
Maturity Onset Diabetes of the Young—New Approaches for Disease Modelling


Abstract
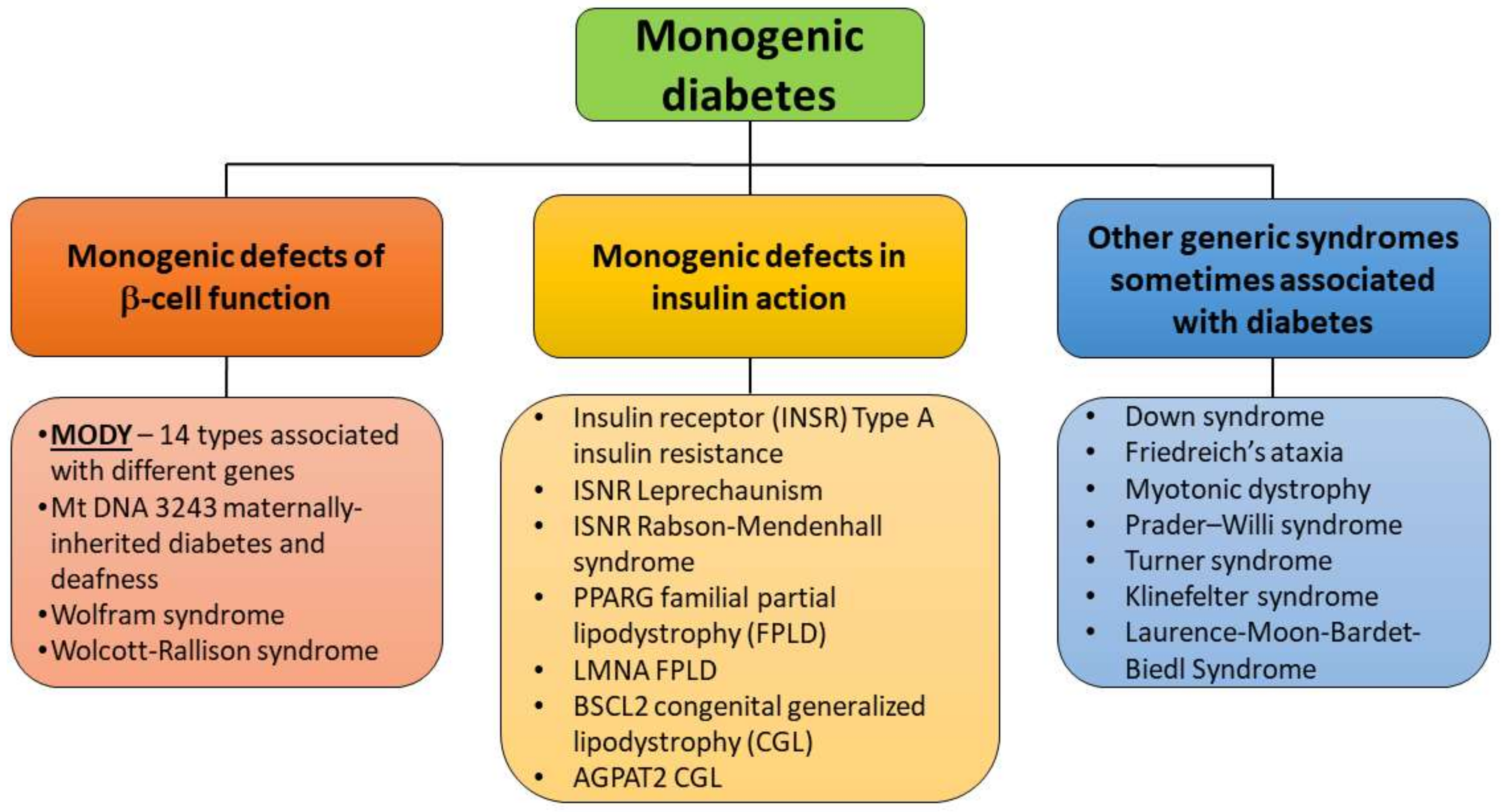
1. Diabetes—Current Classification and Place for Maturity-Onset Diabetes of the Young (MODY)
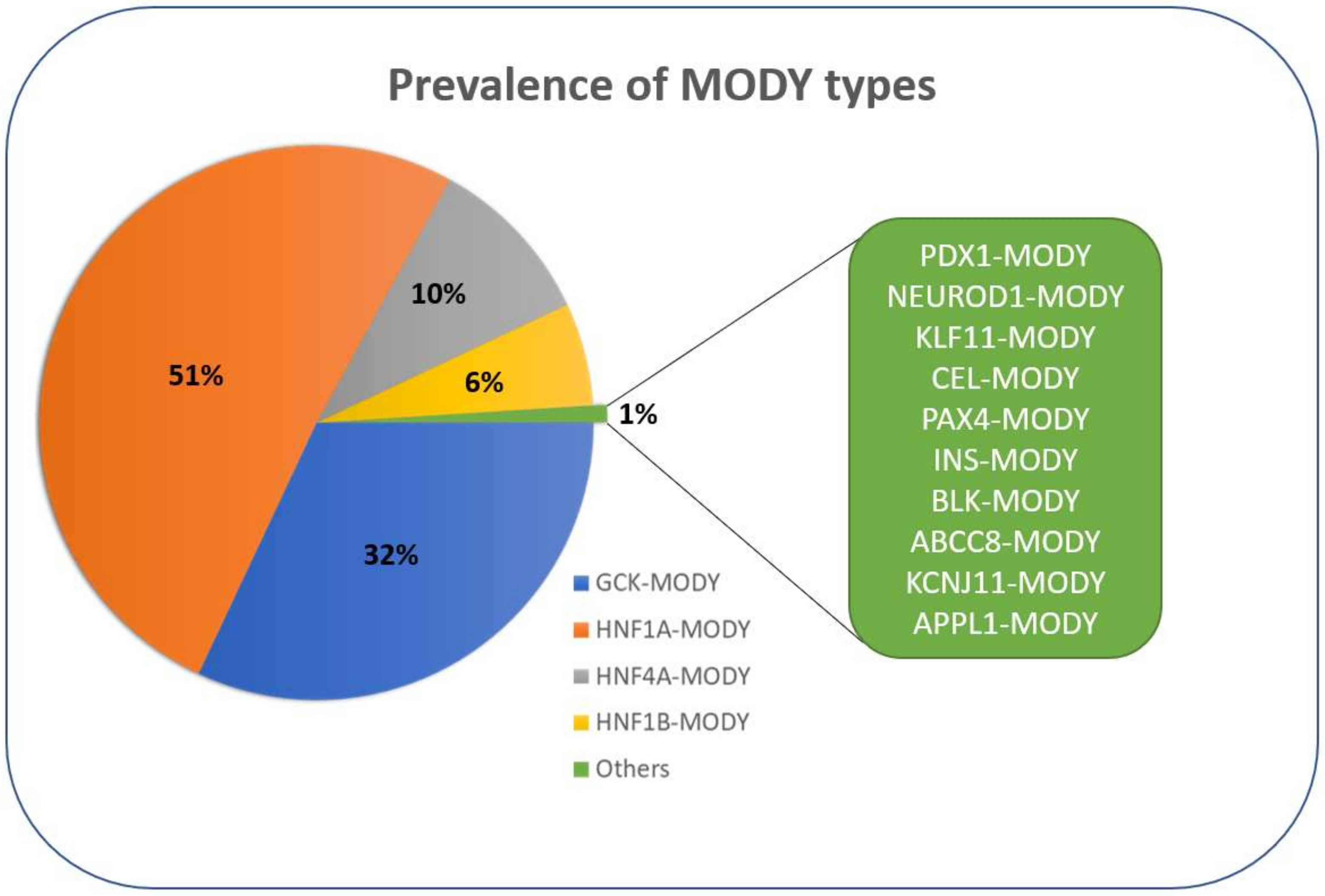
2. MODY Types
3. Molecular Pathophysiology of the Most Common MODY Subtypes
3.1. HNF1A MODY
3.2. HNF4A-MODY
3.3. GCK-MODY
3.4. HNF1B-MODY
3.5. Other MODY Types
4. Diagnosis and Current Treatment Options
4.1. Diagnosis of MODY Patients
4.2. Current Treatment Options
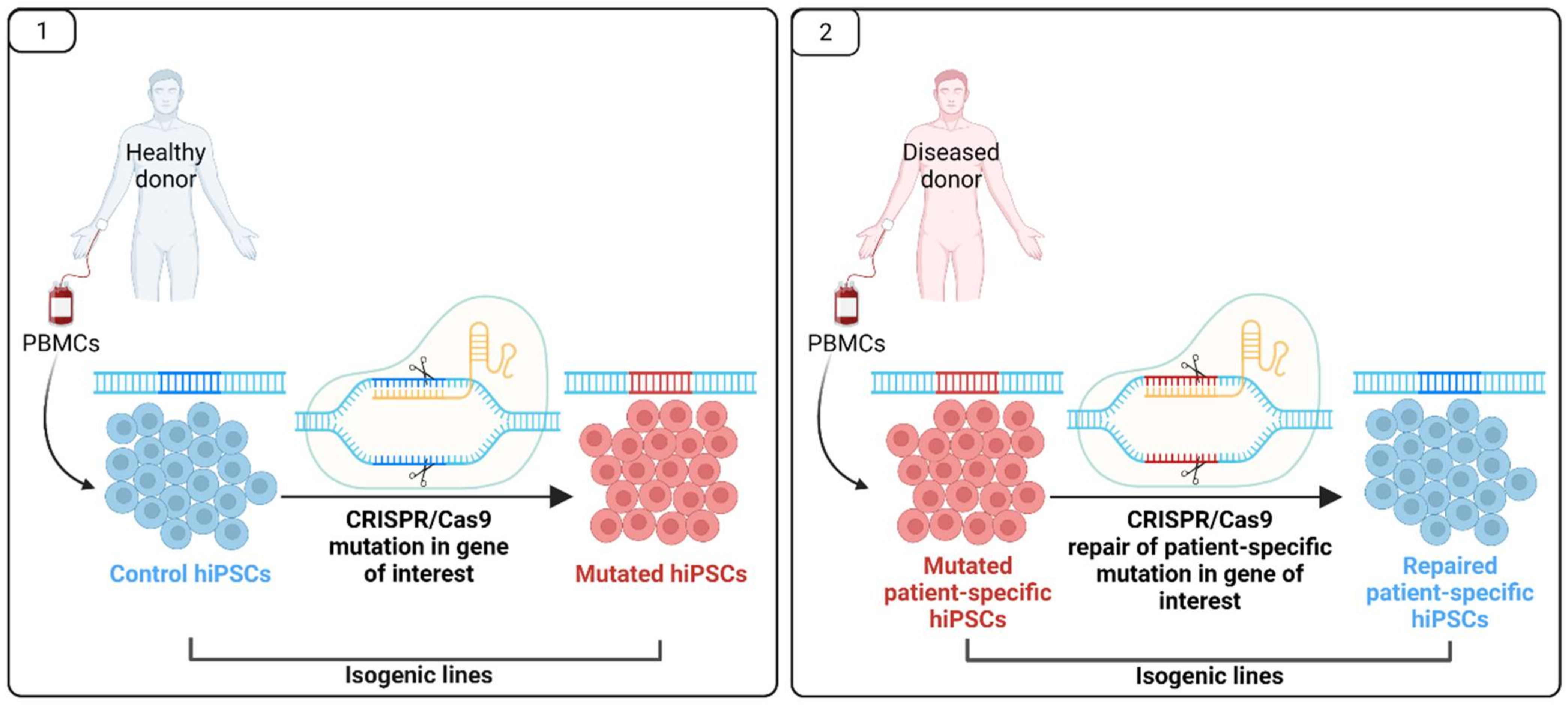
5. Pluripotent Stem Cells for MODY Disease Modelling and Drug Research
6. Final Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO Expert Committee on Diabetes Mellitus; World Health Organization. Diabetes Mellitus: Report of a WHO Expert Committee [Meeting Held in Geneva from 24 to 30 November 1964]; World Health Organization: Geneva, Switzerland, 1965. [Google Scholar]
- World Health Organization. Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications: Report of a WHO Consultation. Part. 1, Diagnosis and Classification of Diabetes Mellitus; World Health Organization: Geneva, Switzerland, 1999. [Google Scholar]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef]
- Malecki, M.T.; Mlynarski, W. Monogenic diabetes: Implications for therapy of rare types of disease. Diabetes Obes. Metab. 2008, 10, 607–616. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Classification of Diabetes Mellitus; World Health Organization: Geneva, Switzerland, 2019; p. 40. [Google Scholar]
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.A.; Gavin, J.R.; Aguilar, R.B. The Time is Right for a New Classification System for Diabetes: Rationale and Implications of the β-Cell–Centric Classification Schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef]
- Hattersley, A.; Bruining, J.; Shield, J.; Njolstad, P.; Donaghue, K.; International Society for Pediatric and Adolescent Diabetes. ISPAD Clinical Practice Consensus Guidelines 2006–2007. The diagnosis and management of monogenic diabetes in children. Pediatr. Diabetes 2006, 7, 352–360. [Google Scholar] [CrossRef]
