
Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues


Abstract
1. Introduction
1.1. Insulin Resistance
1.2. Adipose Tissue and the Development of Insulin Resistance
1.3. Immune and Endocrine Functions of Adipose Tissue
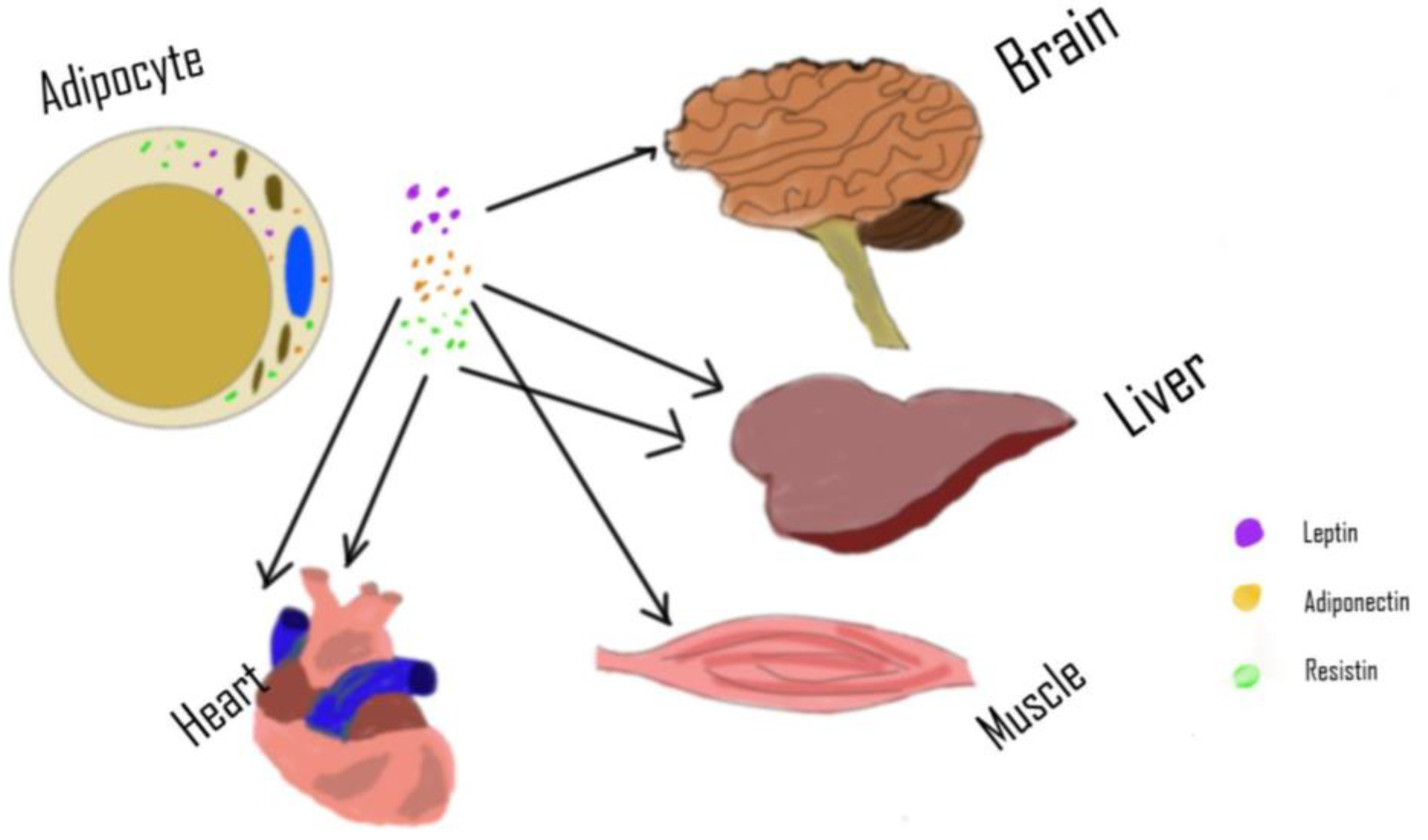
2. Adipose Tissue as an Endocrine Organ
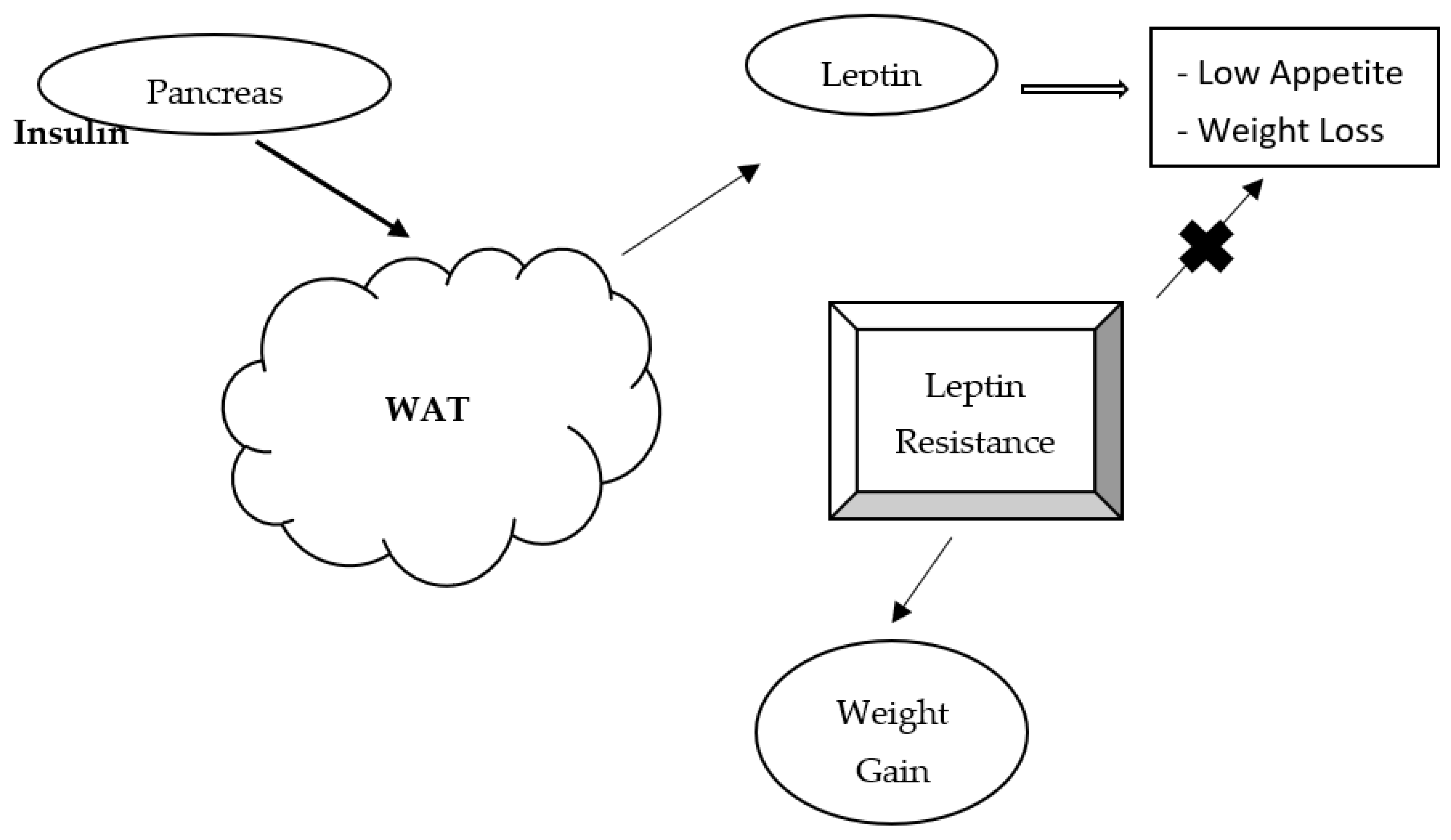
3. Leptin
4. Adiponectin
5. Resistin
