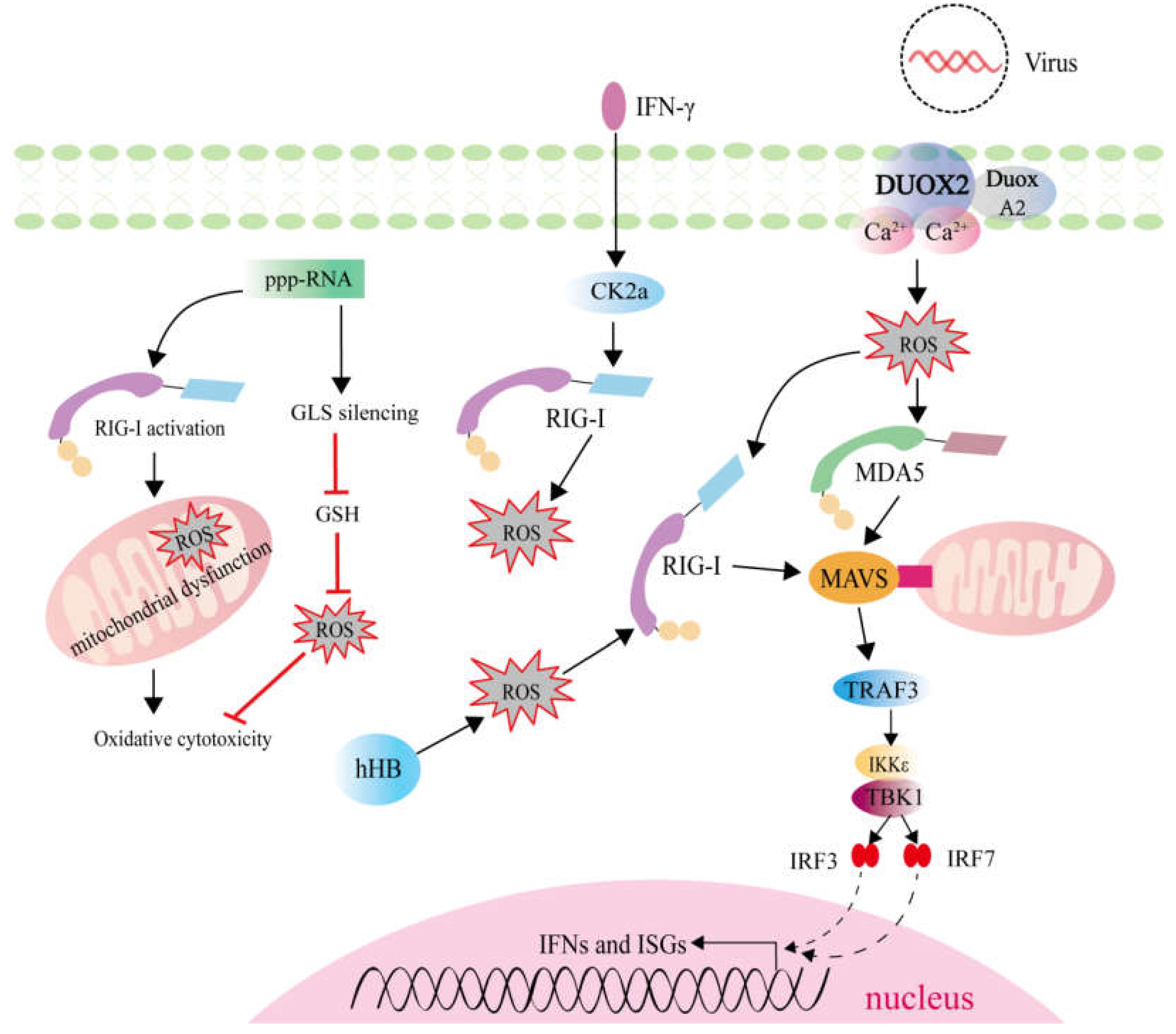
2.3. ROS Generation Mediated by RLRs Activation
3. Therapeutic Targets for Suppressing Inflammatory Diseases Based on the Correlations between ROS Regulation and PRR-Mediated Signaling Pathways
3.1. Therapeutic Targets for Inflammatory Vascular Diseases
3.2. Therapeutic Targets for Inflammatory Neurological Diseases
3.3. Therapeutic Targets for Inflammatory Bowel Diseases
4. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Tate, M.D.; Ong, J.D.H.; Dowling, J.K.; McAuley, J.L.; Robertson, A.B.; Latz, E.; Drummond, G.R.; Cooper, M.A.; Hertzog, P.J.; Mansell, A. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci. Rep. 2016, 6, 27912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.S.; Marques, G.G.; Dellama, J.E.; Zamboni, D.S. The Nlrc4 Inflammasome Contributes to Restriction of Pulmonary Infection by Flagellated Legionella spp. that Trigger Pyroptosis. Front. Microbiol. 2011, 2, 33. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.S.; Morgantetti, G.F.; Massis, L.M.; Horta, C.V.; Hori, J.I.; Zamboni, D.S. Activation of NLRC4 by flagellated bacteria triggers caspase-1-dependent and -independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. J. Immunol. 2011, 187, 6447–6455. [Google Scholar] [CrossRef] [Green Version]
- Semper, R.P.; Vieth, M.; Gerhard, M.; Mejías-Luque, R. Helicobacter pylori Exploits the NLRC4 Inflammasome to Dampen Host Defenses. J. Immunol. 2019, 203, 2183–2193. [Google Scholar] [CrossRef]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef] [Green Version]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant therapy: Current status and future prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharm. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Dryden, M. Reactive oxygen species: A novel antimicrobial. Int. J. Antimicrob. Agents 2018, 51, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; He, X.; Wang, H.; Wang, Z.; Kelly, G.T.; Wang, X.; Chen, Y.; Wang, T.; Qian, Z. TLR4-NOX2 axis regulates the phagocytosis and killing of Mycobacterium tuberculosis by macrophages. BMC Pulm. Med. 2017, 17, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Acker, H.; Coenye, T. The Role of Reactive Oxygen Species in Antibiotic-Mediated Killing of Bacteria. Trends Microbiol. 2017, 25, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Prince, L.R.; Whyte, M.K.; Sabroe, I.; Parker, L.C. The role of TLRs in neutrophil activation. Curr. Opin. Pharm. 2011, 11, 397–403. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Palmai-Pallag, T.; Bachrati, C.Z. Inflammation-induced DNA damage and damage-induced inflammation: A vicious cycle. Microbes Infect. 2014, 16, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Lim, K.H.; Staudt, L.M. Toll-like receptor signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a011247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leifer, C.A.; Medvedev, A.E. Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Deng, S.L.; Lian, Z.X.; Yu, K. Roles of Toll-Like Receptors in Nitroxidative Stress in Mammals. Cells 2019, 8, 576. [Google Scholar] [CrossRef] [Green Version]
- Ogier-Denis, E.; Mkaddem, S.B.; Vandewalle, A. NOX enzymes and Toll-like receptor signaling. Semin. Immunopathol. 2008, 30, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Echizen, K.; Oshima, H.; Nakayama, M.; Oshima, M. The inflammatory microenvironment that promotes gastrointestinal cancer development and invasion. Adv. Biol. Regul. 2018, 68, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Joo, J.H.; Kim, J.; Lim, H.J.; Kim, S.; Curtiss, L.; Seong, J.K.; Cui, W.; Yabe-Nishimura, C.; Bae, Y.S. Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc. Res. 2013, 99, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pei, C.; Yan, S.; Liu, G.; Liu, G.; Chen, W.; Cui, Y.; Liu, Y. NADPH oxidase 1-dependent ROS is crucial for TLR4 signaling to promote tumor metastasis of non-small cell lung cancer. Tumour Biol. 2015, 36, 1493–1502. [Google Scholar] [CrossRef]