- Hattersley, A.; Bruining, J.; Shield, J.; Njolstad, P.; Donaghue, K.C. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 2009, 10 (Suppl. 12), 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H. Maturity-Onset Diabetes of the Young: What Do Clinicians Need to Know? Diabetes Metab. J. 2015, 39, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Urakami, T. Maturity-onset diabetes of the young (MODY): Current perspectives on diagnosis and treatment. Diabetes Metab. Syndr. Obes. 2019, 12, 1047–1056. [Google Scholar] [CrossRef]
- Hattersley, A.T.; Greeley, S.A.W.; Polak, M.; Rubio-Cabezas, O.; Njølstad, P.R.; Mlynarski, W.; Castano, L.; Carlsson, A.; Raile, K.; Chi, D.V.; et al. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 2018, 19 (Suppl. 27), 47–63. [Google Scholar] [CrossRef] [PubMed]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin. Diabetes Endocrinol. 2020, 6. [Google Scholar] [CrossRef]
- Heuvel-Borsboom, H.; de Valk, H.W.; Losekoot, M.; Westerink, J. Maturity onset diabetes of the young: Seek and you will find. Neth J. Med. 2016, 74, 193–200. [Google Scholar]
- Kavvoura, F.K.; Owen, K.R. Maturity onset diabetes of the young: Clinical characteristics, diagnosis and management. Pediatr. Endocrinol. Rev. 2012, 10, 234–242. [Google Scholar]
- Yorifuji, T.; Fujimaru, R.; Hosokawa, Y.; Tamagawa, N.; Shiozaki, M.; Aizu, K.; Jinno, K.; Maruo, Y.; Nagasaka, H.; Tajima, T.; et al. Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus. Pediatr. Diabetes 2012, 13, 26–32. [Google Scholar] [CrossRef]
- Shepherd, M.H.; Shields, B.M.; Hudson, M.; Pearson, E.R.; Hyde, C.; Ellard, S.; Hattersley, A.T.; Patel, K.A. UNITED study A UK nationwide prospective study of treatment change in MODY: Genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia 2018, 61, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Irgens, H.U.; Molnes, J.; Johansson, B.B.; Ringdal, M.; Skrivarhaug, T.; Undlien, D.E.; Søvik, O.; Joner, G.; Molven, A.; Njølstad, P.R. Prevalence of monogenic diabetes in the population-based Norwegian Childhood Diabetes Registry. Diabetologia 2013, 56, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.B.; Irgens, H.U.; Molnes, J.; Sztromwasser, P.; Aukrust, I.; Juliusson, P.B.; Søvik, O.; Levy, S.; Skrivarhaug, T.; Joner, G.; et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia 2017, 60, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, S.S.; Bosma, A.; Henneman, L.; Rigter, T.; Spruijt, C.M.; Grimbergen, A.J.; Breuning, M.H.; de Koning, E.J.; Losekoot, M.; Cornel, M.C. A decade of molecular genetic testing for MODY: A retrospective study of utilization in The Netherlands. Eur. J. Hum. Genet. 2015, 23, 29–33. [Google Scholar] [CrossRef]
- Fendler, W.; Borowiec, M.; Baranowska-Jazwiecka, A.; Szadkowska, A.; Skala-Zamorowska, E.; Deja, G.; Jarosz-Chobot, P.; Techmanska, I.; Bautembach-Minkowska, J.; Mysliwiec, M.; et al. Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia 2012, 55, 2631–2635. [Google Scholar] [CrossRef] [PubMed]
- Matsha, T.E.; Raghubeer, S.; Tshivhase, A.M.; Davids, S.F.G.; Hon, G.M.; Bjørkhaug, L.; Erasmus, R.T. Incidence of HNF1A and GCK MODY Variants in a South African Population. Appl. Clin. Genet. 2020, 13, 209–219. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef]
- Bansal, V.; Gassenhuber, J.; Phillips, T.; Oliveira, G.; Harbaugh, R.; Villarasa, N.; Topol, E.J.; Seufferlein, T.; Boehm, B.O. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Med. 2017, 15, 213. [Google Scholar] [CrossRef]
- Flannick, J.; Mercader, J.M.; Fuchsberger, C.; Udler, M.S.; Mahajan, A.; Wessel, J.; Teslovich, T.M.; Caulkins, L.; Koesterer, R.; Barajas-Olmos, F.; et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 2019, 570, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Boissel, M.; Bolze, A.; Durand, E.; Toussaint, B.; Vaillant, E.; Gaget, S.; Graeve, F.D.; Dechaume, A.; Allegaert, F.; et al. Pathogenic variants in actionable MODY genes are associated with type 2 diabetes. Nat. Metab. 2020, 2, 1126–1134. [Google Scholar] [CrossRef]
- Oliveira, S.C.; Neves, J.S.; Pérez, A.; Carvalho, D. Maturity-onset diabetes of the young: From a molecular basis perspective toward the clinical phenotype and proper management. Endocrinol. Diabetes Nutr. 2020, 67, 137–147. [Google Scholar] [CrossRef]