6. Pathogenesis of Diabetic Complications
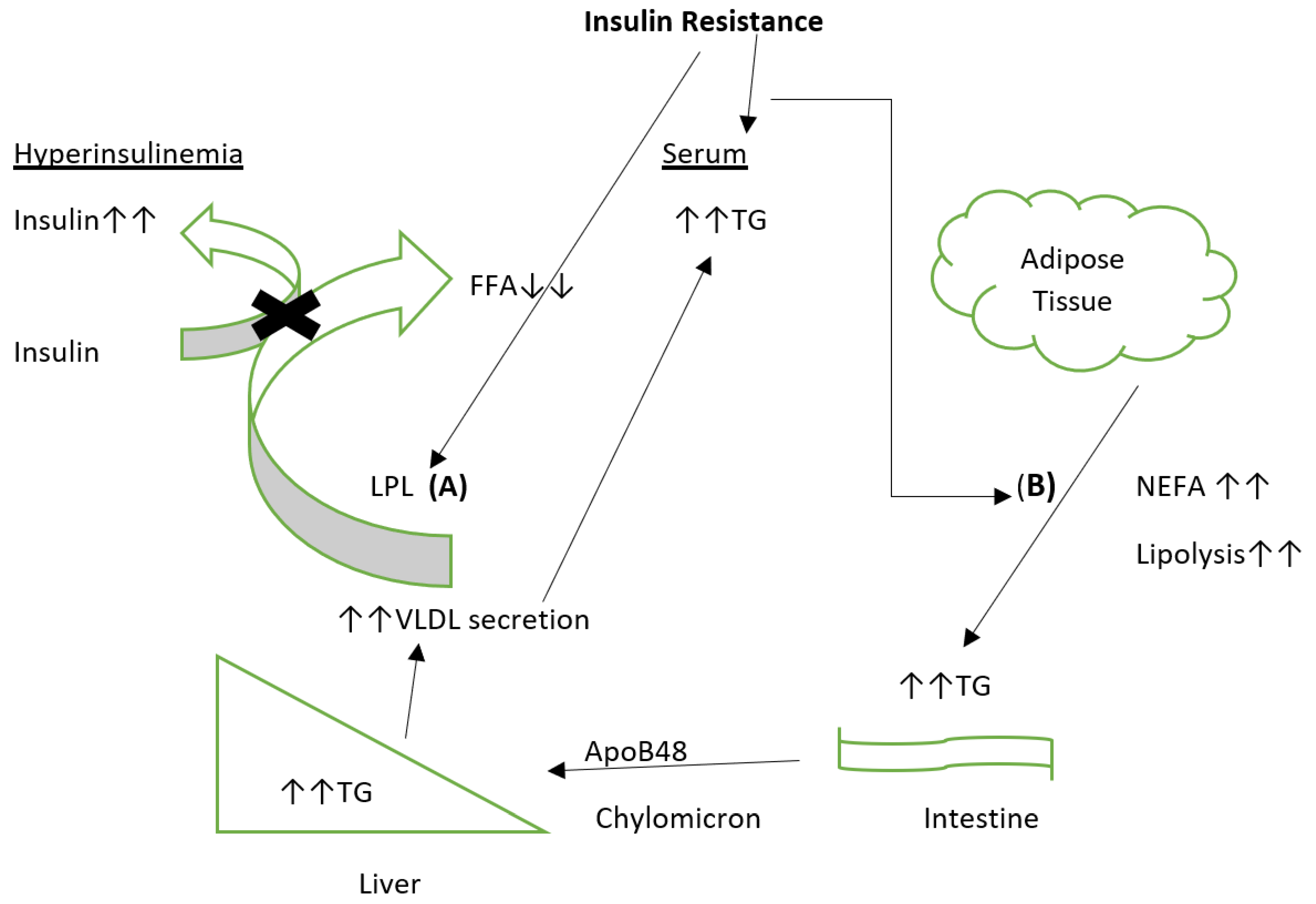
7. Dyslipidemia in Type 2 Diabetes Mellitus
8. Adipose Tissue and Metabolic Dysfunction
9. Lipokines and Adipose Tissue
10. Adipose Tissue, Cholesterol Metabolism and Ethnicity
- Via the LDL receptor,
- Through receptor-independent uptake of cholesterol,
- Via the VLDL receptor,
- Through the hydrolysis of triglyceride-rich lipoproteins.