- Lee, A.J.; Cho, K.J.; Kim, J.H. MyD88-BLT2-dependent cascade contributes to LPS-induced interleukin-6 production in mouse macrophage. Exp. Mol. Med. 2015, 47, e156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Yeo, S.; Shin, D.G.; Bae, Y.S.; Lee, J.J.; Chin, B.R.; Lee, C.H.; Baek, S.H. Glycogen synthase kinase 3beta and beta-catenin pathway is involved in toll-like receptor 4-mediated NADPH oxidase 1 expression in macrophages. FEBS J. 2010, 277, 2830–2837. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeong, J.M.; Kim, S.J.; Seo, W.; Kim, M.H.; Choi, W.M.; Yoo, W.; Lee, J.H.; Shim, Y.R.; Yi, H.S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun. 2017, 8, 2247. [Google Scholar] [CrossRef] [PubMed]
- Trevelin, S.C.; Shah, A.M.; Lombardi, G. Beyond bacterial killing: NADPH oxidase 2 is an immunomodulator. Immunol. Lett. 2020, 221, 39–48. [Google Scholar] [CrossRef]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Nie, S.P.; Wang, J.Q.; Yin, P.F.; Huang, D.F.; Li, W.J.; Xie, M.Y. Toll-like receptor 4-mediated ROS signaling pathway involved in Ganoderma atrum polysaccharide-induced tumor necrosis factor-α secretion during macrophage activation. Food Chem. Toxicol. 2014, 66, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Burgueno, J.F.; Fritsch, J.; Gonzalez, E.E.; Landau, K.S.; Santander, A.M.; Fernandez, I.; Hazime, H.; Davies, J.M.; Santaolalla, R.; Phillips, M.C.; et al. Epithelial TLR4 Signaling Activates DUOX2 to Induce Microbiota-Driven Tumorigenesis. Gastroenterology 2021, 160, 797–808.e6. [Google Scholar] [CrossRef]
- Wang, J.; Chai, J. Structural Insights into the Plant Immune Receptors PRRs and NLRs. Plant Physiol. 2020, 182, 1566–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutermarsh-Ott, S.; Eden, K.; Allen, I.C. Beyond the inflammasome: Regulatory NOD-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol. 2016, 97, 825–838. [Google Scholar] [CrossRef]
- Tan, X.; Li, W.W.; Guo, J.; Zhou, J.Y. Down-regulation of NOD1 in neutrophils of periparturient dairy cows. Vet. Immunol. Immunopathol. 2012, 150, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, S.; Ahmad, S.; Gulzar, F.; Schertzer, J.D.; Tamrakar, A.K. NOD1 activation induces oxidative stress via NOX1/4 in adipocytes. Free Radic. Biol. Med. 2021, 162, 118–128. [Google Scholar] [CrossRef]
- Ke, K.; Sul, O.J.; Chung, S.W.; Suh, J.H.; Choi, H.S. Lack of NOD2 attenuates ovariectomy-induced bone loss via inhibition of osteoclasts. J. Endocrinol. 2017, 235, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, S.; Till, A.; Sina, C.; Arlt, A.; Grasberger, H.; Schreiber, S.; Rosenstiel, P. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci. 2009, 122, 3522–3530. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, S.; Petersen, B.S.; Barann, M.; Piecyk, A.; Tran, F.; Mayr, G.; Jentzsch, M.; Aden, K.; Stengel, S.T.; Klostermeier, U.C.; et al. Missense variants in NOX1 and p22phox in a case of very-early-onset inflammatory bowel disease are functionally linked to NOD2. Cold Spring Harb. Mol. Case Stud. 2019, 5. [Google Scholar] [CrossRef]