- Peixoto-Barbosa, R.; Reis, A.F.; Giuffrida, F.M.A. Update on clinical screening of maturity-onset diabetes of the young (MODY). Diabetol. Metab. Syndr. 2020, 12, 50. [Google Scholar] [CrossRef]
- Mohan, V.; Radha, V.; Nguyen, T.T.; Stawiski, E.W.; Pahuja, K.B.; Goldstein, L.D.; Tom, J.; Anjana, R.M.; Kong-Beltran, M.; Bhangale, T.; et al. Comprehensive genomic analysis identifies pathogenic variants in maturity-onset diabetes of the young (MODY) patients in South India. BMC Med. Genet. 2018, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, J. A genetic switch in pancreatic beta-cells: Implications for differentiation and haploinsufficiency. Diabetes 2002, 51, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Lee, I.; Marcotte, E.M.; Hurles, M.E. Characterising and Predicting Haploinsufficiency in the Human Genome. PLoS Genet. 2010, 6, e1001154. [Google Scholar] [CrossRef] [PubMed]
- Dang, V.T.; Kassahn, K.S.; Marcos, A.E.; Ragan, M.A. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur. J. Hum. Genet. 2008, 16, 1350–1357. [Google Scholar] [CrossRef]
- Misra, S.; Hassanali, N.; Bennett, A.J.; Juszczak, A.; Caswell, R.; Colclough, K.; Valabhji, J.; Ellard, S.; Oliver, N.S.; Gloyn, A.L. Homozygous Hypomorphic HNF1A Alleles Are a Novel Cause of Young-Onset Diabetes and Result in Sulfonylurea-Sensitive Diabetes. Diabetes Care 2020, 43, 909–912. [Google Scholar] [CrossRef]
- Mendel, D.B.; Crabtree, G.R. HNF-1, a member of a novel class of dimerizing homeodomain proteins. J. Biol. Chem. 1991, 266, 677–680. [Google Scholar] [CrossRef]
- Blumenfeld, M.; Maury, M.; Chouard, T.; Yaniv, M.; Condamine, H. Hepatic nuclear factor 1 (HNF1) shows a wider distribution than products of its known target genes in developing mouse. Development 1991, 113, 589–599. [Google Scholar] [CrossRef]
- Harries, L.W.; Ellard, S.; Stride, A.; Morgan, N.G.; Hattersley, A.T. Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum. Mol. Genet. 2006, 15, 2216–2224. [Google Scholar] [CrossRef]
- Harries, L.W.; Brown, J.E.; Gloyn, A.L. Species-Specific Differences in the Expression of the HNF1A, HNF1B and HNF4A Genes. PLoS ONE 2009, 4, e7855. [Google Scholar] [CrossRef] [PubMed]