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, Y.; He, Z.; King, G.L. Introduction of hyperglycemia and dyslipidemia in the pathogenesis of diabetic vascular complications. Curr. Diabetes Rep. 2005, 5, 91–97. [Google Scholar] [CrossRef]
- Watala, C.; Golanski, J.; Boncler, M.A.; Pietrucha, T.; Gwozdzinski, K. Membrane Lipid Fluidity of Blood Platelets: A Common Denominator Underlying the Opposite Actions of Various Agents Affecting Platelet Activation in Whole Blood. Platelets 1998, 9, 315–327. [Google Scholar] [CrossRef]
- Zheng, F.; Lu, W.; Jia, C.; Li, H.; Wang, Z.; Jia, W. Relationships between Glucose Excursion and the Activation of Oxidative Stress in Patients with Newly Diagnosed Type 2 Diabetes or Impaired Glucose Regulation. Endocrine 2010, 37, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Augustyniak, K.; Zavodnik, I.; Palecz, D.; Szosland, K.; Bryszewska, M. The Effect of Oxidizing Agent and Diabetes Mellitus on the Human Red Blood Cell Membrane Potential. Clin. Biochem. 1996, 29, 283–286. [Google Scholar] [CrossRef]
- Zavodnik, I.B.; Szosaland, K.; Bryszewska, M. Human Red Blood Cell membrane potential and fluidity in glucose solutions. Scand. J. Clin. Lab. Investig. 1997, 57, 59–63. [Google Scholar] [CrossRef]
- Sözmen, E.Y.; Sözmen, B.; Delen, Y.; Onat, T. Catalase/Superoxide Dismutase (SOD) and Catalase/Paraoxonase (PON) Ratios May Implicate Poor Glycemic Control. Arch. Med. Res. 2001, 32, 283–287. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Langenberg, C.; Lotta, L.A. Genomic insights into the causes of type 2 diabetes. Lancet 2018, 391, 2463–2474. [Google Scholar] [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas—7th Edition. Available online: http://www.diabetesatlas.org/ (accessed on 19 January 2020).
- Vladu, M.; Clenciu, D.; Efrem, I.C.; Forțofoiu, M.-C.; Amzolini, A.; Micu, S.T.; Moţa, M.; Forțofoiu, M. Insulin Resistance and Chronic Kidney Disease in Patients with Type 1 Diabetes Mellitus. J Nutr. Metab. 2017. [CrossRef]
- Reaven, G. Metabolic syndrome: Pathophysiology and implications for management of cardiovascular disease. Circulation 2002, 106, 286–288. [Google Scholar] [CrossRef]
- Bluher, M. Adipose tissue inflammation: A cause or consequence of obesity-related insulin resistance? Clin. Sci. 2016, 130, 1603–1614. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 2010, 21, 589–598. [Google Scholar] [CrossRef]
- Flamment, M.; Hajduch, E.; Ferre, P.; Foufelle, F. New insights into ER stress induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Zatalia, S.R.; Sanusi, H. The role of antioxidants in the pathophysiology, complications, and management of diabetes mellitus. Acta. Med. Indones. 2013, 45, 141–147. Available online: https://europepmc.org/article/med/23770795 (accessed on 23 April 2021). [PubMed]
- Akash, H.; Sajid, M.; Rehman, K. Tumor necrosis factor-alpha: Role in development of insulin resistance and pathogenesis of type 2 diabetes mellitus. J. Cell. Biochem. 2018, 119, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sosa, M.; Cabellos-Avelar, T.; Sanchez-Zamora, Y.; Juárez-Avelar, I.; García-Reyes, E.; Lira-León, A.; Benítez-Flores, J.C.; Pacheco-Fernández, T.; Hiriart, M.; Gutiérrez-Cirlos, E.B. Proinflammatory cytokine MIF plays a role in the pathogenesis of type-2 diabetes mellitus but does not affect hepatic mitochondrial function. Cytokine 2017, 99, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Coope, A.; Torsoni, A.S.; Velloso, L.A. Mechanisms in endocrinology: Metabolic and inflammatory pathways on the pathogenesis of type 2 diabetes. Eur. J. Endocrinol. 2016, 174, 175–187. [Google Scholar] [CrossRef]
- Meshkani, R.; Vakili, S. Tissue resident macrophages: Key players in the pathogenesis of type 2 diabetes and its complications. Clin. Chim. Acta 2016, 462, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.T.; Navidi, A.A.; Rezaei, M.; Babahmadi-Rezaei, H. Oxidative damage to DNA and lipids: Correlation with protein glycation in patients with type 1 diabetes. J. Clin. Lab. Anal. 2010, 24, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Li, Y.; Wang, M.; Tang, Z.; Wang, T.; Liu, C.; Wang, C.; Zhao, B. Progress in Metabonomics of Type 2 Diabetes Mellitus. Molecules 2018, 23, 1834. [Google Scholar] [CrossRef] [PubMed]
- Matough, F.A.; Budin, S.B.; Hamid, Z.A.; Alwahaibi, N.; Mohamed, J. The role of oxidative stress and antioxidants in diabetic complications. Sultan Qaboos Univ. Med. J. 2012, 12, 556–569. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. Available online: https://www.ahajournals.org/doi/full/10.1161/CIRCRESAHA.110.223545 (accessed on 14 May 2021). [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengellér, Z.; Szabó, C.; Brownlee, M. Inhibition of GAPDH activity by poly (ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Invesig. 2003, 112, 1049. Available online: https://www.jci.org/articles/view/18127 (accessed on 14 May 2021). [CrossRef] [PubMed]
- Easterday, A.; Keil, N.; Subramaniam, R. Mechanism of inhibition of glyceraldehyde-3-phosphate dehydrogenase activity by glucose. FASEB J. 2007, 21, A1015. [Google Scholar] [CrossRef]
- Camacho-Ruiz, A.; Esteban-Méndex, M. Diabetes y radicales libres. In Diabetes, 2nd ed.; Morales-González, J.A., Madrigal-Santillán, E.O., Nava-Chapa, G., Durante-Montiel, I., Jongitud-Falcón, A., Esquivel-Soto, J., Eds.; Universidad Autónoma del Estado de Hidalgo: Pachuca, Hidalgo, México, 2010; Available online: http://refhub.elsevier.com/S0753-3322(18)35789-5/sbref0175 (accessed on 14 May 2021).