- Liu, H.; Wei, X.; Kong, L.; Liu, X.; Cheng, L.; Yan, S.; Zhang, X.; Chen, L. NOD2 is involved in the inflammatory response after cerebral ischemia-reperfusion injury and triggers NADPH oxidase 2-derived reactive oxygen species. Int. J. Biol. Sci. 2015, 11, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.J.; Liu, X.Q.; Xue, Y.; Gao, W.; Lv, Q.Z. Muramyl Dipeptide Induces Reactive Oxygen Species Generation Through the NOD2/COX-2/NOX4 Signaling Pathway in Human Umbilical Vein Endothelial Cells. J. Cardiovasc. Pharm. 2018, 71, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Maurya, C.K.; Arha, D.; Rai, A.K.; Kumar, S.K.; Pandey, J.; Avisetti, D.R.; Kalivendi, S.V.; Klip, A.; Tamrakar, A.K. NOD2 activation induces oxidative stress contributing to mitochondrial dysfunction and insulin resistance in skeletal muscle cells. Free Radic. Biol. Med. 2015, 89, 158–169. [Google Scholar] [CrossRef]
- Levy, A.; Stedman, A.; Deutsch, E.; Donnadieu, F.; Virgin, H.W.; Sansonetti, P.J.; Nigro, G. Innate immune receptor NOD2 mediates LGR5(+) intestinal stem cell protection against ROS cytotoxicity via mitophagy stimulation. Proc. Natl. Acad. Sci. USA 2020, 117, 1994–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, S.D.; Dikshit, N.; Kumar, P.; Balamuralidhar, V.; Khameneh, H.J.; Bin Abdul Malik, N.; Koh, T.H.; Tan, G.G.Y.; Tan, T.T.; Mortellaro, A.; et al. Nod2 is required for the early innate immune clearance of Acinetobacter baumannii from the lungs. Sci. Rep. 2017, 7, 17429. [Google Scholar] [CrossRef] [Green Version]
- Tattoli, I.; Carneiro, L.A.; Jéhanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293–300. [Google Scholar] [CrossRef]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, S.C.; Huang, P.R.; Almeida-da-Silva, C.L.C.; Atanasova, K.R.; Yilmaz, O.; Ojcius, D.M. NLRX1 modulates differentially NLRP3 inflammasome activation and NF-κB signaling during Fusobacterium nucleatum infection. Microbes Infect. 2018, 20, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Soares, F.; Tattoli, I.; Castanier, C.; Philpott, D.J.; Girardin, S.E. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 2009, 122, 3161–3168. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Poteryakhina, A.; Zheltukhin, A.; Bhatelia, K.; Prajapati, P.; Sripada, L.; Tomar, D.; Singh, R.; Singh, A.K.; Chumakov, P.M.; et al. NLRX1 acts as tumor suppressor by regulating TNF-α induced apoptosis and metabolism in cancer cells. Biochim. Biophys. Acta 2015, 1853, 1073–1086. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Yang, Q.; Cao, Z.; Li, H.; Yu, Z.; Zhang, G.; Sun, G.; Man, R.; Wang, H.; Li, J. Activation of NLRX1-mediated autophagy accelerates the ototoxic potential of cisplatin in auditory cells. Toxicol. Appl. Pharm. 2018, 343, 16–28. [Google Scholar] [CrossRef]
- Ma, S.R.; Xie, X.W. NLRC5 deficiency promotes myocardial damage induced by high fat diet in mice through activating TLR4/NF-κB. Biomed. Pharm. 2017, 91, 755–766. [Google Scholar] [CrossRef]
- Li, L.; Yu, M.; Pang, H.; Chen, L.; Liu, J.; Hou, S. NLRC5 protects neurons from oxygen-glucose deprivation-induced injury through activating the Nrf2/HO-1 pathway. J. Recept. Signal. Transduct. Res. 2021, 41, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, Y.; Chen, X. NLRC5 alleviated OGD/R-induced PC12-cell injury by inhibiting activation of the TLR4/MyD88/NF-κB pathway. J. Int. Med. Res. 2020, 48, 300060520940455. [Google Scholar] [CrossRef] [PubMed]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Li, C.; Liu, Q.; Xie, L. Suppressing NLRP2 expression accelerates hepatic steatosis: A mechanism involving inflammation and oxidative stress. Biochem. Biophys. Res. Commun. 2018, 507, 22–29. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Nunez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Alyaseer, A.A.A.; de Lima, M.H.S.; Braga, T.T. The Role of NLRP3 Inflammasome Activation in the Epithelial to Mesenchymal Transition Process during the Fibrosis. Front. Immunol. 2020, 11, 883. [Google Scholar] [CrossRef]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharm. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Loeches, S.; Urena-Peralta, J.R.; Morillo-Bargues, M.J.; Oliver-De La Cruz, J.; Guerri, C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell Neurosci. 2014, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Tan, X.; Zhou, Q.; Zhu, Y.; Tian, Y.; Yu, H.; Kijlstra, A.; Yang, P. IL-1beta triggered by peptidoglycan and lipopolysaccharide through TLR2/4 and ROS-NLRP3 inflammasome-dependent pathways is involved in ocular Behcet’s disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 402–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Zuo, D.; Yu, L.; Zhang, L.; Tang, J.; Cui, C.; Bao, L.; Zan, K.; Zhang, Z.; Yang, X.; et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017, 485, 499–505. [Google Scholar] [CrossRef]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R., Jr.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Kern, L.; Elinav, E. The NLRP6 inflammasome. Immunology 2021, 162, 281–289. [Google Scholar] [CrossRef]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Núñez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664.e1614. [Google Scholar] [CrossRef] [Green Version]
- Tuladhar, S.; Kanneganti, T.D. NLRP12 in innate immunity and inflammation. Mol. Asp. Med. 2020, 76, 100887. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, F.; Zhang, M.; Zhou, P.; Xu, C.; Li, Y.; Bian, L.; Liu, Y.; Yao, Y.; Wang, F.; et al. NLRP12 Promotes Mouse Neutrophil Differentiation through Regulation of Non-canonical NF-κB and MAPK(ERK1/2) Signaling. Int. J. Biol. Sci. 2018, 14, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Chen, H.; Jiang, R.; Zhang, L.; Wang, L.; Gan, H.; Jiang, N.; Zhao, J.; Zhai, X.; Liang, P. NLRP6 contributes to inflammation and brain injury following intracerebral haemorrhage by activating autophagy. J. Mol. Med. 2020, 98, 1319–1331. [Google Scholar] [CrossRef]
- Ghimire, L.; Paudel, S.; Jin, L.; Baral, P.; Cai, S.; Jeyaseelan, S. NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. PLoS Pathog. 2018, 14, e1007308. [Google Scholar] [CrossRef]
- Borghini, S.; Tassi, S.; Chiesa, S.; Caroli, F.; Carta, S.; Caorsi, R.; Fiore, M.; Delfino, L.; Lasigliè, D.; Ferraris, C.; et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. 2011, 63, 830–839. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Asdonk, T.; Motz, I.; Werner, N.; Coch, C.; Barchet, W.; Hartmann, G.; Nickenig, G.; Zimmer, S. Endothelial RIG-I activation impairs endothelial function. Biochem. Biophys. Res. Commun. 2012, 420, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, R.; Sen, E. CK2 induced RIG-I drives metabolic adaptations in IFNγ-treated glioma cells. Cytokine 2017, 89, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Xia, M.; Xu, C.; Yuan, D.; Schnurr, M.; Wei, J. Multifunctional antitumor molecule 5’-triphosphate siRNA combining glutaminase silencing and RIG-I activation. Int. J. Cancer 2014, 134, 1958–1971. [Google Scholar] [CrossRef]