- Odom, D.T.; Zizlsperger, N.; Gordon, D.B.; Bell, G.W.; Rinaldi, N.J.; Murray, H.L.; Volkert, T.L.; Schreiber, J.; Rolfe, P.A.; Gifford, D.K.; et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004, 303, 1378–1381. [Google Scholar] [CrossRef] [PubMed]
- Luni, C.; Marth, J.D.; Doyle, F.J. Computational modeling of glucose transport in pancreatic β-cells identifies metabolic thresholds and therapeutic targets in diabetes. PLoS ONE 2012, 7, e53130. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, A.; Bluteau, O.; Garcia-Gonzalez, M.A.; Gresh, L.; Doyen, A.; Garbay, S.; Robine, S.; Pontoglio, M. Hepatocyte nuclear factor 1alpha and beta control terminal differentiation and cell fate commitment in the gut epithelium. Development 2010, 137, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Valkovicova, T.; Skopkova, M.; Stanik, J.; Gasperikova, D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr. Regul. 2019, 53, 110–134. [Google Scholar] [CrossRef]
- Fu, J.; Wang, T.; Zhai, X.; Xiao, X. Primary hepatocellular adenoma due to biallelic HNF1A mutations and its co-occurrence with MODY 3: Case-report and review of the literature. Endocrine 2020, 67, 544–551. [Google Scholar] [CrossRef]
- Pontoglio, M.; Barra, J.; Hadchouel, M.; Doyen, A.; Kress, C.; Bach, J.P.; Babinet, C.; Yaniv, M. Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell 1996, 84, 575–585. [Google Scholar] [CrossRef]
- Pontoglio, M.; Sreenan, S.; Roe, M.; Pugh, W.; Ostrega, D.; Doyen, A.; Pick, A.J.; Baldwin, A.; Velho, G.; Froguel, P.; et al. Defective insulin secretion in hepatocyte nuclear factor 1alpha-deficient mice. J. Clin. Investig. 1998, 101, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.V.; Goto, M.; Magnuson, B.; Abraham, S.; Ramanathan, N.; Hotaling, E.; Alaniz, A.A.; Kumar-Sinha, C.; Dziubinski, M.L.; Urs, S.; et al. HNF1A is a novel oncogene that regulates human pancreatic cancer stem cell properties. eLife 2018, 7, e33947. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-T.; Liu, S.; Liu, L.; Ma, R.-R.; Gao, P. EGR1-Mediated Transcription of lncRNA-HNF1A-AS1 Promotes Cell-Cycle Progression in Gastric Cancer. Cancer Res. 2018, 78, 5877–5890. [Google Scholar] [CrossRef]