- Cho, S.J.; Roman, G.; Yeboah, F.; Konishi, Y. The road to advanced glycation end products: A mechanistic perspective. Curr. Med. Chem. 2007, 14, 1653–1671. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662. [Google Scholar] [CrossRef]
- Ma, X.; Chen, Z.; Wang, L.; Wang, G.; Wang, Z.; Dong, X.; Wen, B.; Zhang, Z. The Pathogenesis of Diabetes Mellitus by Oxidative Stress and Inflammation: Its Inhibition by Berberine. Front. Pharm. 2018, 9, 782. [Google Scholar] [CrossRef]
- Lubkowska, A.; Szymański, S.; Chudecka, M. Surface Body Temperature of Full-Term Healthy Newborns Immediately after Birth-Pilot Study. Int. J. Environ. Res. Public Health 2019, 16, 1312. [Google Scholar] [CrossRef]
- Solmonson, A.; Mills, E.M. Uncoupling Proteins and the Molecular Mechanisms of Thyroid Thermogenesis. Endocrinology 2016, 157, 455–462. [Google Scholar] [CrossRef]
- Bianco, A.; McAninch, E. The role of thyroid hormone and brown adipose tissue in energy homoeostasis. Lancet Diabetes Endo 2013, 1, 250–258. [Google Scholar] [CrossRef]
- Saely, C.H.; Geiger, K.; Drexel, H. Brown versus White Adipose Tissue: A Mini-Review. Gerontology 2012, 58, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell. Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc. 2001, 60, 329–339. [Google Scholar] [CrossRef]
- Kuda, O.; Rossmeisl, M.; Kopecky, J. Omega-3 fatty acids and adipose tissue biology. Mol. Aspects. Med. 2018, 64, 147–160. [Google Scholar] [CrossRef]
- Konige, M.; Wang, H.; Sztalryd, C. Role of adipose specific lipid droplet proteins in maintaining whole body energy homeostasis. Biochim. Biophys. Acta. 2014, 1842, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Zoico, E.; Rubele, S.; De Caro, A.; Nori, N.; Mazzali, G.; Fantin, F.; Rossi, A.; Zamboni, M. Brown and Beige Adipose Tissue and Aging. Front. Endocrinol. 2019, 10, 368. [Google Scholar] [CrossRef]
- Xue, B.; Rim, J.-S.; Hogan, J.C.; Coulter, A.A.; Koza, R.A.; Kozak, L.P. Genetic variability affects the development of brown adipocytes in white fat but not in interscapular brown fat. J. Lipid Res. 2012, 48, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Roh, H.C.; Tsai, L.T.Y.; Shao, M.; Tenen, D.; Shen, Y.; Kumari, M.; Lyubetskaya, A.; Jacobs, C.; Dawes, B.; Gupta, R.K.; et al. Warming induces significant reprogramming of beige, but not brown, adipocyte cellular identity. Cell. Metab. 2018, 27, 1121–1137.e5. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Vidal-Puig, A.J. Thematic review series: Adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007, 48, 1253–1262. [Google Scholar] [CrossRef]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose Tissue: Physiology to Metabolic Dysfunction. (Updated 4 April 2020). In Feingold KR.; Anawalt, B., Boyce, A., Feingold, K.R., Chrousos, G., Herder, W.W.D., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Eds.; Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK555602/ (accessed on 14 May 2021).
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Parimisetty, A.; Dorsemans, A.C.; Awada, R.; Ravanan, P.; Diotel, N.; Lefebvre d’Hellencourt, C. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J. Neuroinflamm. 2016, 13. [Google Scholar] [CrossRef]
- Kohlgruber, A.; Lynch, L. Adipose Tissue Inflammation in the Pathogenesis of Type 2 Diabetes. Curr. Diab. Rep. 2015, 15. [Google Scholar] [CrossRef]
- Seijkens, T.; Engel, D.; Tjwa, M. The role of CD154 in haematopoietic development. Thromb. Haemost. 2010, 104, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Engel, D.; Seijkens, T.; Poggi, M. The immunobiology of CD154-CD40-TRAF interactions in atherosclerosis. Semin. Immunol 2009, 21, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Missiou, A.; Wolf, D.; Platzer, I. CD40L induces inflammation and adipogenesis in adipose cells—A potential link between metabolic and cardiovascular disease. Thromb. Haemost. 2010, 103, 788–796. [Google Scholar] [CrossRef]
- Lievens, D.; Zernecke, A.; Seijkens, T. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 2010, 116, 4317–4327. [Google Scholar] [CrossRef]
- Oliver, S.R.; Flores, R.L.; Pontello, A.M. Acute suppression of circulating sCD40L during hyperglycemia and euglycemic-hyperinsulinemia in healthy young males. J. Investig. Med. 2008, 56, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Poggi, M.; Jager, J.; Paulmyer-Lacroix, O.; Peiretti, F.; Gremeaux, T.; Verdier, M.; Grino, M.; Stepanian, A.; Msika, S.; Burcelin, R.; et al. The inflammatory receptor CD40 is expressed on human adipocytes: Contribution to crosstalk between lymphocytes and adipocytes. Diabetologia 2009, 52, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Seijkens, T.; Kusters, P.; Engel, D.; Lutgens, E. CD40–CD40L: Linking pancreatic, adipose tissue and vascular inflammation in type 2 diabetes and its complications. Diab. Vasc. Dis. Res. 2013, 10, 115–122. [Google Scholar] [CrossRef]
- Ahima, R.S. Central actions of adipocyte hormones. Trends Endocrinol. Metab. 2005, 16, 307–313. [Google Scholar] [CrossRef]
- Gregoire, F.M. Adipocyte Differentiation: From Fibroblast to Endocrine Cell. Exp. Biol. Med. 2001, 226, 997–1002. [Google Scholar] [CrossRef]
- Cao, H. Adipocytokines in obesity and metabolic disease. J. Endocrinol. 2014, 220, T47–T59. [Google Scholar] [CrossRef]
- Halberg, N.; Wernstedt-Asterholm, I.; Scherer, P.E. The Adipocyte as an Endocrine Cell. Endocrin Metab. Clin. N. Am. 2008, 37, 753–768. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2008, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Moustaid-Moussa, N. Secretory, Endocrine and Autocrine/Paracrine Function of the Adipocyte. J. Nutr. 2000, 130. [Google Scholar] [CrossRef] [PubMed]
- Ronti, T.; Lupattelli, G.; Mannarino, E. The endocrine function of adipose tissue: An update. Clin. Endocrinol. 2006, 64, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Prins, J.B. Adipose tissue as an endocrine organ. Best. Pract. Res. Clin. Endocrinol. Metab. 2002, 16, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Montez, J.M.; Soukas, A.; Asilmaz, E.; Fayzikhodjaeva, G.; Fantuzzi, G.; Friedman, J.M. Acute leptin deficiency, leptin resistance, and the physiologic response to leptin withdrawal. Proc. Natl. Acad. Sci. USA 2005, 102, 2537–2542. [Google Scholar] [CrossRef]
- Bi, X.; Loo, Y.T.; Henry, C.J. Does circulating leptin play a role in energy expenditure? Nutrition 2019, 60, 6–10. [Google Scholar] [CrossRef]
- Duvnjak, L.; Duvnjak, M. The metabolic syndrome-An ongoing story. J. Physiol. Pharm. 2009, 60 (Suppl. 7), 19–24. Available online: www.jpp.krakow.pl (accessed on 23 April 2021).