- Yang, Q.; Bai, S.Y.; Li, L.F.; Li, S.; Zhang, Y.; Munir, M.; Qiu, H.J. Human Hemoglobin Subunit Beta Functions as a Pleiotropic Regulator of RIG-I/MDA5-Mediated Antiviral Innate Immune Responses. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, C.H.; Kim, M.J.; Ryu, J.H.; Seong, S.Y.; Kim, S.; Lim, S.J.; Holtzman, M.J.; Yoon, J.H. The Induction of Pattern-Recognition Receptor Expression against Influenza A Virus through Duox2-Derived Reactive Oxygen Species in Nasal Mucosa. Am. J. Respir. Cell Mol. Biol. 2015, 53, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Deng, J.; Ye, Z.; Wang, J.; Gou, H.; Liu, W.; Zhao, M.; Liao, M.; Yi, L.; Chen, J. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy 2016, 12, 1738–1758. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jin, Y.; Zeng, N.; Ruan, Q.; Qian, F. SOD2 Facilitates the Antiviral Innate Immune Response by Scavenging Reactive Oxygen Species. Viral Immunol. 2017, 30, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Peng, T.; Zhu, H.; Zheng, X.; Zhang, X.; Jiang, N.; Cheng, X.; Lai, X.; Shunnar, A.; Singh, M.; et al. Prevention of hyperglycemia-induced myocardial apoptosis by gene silencing of Toll-like receptor-4. J. Transl. Med. 2010, 8, 133. [Google Scholar] [CrossRef]
- Pahwa, R.; Nallasamy, P.; Jialal, I. Toll-like receptors 2 and 4 mediate hyperglycemia induced macrovascular aortic endothelial cell inflammation and perturbation of the endothelial glycocalyx. J. Diabetes Complicat. 2016, 30, 563–572. [Google Scholar] [CrossRef]
- Zabad, O.M.; Samra, Y.A.; Eissa, L.A. P-Coumaric acid alleviates experimental diabetic nephropathy through modulation of Toll like receptor-4 in rats. Life Sci. 2019, 238, 116965. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Y.; Mu, N.; Lou, X.; Li, W.; Chen, Y.; Fan, D.; Tan, H. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab. Investig. 2017, 97, 922–934. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, M.; Wang, X.D.; Lu, Z.Y.; Ni, X.L. Peperomin E (PepE) protects against high fat diet-induced atherosclerosis in Apolipoprotein E deficient (ApoE(−/−)) mice through reducing inflammation via the suppression of NLRP3 signaling pathway. Biomed. Pharm. 2018, 105, 862–869. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.M. Parkinson’s disease: A review. Front. Biosci. 2014, 6, 65–74. [Google Scholar] [CrossRef]
- Yu, S.; Wang, X.; He, X.; Wang, Y.; Gao, S.; Ren, L.; Shi, Y. Curcumin exerts anti-inflammatory and antioxidative properties in 1-methyl-4-phenylpyridinium ion (MPP(+))-stimulated mesencephalic astrocytes by interference with TLR4 and downstream signaling pathway. Cell Stress Chaperones 2016, 21, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Maghimaa, M.; Chinnathambi, A.; Alharbi, S.A.; Veeraraghavan, V.P.; Mohan, S.K.; Hussain, S.; Rengarajan, T. Tomentosin Reduces Behavior Deficits and Neuroinflammatory Response in MPTP-Induced Parkinson’s Disease in Mice. J. Env. Pathol. Toxicol. Oncol. 2021, 40, 75–84. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 30. [Google Scholar] [CrossRef]
- Lawana, V.; Singh, N.; Sarkar, S.; Charli, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. Involvement of c-Abl Kinase in Microglial Activation of NLRP3 Inflammasome and Impairment in Autolysosomal System. J. Neuroimmune Pharm. 2017, 12, 624–660. [Google Scholar] [CrossRef]
- Yang, C.; Mo, Y.; Xu, E.; Wen, H.; Wei, R.; Li, S.; Zheng, J.; Li, W.; Le, B.; Chen, Y.; et al. Astragaloside IV ameliorates motor deficits and dopaminergic neuron degeneration via inhibiting neuroinflammation and oxidative stress in a Parkinson’s disease mouse model. Int. Immunopharmacol. 2019, 75, 105651. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Fao, L.; Mota, S.I.; Rego, A.C. Shaping the Nrf2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing Res. Rev. 2019, 54, 100942. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Franchimont, D.; Vermeire, S.; El Housni, H.; Pierik, M.; Van Steen, K.; Gustot, T.; Quertinmont, E.; Abramowicz, M.; Van Gossum, A.; Devière, J.; et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut 2004, 53, 987–992. [Google Scholar] [CrossRef] [Green Version]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Muise, A.M.; Xu, W.; Guo, C.H.; Walters, T.D.; Wolters, V.M.; Fattouh, R.; Lam, G.Y.; Hu, P.; Murchie, R.; Sherlock, M.; et al. NADPH oxidase complex and IBD candidate gene studies: Identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut 2012, 61, 1028–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.L.; Hollis-Moffatt, J.E.; Gearry, R.B.; Kennedy, M.A.; Barclay, M.L.; Merriman, T.R. Confirmation of association of IRGM and NCF4 with ileal Crohn’s disease in a population-based cohort. Genes Immun. 2008, 9, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brumell, J.H. NADPH oxidases contribute to autophagy regulation. Autophagy 2009, 5, 887–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Zhou, X.; Zhang, Y.; Pu, W.; Yang, Y.; Wei, F.; Zhou, Q.; Zhang, L.; Du, Z.; Wu, J. Xi Lei San Attenuates Dextran Sulfate Sodium-Induced Colitis in Rats and TNF-α-Stimulated Colitis in CACO2 Cells: Involvement of the NLRP3 Inflammasome and Autophagy. Mediat. Inflamm. 2021, 2021, 1610251. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, W.; Zhang, X.; Lu, P.; Du, Q.; Tao, L.; Ding, Y.; Wang, Y.; Hu, R. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem. Pharm. 2016, 112, 37–49. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Qian, C.; Tang, J.; Zhou, W.; Liu, X.; You, Q.; Hu, R. 3-(2-Oxo-2-phenylethylidene)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (compound 1), a novel potent Nrf2/ARE inducer, protects against DSS-induced colitis via inhibiting NLRP3 inflammasome. Biochem. Pharm. 2016, 101, 71–86. [Google Scholar] [CrossRef]
- Gong, Z.; Zhao, S.; Zhou, J.; Yan, J.; Wang, L.; Du, X.; Li, H.; Chen, Y.; Cai, W.; Wu, J. Curcumin alleviates DSS-induced colitis via inhibiting NLRP3 inflammsome activation and IL-1β production. Mol. Immunol. 2018, 104, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; You, Q.; Hu, L.; Gao, J.; Meng, Q.; Liu, W.; Wu, X.; Xu, Q. The Antioxidant Procyanidin Reduces Reactive Oxygen Species Signaling in Macrophages and Ameliorates Experimental Colitis in Mice. Front. Immunol. 2017, 8, 1910. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yang, Y.; Zhang, J.; Wang, R.; Cheng, B.; Kalambhe, D.; Wang, Y.; Gu, Z.; Chen, D.; Wang, B.; et al. Lactoferrin-mediated macrophage targeting delivery and patchouli alcohol-based therapeutic strategy for inflammatory bowel diseases. Acta Pharm. Sin. B 2020, 10, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mentella, M.C.; Scaldaferri, F.; Pizzoferrato, M.; Gasbarrini, A.; Miggiano, G.A.D. Nutrition, IBD and Gut Microbiota: A Review. Nutrients 2020, 12, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battistini, C.; Ballan, R.; Herkenhoff, M.E.; Saad, S.M.I.; Sun, J. Vitamin D Modulates Intestinal Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2020, 22, 362. [Google Scholar] [CrossRef]
- Castro, F.; de Souza, H.S.P. Dietary Composition and Effects in Inflammatory Bowel Disease. Nutrients 2019, 11, 1398. [Google Scholar] [CrossRef] [Green Version]
- Sugihara, K.; Morhardt, T.L.; Kamada, N. The Role of Dietary Nutrients in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 3183. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Zambetti, L.P.; Mortellaro, A. NLRPs, microbiota, and gut homeostasis: Unravelling the connection. J. Pathol. 2014, 233, 321–330. [Google Scholar] [CrossRef]