- Bellanné-Chantelot, C.; Carette, C.; Riveline, J.-P.; Valéro, R.; Gautier, J.-F.; Larger, E.; Reznik, Y.; Ducluzeau, P.-H.; Sola, A.; Hartemann-Heurtier, A.; et al. The Type and the Position of HNF1A Mutation Modulate Age at Diagnosis of Diabetes in Patients with Maturity-Onset Diabetes of the Young (MODY)-3. Diabetes 2008, 57, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Çubuk, H.; Yalçın Çapan, Ö. A Review of Functional Characterization of Single Amino Acid Change Mutations in HNF Transcription Factors in MODY Pathogenesis. Protein J. 2021, 40, 348–360. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Vaxillaire, M.; Abderrahmani, A.; Boutin, P.; Bailleul, B.; Froguel, P.; Yaniv, M.; Pontoglio, M. Anatomy of a homeoprotein revealed by the analysis of human MODY3 mutations. J. Biol. Chem. 1999, 274, 35639–35646. [Google Scholar] [CrossRef] [PubMed]
- Malikova, J.; Kaci, A.; Dusatkova, P.; Aukrust, I.; Torsvik, J.; Vesela, K.; Kankova, P.D.; Njølstad, P.R.; Pruhova, S.; Bjørkhaug, L. Functional Analyses of HNF1A-MODY Variants Refine the Interpretation of Identified Sequence Variants. J. Clin. Endocrinol. Metab. 2020, 105, e1377–e1386. [Google Scholar] [CrossRef]
- Locke, J.M.; Saint-Martin, C.; Laver, T.W.; Patel, K.A.; Wood, A.R.; Sharp, S.A.; Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T.; Harries, L.W.; et al. The Common HNF1A Variant I27L Is a Modifier of Age at Diabetes Diagnosis in Individuals with HNF1A-MODY. Diabetes 2018, 67, 1903–1907. [Google Scholar] [CrossRef]
- Najmi, L.A.; Aukrust, I.; Flannick, J.; Molnes, J.; Burtt, N.; Molven, A.; Groop, L.; Altshuler, D.; Johansson, S.; Bjørkhaug, L.; et al. Functional Investigations of HNF1A Identify Rare Variants as Risk Factors for Type 2 Diabetes in the General Population. Diabetes 2017, 66, 335–346. [Google Scholar] [CrossRef]
- Althari, S.; Najmi, L.A.; Bennett, A.J.; Aukrust, I.; Rundle, J.K.; Colclough, K.; Molnes, J.; Kaci, A.; Nawaz, S.; van der Lugt, T.; et al. Unsupervised Clustering of Missense Variants in HNF1A Using Multidimensional Functional Data Aids Clinical Interpretation. Am. J. Hum. Genet. 2020, 107, 670–682. [Google Scholar] [CrossRef]