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. Available online: Annualreviews.org (accessed on 23 April 2021). [CrossRef] [PubMed]
- Misra, A.; Garg, A. Leptin, its receptor and obesity. J. Investig. Med. 1996, 44, 540–548. Available online: Utsouthwestern.pure.elsevier.com (accessed on 23 April 2021).
- Lundasen, T.; Liao, W.; Angelin, B.; Rudling, M. Leptin induces the hepatic high density lipoprotein receptor scavenger receptor b type I (SR-BI) but not cholesterol 7alpha-hydroxylase (Cyp7a1) in leptin-deficient (ob/ob) mice. J. Biol. Chem. 2003, 278, 43224–43228. [Google Scholar] [CrossRef]
- Ekmen, N.; Helvaci, A.; Gunaldi, M.; Sasani, H.; Yildirmak, S.T. Leptin as an important link between obesity and cardiovascular risk factors in men with acute myocardial infarction. Indian Heart J. 2016, 68, 132–137. [Google Scholar] [CrossRef]
- D’Elia, L.; Strazzullo, P.; Iacone, R.; Russo, O.; Galletti, F. Leptin levels predict the development of insulin resistance in a sample of adult men–The Olivetti Heart Study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 39–44. [Google Scholar] [CrossRef]
- Segal, K.R.; Landt, M.; Klein, S. Relationship Between Insulin Sensitivity and Plasma Leptin Concentration in Lean and Obese Men. Diabetes 1996, 45, 988–991. [Google Scholar] [CrossRef] [PubMed]
- German, J.P.; Thaler, J.P.; Wisse, B.E.; Oh-I, S.; Sarruf, D.A.; Matsen, M.E.; Fischer, J.D.; Taborsky, G.J., Jr.; Schwartz, M.W.; Morton, G.J. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology 2011, 152, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Hedbacker, K.; Birsoy, K.; Wysocki, R.W.; Asilmaz, E.; Ahima, R.S.; Farooqi, I.S.; Friedman, J.M. Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell. Metab. 2010, 11, 11–22. [Google Scholar] [CrossRef]
- Hui, H.X.; Feng, T. Adipose Tissue as an Endocrine Organ. Adipose Tissue, Leszek Szablewski. IntechOpen, 2018. Available online: https://www.intechopen.com/books/adipose-tissue/adipose-tissue-as-an-endocrine-organ (accessed on 23 April 2021). [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 30. [Google Scholar] [CrossRef]
- Halberg, N.; Henriksen, M.; Söderhamn, N.; Stallknecht, B.; Ploug, T.; Schjerling, P.; Dela, F. Effect of intermittent fasting and refeeding on insulin action in healthy men. J. Appl. Physiol. 2005, 99, 2128–2136. [Google Scholar] [CrossRef]
- Boden, G.; Chen, X.; Mozzoli, M.; Ryan, I. Effect of fasting on serum leptin in normal human subjects. J. Clin. Endocrinol. Metab. 1996, 81, 3419–3423. [Google Scholar] [CrossRef]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in obesity: Mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef]
- Berg, A.H.; Coombs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Cheng, K.K.; Lam, K.S.; Wang, B.; Xu, A. Signaling mechanisms underlying the insulin-sensitizing effects of adiponectin. Best. Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 3–13. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, R.W.; Kunutsor, S.K.; Chowdhury, R.; Yuan, J.M.; Koh, W.P.; Pan, A. Plasma adiponectin levels and type 2 diabetes risk: A nested case-control study in a Chinese population and an updated meta-analysis. Sci. Rep. 2018, 8, 406. [Google Scholar] [CrossRef]
- Facey, A.; Dilworth, L.; Wright-Pascoe, R.; Walker, M.; Irving, R. The Metabolic Hormones Leptin, Adiponectin and Troponin T in Type 2 Diabetes Patients and Athletes in Jamaica. Int. J. Diabetes Res. 2017, 6, 35–40. [Google Scholar]
- Zoccali, C.; Mallamaci, F.; Tripepi, G.; Benedetto, F.A.; Cutruoi, S.; Parlongo, S.; Malatino, L.S. Adiponectin, metabolic risk factors, and cardiovascular events among patients with end-stage renal disease. J. Am. Soc. Nephrol. 2002, 13, 134–141. [Google Scholar] [CrossRef]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef]
- Marette, A.; Liu, Y.; Sweeney, G. Skeletal muscle glucose metabolism and inflammation in the development of the metabolic syndrome. Rev. Endocr. Metab. Dis. 2014, 15, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Ahlstrom, P.; Rai, E.; Chakma, S.; Cho, H.; Rengasamy, P.; Sweeney, G. Adiponectin improves insulin sensitivity via activation of autophagic flux. J. Mol. Endocrinol. 2017, 59, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Dong, L.Q. APPL1: Role in adiponectin signaling and beyond. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E22–E36. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, A Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- McTernan, C.L.; McTernan, P.G.; Harte, A.L.; Levick, P.L.; Barnett, A.H.; Kumar, S. Resistin, central obesity, and type 2 diabetes. Lancet 2002, 359, 46–47. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Kim, Y.; Peroni, O.D.; Fryer, L.G.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Steppan, C.M.; Brown, E.J.; Wright, C.M.; Bhat, S.; Banerjee, R.R.; Dai, C.Y.; Enders, G.H.; Silberg, D.G.; Wen, X.; Wu, G.D.; et al. A family of tissue-specific resistin-like molecules. Proc. Nat. Acad. Sci. USA 2001, 98, 502–506. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Hlavatý, P.; Kunesová, M. Acylation stimulating protein—Its role in control of metabolism in the adipose tissue. Casopis Lekaru Ceskych 2006, 145, 8–14. Available online: https://pubmed.ncbi.nlm.nih.gov/16468236/ (accessed on 23 April 2021).