- Ringel-Scaia, V.M.; McDaniel, D.K.; Allen, I.C. The Goldilocks Conundrum: NLR Inflammasome Modulation of Gastrointestinal Inflammation during Inflammatory Bowel Disease. Crit. Rev. Immunol. 2016, 36, 283–314. [Google Scholar] [CrossRef]
- Lin, R.; Wang, Z.; Cao, J.; Gao, T.; Dong, Y.; Chen, Y. Role of melatonin in murine “restraint stress”-induced dysfunction of colonic microbiota. J. Microbiol. 2021, 59, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Shan, C.; Ma, T.; Geng, S.; Ning, D. Walnut oil alleviates DSS-induced colitis in mice by inhibiting NLRP3 inflammasome activation and regulating gut microbiota. Microb. Pathog. 2021, 154, 104866. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, S.; Mandrekar, P. Cellular stress response and innate immune signaling: Integrating pathways in host defense and inflammation. J. Leukoc. Biol. 2013, 94, 1167–1184. [Google Scholar] [CrossRef] [Green Version]
- Charlton, A.; Garzarella, J.; Jandeleit-Dahm, K.A.M.; Jha, J.C. Oxidative Stress and Inflammation in Renal and Cardiovascular Complications of Diabetes. Biology 2020, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Spychalowicz, A.; Wilk, G.; Śliwa, T.; Ludew, D.; Guzik, T.J. Novel therapeutic approaches in limiting oxidative stress and inflammation. Curr. Pharm. Biotechnol. 2012, 13, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct Target. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Kahroba, H.; Ramezani, B.; Maadi, H.; Sadeghi, M.R.; Jaberie, H.; Ramezani, F. The role of Nrf2 in neural stem/progenitors cells: From maintaining stemness and self-renewal to promoting differentiation capability and facilitating therapeutic application in neurodegenerative disease. Ageing Res. Rev. 2021, 65, 101211. [Google Scholar] [CrossRef]
- Ramesh, J.; Ronsard, L.; Gao, A.; Venugopal, B. Autophagy Intertwines with Different Diseases-Recent Strategies for Therapeutic Approaches. Diseases 2019, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Onyango, I.G. Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease. Neural Regen Res. 2018, 13, 19–25. [Google Scholar] [CrossRef]
- Nandi, A.; Bishayi, B. Host antioxidant enzymes and TLR-2 neutralization modulate intracellular survival of Staphylococcus aureus: Evidence of the effect of redox balance on host pathogen relationship during acute staphylococcal infection. Microb. Pathog. 2015, 89, 114–127. [Google Scholar] [CrossRef]
- Moghimi, N.; Eslami Farsani, B.; Ghadipasha, M.; Mahmoudiasl, G.R.; Piryaei, A.; Aliaghaei, A.; Abdi, S.; Abbaszadeh, H.A.; Abdollahifar, M.A.; Forozesh, M. COVID-19 disrupts spermatogenesis through the oxidative stress pathway following induction of apoptosis. Apoptosis 2021, 1–16. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.P.; Kolli, D.; Casola, A. Respiratory syncytial virus infection: Mechanisms of redox control and novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 186–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, D.; Hildt, E. Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. Int. J. Mol. Sci. 2019, 20, 4659. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | Class | Targets | Function | Reference |
|---|---|---|---|---|
| Therapeutic targets for inflammatory vascular diseases | ||||
| N-acetyl cysteine (NAC) | antioxidant | TLR2/TLR4 | antioxidants treatment reduced TLR2/4 expression and downstream inflammatory biomediators | [98] |
| P-Coumaric Acid (P-CA) | hydroxycinnamic acid | TLR4 | PCA alleviates experimental diabetic nephropathy | [99] |
| NAC | antioxidant | NLRP3 inflammasome | NAC suppressed NLRP3 inflammasome activation and improved HHcy-induced atherosclerosis | [100] |