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef]
- Papatheodorou, K.; Papanas, N.; Banach, M.; Papazoglou, D.; Edmonds, M. Complications of diabetes 2016. J. Diabetes Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Chilelli, N.C.; Burlina, S.; Lapolla, A. AGEs, rather than hyperglycemia, are responsible for microvascular complications in diabetes: A ‘glycoxidation-centric’ point of view. Nutr. Metab. Cardiovas. Dis. 2013, 23, 913–919. [Google Scholar] [CrossRef]
- Nguyen, D.V.; Shaw, L.C.; Grant, M.B. Inflammation in the pathogenesis of microvascular complications in diabetes. Front. Endocrinol. 2012, 3, 170. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Theriault, R.; Watkins, S.C.; Kelley, D.E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism 2000, 49, 467–472. [Google Scholar] [CrossRef]
- Greco, A.V.; Mingrone, G.; Giancaterini, A.; Manco, M.; Morroni, M.; Cinti, S.; Granzotto, M.; Vettor, R.; Camastra, S.; Ferrannini, E. Insulin Resistance in Morbid Obesity-Reversal with Intramyocellular Fat Depletion. Diabetes 2002, 51, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Ryysy, L.; Hakkinen, A.M.; Goto, T.; Vehkavaara, S.; Westerbacka, J.; Halavaara, J.; Yki-Jarvinen, H. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 2000, 49, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Yegin, A.; Ozben, T.; Yegin, H. Glycation of lipoproteins and accelerated atherosclerosis in non-insulin-dependent diabetes mellitus. Int. J. Clin. Lab. Res. 1995, 25, 157–161. [Google Scholar] [CrossRef]
- Numano, F.; Tanaka, A.; Makita, T.; Kishi, Y. Glycated lipoprotein and atherosclerosis. Ann. N. Y. Acad. Sci. 1997, 811, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Shah, R. Assessing the risk of diabetes. BMJ 2015, 351, h4525. [Google Scholar] [CrossRef] [PubMed]
- Medical Research Council. Diabetes: Facts and Stats. Available online: https://www.mrc.ac.uk/documents/pdf/diabetes-uk-facts-and-stats-june-2015 (accessed on 23 April 2021).
- Sheikh, A.B.; Nasrullah, A.; Haq, S.; Akhtar, A.; Ghazanfar, H.; Nasir, A.; Afzal, R.M.; Bukhari, M.M.; Chaudhary, A.Y.; Naqvi, S.W. The Interplay of Genetics and Environmental Factors in the Development of Obesity. Cureus 2017, 9, e1435. [Google Scholar] [CrossRef]
- Machinal, F.; Dieudonne, M.N.; Leneveu, M.C.; Pecquery, R.; Giudicelli, Y. In Vivo and in Vitro ob Gene Expression and Leptin Secretion in Rat Adipocytes: Evidence for a Regional Specific Regulation by Sex Steroid Hormones. Endocrinology 1999, 140, 1567–1574. [Google Scholar] [CrossRef]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef]
- Hoenig, M.R. Implications of the obesity epidemic for lipid-lowering therapy: Non-HDL cholesterol should replace LDL cholesterol as the primary therapeutic target. Vasc. Health Risk Manag. 2008, 4, 143–156. [Google Scholar] [CrossRef]
- Makhoul, Z.; Kristal, A.R.; Gulati, R.; Luick, B.; Bersamin, A.; O’Brien, D.; Hopkins, S.E.; Stephensen, C.B.; Stanhope, K.L.; Havel, P.J.; et al. Associations of obesity with triglycerides and C-reactive protein are attenuated in adults with high red blood cell eicosapentaenoic and docosahexaenoic acids. Eur. J. Clin. Nutr. 2011, 65, 808–817. [Google Scholar] [CrossRef]
- Bays, H.E.; Toth, P.P.; Kris-Etherton, P.M.; Abate, N.; Aronne, L.J.; Brown, W.V.; Gonzalez-Campoy, J.M.; Jones, S.R.; Kumar, R.; La Forge, R.; et al. Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 304–383. [Google Scholar] [CrossRef]
- Choi, S.M.; Tucker, D.F.; Gross, D.N.; Easton, R.M.; DiPilato, L.M.; Dean, A.S.; Monks, B.R.; Birnbaum, M.J. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol. Cell. Biol. 2010, 30, 5009–5020. [Google Scholar] [CrossRef]
- Yu, Y.H.; Ginsberg, H.N. Adipocyte signaling and lipid homeostasis: Sequelae of insulin-resistant adipose tissue. Circ. Res. 2005, 96, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Christopoulou, E.; Tsimihodimos, V.; Filippatos, T.; Elisaf, M. Apolipoprotein CIII and diabetes. Is there a link? Diabetes Metab. Res. Rev. 2019, 35, e3118. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C. Introduction to Lipids and Lipoproteins. In Feingold; Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., Korbonits, M., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2018; Available online: https://europepmc.org/article/NBK/nbk305896 (accessed on 6 July 2021).