| NAC | antioxidant | NLRP3 inflammasome | NAC diminished both inflammasome activation and inflammatory cytokine maturation, and subsequently pyroptotic death induced by nicotine | [101] |
| Peperomin E (PepE) | antioxidant | NLRP3 inflammasome | PepE inhibited atherosclerosis development in HFD-fed ApoE−/− mice | [102] |
| EUK134 | ROS scavenger | NLRP3 inflammasome | EUK134 decreased the MI/R-induced interaction between Txnip and NLRP3, and lowered MI/R-induced IL-1β mRNA and protein expression | [103] |
| Mito-TEMPO | mitochondrial ROS scavenger | NLRP3 inflammasome | Mito-TEMPO mitigated mROS levels and inhibited TMAO-induced activation of the NLRP3 inflammasome | [104] |
| Therapeutic targets for inflammatory neurological diseases | ||||
| Curcumin | polyphenolic compound | TLR4 | Curcumin exerted anti-inflammatory action in MPP+-induced astrocytes | [106] |
| Tomentosin | active compound | TLR4 | Tomentosin reduced behavior deficits and neuroinflammatory response in MPTP-induced Parkinson’s disease | [107] |
| Mito-TEMPO | mitochondrial ROS scavenger | NLRP3 inflammasome | Mito-TEMPO attenuated NLRP3 inflammasome activation | [109] |
| Astragaloside IV | active compound of Astragalus membranaceus | NLRP3 inflammasome | Astragaloside IV protected dopaminergic neuron from neuroinflammation and oxidative stress | [110] |
| Therapeutic targets for inflammatory bowel diseases | ||||
| Xi Lei San | Chinese patent medicine | NLRP3 Inflammasome | Xi Lei San ameliorated IBD by inhibiting NLRP3 inflammasome, autophagy and oxidative stress | [120] |
| Dimethyl fumarate (DMF) | Nrf2 activator | Nrf2 and NLRP3 inflammasome | DMF ameliorated DSS-induced murine experimental colitis | [121] |
| 3-(2-oxo-2-phenylethylidene)-2,3,6,7-tetrahydro-1H-pyrazino-[2,1-a] isoquinolin-4(11bH)-one | Nrf2 activator | Nrf2 and NLRP3 inflammasome | Compound 1 protected against DSS-induced colitis | [122] |
| Curcumin | polyphenolic compound | NLRP3 inflammasome | Curcumin alleviated DSS-induced colitis | [123] |
| Procyanidin | antioxidant | NLRP3 inflammasome | Procyanidin attenuated DSS-induced experimental colitis | [124] |
| lactoferrin-modified liposomes (LF-lipo) containing Patchouli alcohol | active compound of Chinese herb patchouli | NLRP3 inflammasome | LF-lipo showed improved therapeutic efficacy in a DSS-induced colitis murine model | [125] |
| Melatonin | hormone | Intestinal microbiota, TLR2/TLR4 | Melatonin mitigated “restraint stress”-induced colonic microbiota dysbiosis and intestinal inflammation | [134] |
| Walnut oil | dietary supplement | NLRP3 inflammasome, gut microbiota | Walnut oil alleviated DSS-induced colitis in mice | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Chang, M. Roles of PRR-Mediated Signaling Pathways in the Regulation of Oxidative Stress and Inflammatory Diseases. Int. J. Mol. Sci. 2021, 22, 7688. https://doi.org/10.3390/ijms22147688
Li P, Chang M. Roles of PRR-Mediated Signaling Pathways in the Regulation of Oxidative Stress and Inflammatory Diseases. International Journal of Molecular Sciences. 2021; 22(14):7688. https://doi.org/10.3390/ijms22147688
Chicago/Turabian StyleLi, Pengwei, and Mingxian Chang. 2021. "Roles of PRR-Mediated Signaling Pathways in the Regulation of Oxidative Stress and Inflammatory Diseases" International Journal of Molecular Sciences 22, no. 14: 7688. https://doi.org/10.3390/ijms22147688
APA StyleLi, P., & Chang, M. (2021). Roles of PRR-Mediated Signaling Pathways in the Regulation of Oxidative Stress and Inflammatory Diseases. International Journal of Molecular Sciences, 22(14), 7688. https://doi.org/10.3390/ijms22147688