- Åvall, K.; Ali, Y.; Leibiger, I.B.; Leibiger, B.; Moede, T.; Paschen, M.; Dicker, A.; Daré, E.; Köhler, M.; Ilegems, E.; et al. Apolipoprotein CIII links islet insulin resistance to β-cell failure in diabetes. Proc. Natl. Acad. Sci. USA 2015, 112, E2611–E2619. [Google Scholar] [CrossRef] [PubMed]
- Juntti-Berggren, L.; Berggren, P.O. Apolipoprotein CIII is a new player in diabetes. Curr. Opin. Lipidol. 2017, 28, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Steiner, G. Acute effects of insulin in the control of VLDL production in humans. Implications for the insulin-resistant state. Diabetes Care 1996, 19, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Taghibiglou, C.; Carpentier, A.; Van Iderstine, S.C.; Chen, B.; Rudy, D.; Aiton, A.; Lewis, G.F.; Adeli, K. Mechanisms of Hepatic Very Low Density Lipoprotein Overproduction in Insulin Resistance. Evidence for enhanced Lipoprotein Assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J. Biol. Chem. 2000, 275, 8416–8425. [Google Scholar] [CrossRef]
- Duez, H.; Lamarche, B.; Valéro, R.; Pavlic, M.; Proctor, S.; Xiao, C.; Szeto, L.; Patterson, B.W.; Lewis, G.F. Both intestinal and hepatic lipoprotein production are stimulated by an acute elevation of plasma free fatty acids in humans. Circulation 2008, 117, 2369–2376. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Bornfeldt, K.E. Lipids and the endothelium: Bidirectional interactions. Curr. Atheroscler. Rep. 2013, 15, 365. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R. Lipoprotein lipase in diabetes. Diabetes Metab Rev. 1987, 3, 551–570. [Google Scholar] [CrossRef]
- Panarotto, D.; Rémillard, P.; Bouffard, L.; Maheux, P. Insulin resistance affects the regulation of lipoprotein lipase in the postprandial period and in an adipose tissue-specific manner. Eur. J. Clin. Investig. 2002, 32, 84–92. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Eckel, R.H.; McPherson, R. Triglycerides and heart disease: Still a hypothesis? Arter. Thromb. Vasc. Biol. 2011, 31, 1716–1725. [Google Scholar] [CrossRef]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Models Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Sethi, J.K. TNF-α and adipocyte biology. FEBS Lett. 2007, 582, 117–131. [Google Scholar] [CrossRef]
- Slutsky, N.; Vatarescu, M.; Haim, Y.; Goldstein, N.; Kirshtein, B.; Harman-Boehm, I.; Gepner, Y.; Shai, I.; Bashan, N.; Blüher, M.; et al. Decreased adiponectin links elevated adipose tissue autophagy with adipocyte endocrine dysfunction in obesity. Int. J. Obes. 2016, 40, 912–920. [Google Scholar] [CrossRef]
- Medina-Gómez, G. Mitochondria and endocrine function of adipose tissue. Best. Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 791–804. [Google Scholar] [CrossRef]
- Klöting, N.; Blüher, M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Dis. 2014, 15, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Saavedra, D.; Stanford, K.I. The Regulation of Lipokines by Environmental Factors. Nutrients 2019, 11, 2422. [Google Scholar] [CrossRef]
- Stanford, K.I.; Goodyear, L.J. Exercise regulation of adipose tissue. Adipocyte 2016, 5, 153–162. [Google Scholar] [CrossRef]
- Dewal, R.S.; Stanford, K.I. Effects of exercise on brown and beige adipocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1864, 71–78. [Google Scholar] [CrossRef]
- Lynes, M.D.; Tseng, Y.H. Deciphering adipose tissue heterogeneity. Ann. N. Y. Acad. Sci. 2018, 1411, 5–20. [Google Scholar] [CrossRef]
- Lynes, M.D.; Shamsi, F.; Sustarsic, E.G.; Leiria, L.O.; Wang, C.H.; Su, S.C.; Huang, T.L.; Gao, F.; Narain, N.R.; Chen, E.Y.; et al. Cold-Activated Lipid Dynamics in Adipose Tissue Highlights a Role for Cardiolipin in Thermogenic Metabolism. Cell Rep. 2018, 24, 781–790. [Google Scholar] [CrossRef]
- Lehnig, A.C.; Dewal, R.S.; Baer, L.A.; Kitching, K.M.; Munoz, V.R.; Arts, P.J.; Sindeldecker, D.A.; May, F.J.; Lauritzen, H.; Goodyear, L.J.; et al. Exercise Training Induces Depot-Specific Adaptations to White and Brown Adipose Tissue. IScience 2019, 11, 425–439. [Google Scholar] [CrossRef]
- Leitner, B.P.; Huang, S.; Brychta, R.J.; Duckworth, C.J.; Baskin, A.S.; McGehee, S.; Tal, I.; Dieckmann, W.; Gupta, G.; Kolodny, G.M.; et al. Mapping of human brown adipose tissue in lean and obese young men. Proc. Natl. Acad. Sci. USA 2017, 114, 8649–8654. [Google Scholar] [CrossRef]
- Poekes, L.; Lanthier, N.; Leclercq, I.A. Brown adipose tissue: A potential target in the fight against obesity and the metabolic syndrome. Clin. Sci. 2015, 129, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Investig. 2013, 123, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Burhans, M.S.; Flowers, M.T.; Ntambi, J.M. Hepatic oleate regulates liver stress response partially through PGC-1alpha during high-carbohydrate feeding. J. Hepatol. 2016, 65, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Burhans, M.S.; Flowers, M.T.; Harrington, K.R.; Bond, L.M.; Guo, C.A.; Anderson, R.M.; Ntambi, J.M. Hepatic oleate regulates adipose tissue lipogenesis and fatty acid oxidation. J. Lipid Res. 2015, 56, 304–318. [Google Scholar] [CrossRef]
- ALJohani, A.M.; Syed, D.N.; Ntambi, J.M. Insights into Stearoyl-CoA Desaturase-1 Regulation of Systemic Metabolism. Trends Endocrinol. Metab. 2017, 28, 831–842. [Google Scholar] [CrossRef]
- Imamura, F.; Micha, R.; Wu, J.H.; de Oliveira Otto, M.C.; Otite, F.O.; Abioye, A.I.; Mozaffarian, D. Effects of Saturated Fat, Polyunsaturated Fat, Monounsaturated Fat, and Carbohydrate on Glucose-Insulin Homeostasis: A Systematic Review and Meta-analysis of Randomised Controlled Feeding Trials. PLoS Med. 2016, 13, e1002087. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. Fat talks, liver and muscle listen. Cell 2008, 134, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Frigolet, M.E.; Gutiérrez-Aguilar, R. The Role of the Novel Lipokine Palmitoleic Acid in Health and Disease. Adv. Nutr. 2017, 8, 173S–181S. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Cao, H.; King, I.B.; Lemaitre, R.N.; Song, X.; Siscovick, D.S.; Hotamisligil, G.S. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am. J. Clin. Nutr. 2010, 92, 1350–1358. [Google Scholar] [CrossRef]
- Stefan, N.; Kantartzis, K.; Celebi, N.; Staiger, H.; Machann, J.; Schick, F.; Cegan, A.; Elcnerova, M.; Schleicher, E.; Fritsche, A.; et al. Circulating palmitoleate strongly and independently predicts insulin sensitivity in humans. Diabetes Care 2010, 33, 405–407. [Google Scholar] [CrossRef]
- Gong, J.; Campos, H.; McGarvey, S.; Wu, Z.; Goldberg, R.; Baylin, A. Adipose tissue palmitoleic acid and obesity in humans: Does it behave as a lipokine? Am. J. Clin. Nutr. 2011, 93, 186–191. [Google Scholar] [CrossRef]
- Li, V.L.; Kim, J.T.; Long, J.Z. Adipose tissue lipokines: Recent progress and future directions. Diabetes 2020, 69, 2541–2548. [Google Scholar] [CrossRef]
- Farkas, J.; Angel, A.; Avigan, M.I. Studies on the compartmentation of lipid in adipose cells. 11. Cholesterol accumulation and distribution in adipose tissue components. J. Lipid Res. 1973, 14, 344–356. Available online: https://pubmed.ncbi.nlm.nih.gov/9704080/ (accessed on 6 July 2021). [CrossRef]
- Gerson, T.; Shorland, F.B.; Dunckley, G.G. The effect of B-sitosterol on the metabolism of cholesterol and lipids in rats on a diet low in fat. Biochem. J. 1964, 92, 385–390. [Google Scholar] [CrossRef]
- Krause, B.R.; Hartman, A.D. Quantification of adipocyte-free and esterified cholesterol using liquid gel chromatography. J. Lipid Res. 1978, 19, 774–777. Available online: https://pubmed.ncbi.nlm.nih.gov/690517 (accessed on 6 July 2021). [CrossRef]
- Krause, B.R.; Hartman, A.D. Adipose tissue and cholesterol metabolism. J. Lipid Res. 1984, 25, 97–110. [Google Scholar] [CrossRef]
- Izem, L.; Morton, R.E. Possible role for intracellular cholesteryl ester transfer protein in adipocyte lipid metabolism and storage. J. Biol. Chem. 2007, 282, 21856–21865. [Google Scholar] [CrossRef] [PubMed]
- Abate, N.; Chandalia, M.; Snell, P.G.; Grundy, S.M. Adipose tissue metabolites and insulin resistance in nondiabetic Asian Indian men. J. Clin. Endocrinol. Metab. 2004, 89, 2750–2755. [Google Scholar] [CrossRef] [PubMed]
- Mente, A.; Razak, F.; Blankenberg, S.; Vuksan, V.; Davis, A.D.; Miller, R.; Teo, K.; Gerstein, H.; Sharma, A.M.; Yusuf, S.; et al. Ethnic variation in adiponectin and leptin levels and their association with adiposity and insulin resistance. Diabetes Care 2010, 33, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Bakker, L.E.; Sleddering, M.A.; Schoones, J.W.; Meinders, A.E.; Jazet, I.M. Pathogenesis of type 2 diabetes in South Asians. Eur. J. Endocrinol. 2013, 169, R99–R114. [Google Scholar] [CrossRef]
- Stults-Kolehmainen, M.A.; Stanforth, P.R.; Bartholomew, J.B. Fat in Android, Trunk, and Peripheral Regions Varies by Ethnicity and Race in College Aged Women. Obesity 2012, 20, 660–665. [Google Scholar] [CrossRef]
- Osei, K.; Schuster, D.P.; Owusu, S.K.; Amoah, A.G. Race and ethnicity determine serum insulin and C-peptide concentrations and hepatic insulin extraction and insulin clearance: Comparative studies of three populations of West African ancestry and white Americans. Metab. Clin. Exp. 1997, 46, 53–58. [Google Scholar] [CrossRef]
- Gower, B.A.; Fowler, L.A. Obesity in African Americans: The role of physiology. J. Intern. Med. 2020, 288, 295–304. [Google Scholar] [CrossRef]
- Eastwood, S.V.; Tillin, T.; Dehbi, H.M.; Wright, A.; Forouhi, N.G.; Godsland, I.; Whincup, P.; Sattar, N.; Hughes, A.D.; Chaturvedi, N. Ethnic differences in associations between fat deposition and incident diabetes and underlying mechanisms: The SABRE study. Obesity 2015, 3, 699–706. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Nono Nankam, P.A.; Nguelefack, T.B.; Goedecke, J.H.; Blüher, M. Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry. Antioxidants 2021, 10, 622. [Google Scholar] [CrossRef] [PubMed]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Fisher, G.; Alvarez, J.A.; Ellis, A.C.; Granger, W.M.; Ovalle, F.; Man, C.D.; Cobelli, C.; Gower, B.A. Race differences in the association of oxidative stress with insulin sensitivity in African and European-American women. Obesity 2012, 20, 972–977. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Adipocytokine | Primary Function | Source |
|---|---|---|
| Leptin | Regulates food intake | Minokoshi et al., 2002 [96] |
| Adiponectin | Regulates insulin sensitivity | Achari and Jain 2017 [94] |
| Resistin | Antagonize insulin action Links obesity with diabetes | Steppan et al., 2001 [97] |
| Interleukin 6 | Involved in immune response to inflammation | Tanaka et al., 2014 [98] |
| Tumor Necrosis Factor | Multifunctional cytokine used by the immune system for cell signaling | Wang and Lin, 2008 [99] |
| Acylation Stimulating Protein | A fat storage factor that stimulates the synthesis and storage of triglycerides in adipocytes | Hlavatý and Kunesová, 2006 [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilworth, L.; Facey, A.; Omoruyi, F. Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues. Int. J. Mol. Sci. 2021, 22, 7644. https://doi.org/10.3390/ijms22147644
Dilworth L, Facey A, Omoruyi F. Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues. International Journal of Molecular Sciences. 2021; 22(14):7644. https://doi.org/10.3390/ijms22147644
Chicago/Turabian StyleDilworth, Lowell, Aldeam Facey, and Felix Omoruyi. 2021. "Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues" International Journal of Molecular Sciences 22, no. 14: 7644. https://doi.org/10.3390/ijms22147644
APA StyleDilworth, L., Facey, A., & Omoruyi, F. (2021). Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues. International Journal of Molecular Sciences, 22(14), 7644. https://doi.org/10.3390/ijms22147644