- Sujjitjoon, J.; Charoensuk, C.; Thanyaphon, T.; Kooptiwut, S.; Thamtarana, P.J.; Tangjittipokin, W.; Chongjaroen, N.; Chanprasert, C.; Abubakar, Z.; Lapbenjakul, S.; et al. Defective functions of HNF1A variants on BCL2L1 transactivation and beta-cell growth. Biochem. Biophys. Res. Commun. 2020, 529, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, A.; Gilligan, L.C.; Hughes, B.A.; Hassan-Smith, Z.K.; McCarthy, M.I.; Arlt, W.; Tomlinson, J.W.; Owen, K.R. Altered cortisol metabolism in individuals with HNF1A-MODY. Clin. Endocrinol. 2020, 93, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Szopa, M.; Wolkow, J.; Matejko, B.; Skupien, J.; Klupa, T.; Wybrańska, I.; Trznadel-Morawska, I.; Kiec-Wilk, B.; Borowiec, M.; Malecki, M.T. Prevalence of Retinopathy in Adult Patients with GCK-MODY and HNF1A-MODY. Exp. Clin. Endocrinol. Diabetes 2015, 123, 524–528. [Google Scholar] [CrossRef]
- Steele, A.M.; Shields, B.M.; Shepherd, M.; Ellard, S.; Hattersley, A.T.; Pearson, E.R. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet. Med. 2010, 27, 157–161. [Google Scholar] [CrossRef]
- Szopa, M.; Klupa, T.; Kapusta, M.; Matejko, B.; Ucieklak, D.; Glodzik, W.; Zapala, B.; Sani, C.M.; Hohendorff, J.; Malecki, M.T.; et al. A decision algorithm to identify patients with high probability of monogenic diabetes due to HNF1A mutations. Endocrine 2019, 64, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Poy, M.N.; Stoffel, M. Apolipoprotein M is required for preβ-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 2005, 11, 418–422. [Google Scholar] [CrossRef]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef]
- Richter, S.; Shih, D.Q.; Pearson, E.R.; Wolfrum, C.; Fajans, S.S.; Hattersley, A.T.; Stoffel, M. Regulation of apolipoprotein M gene expression by MODY3 gene hepatocyte nuclear factor-1alpha: Haploinsufficiency is associated with reduced serum apolipoprotein M levels. Diabetes 2003, 52, 2989–2995. [Google Scholar] [CrossRef]
- Szopa, M.; Osmenda, G.; Wilk, G.; Matejko, B.; Skupien, J.; Zapala, B.; Młynarski, W.; Guzik, T.; Malecki, M.T. Intima-media thickness and endothelial dysfunction in GCK and HNF1A-MODY patients. Eur. J. Endocrinol. 2015, 172, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Kachamakova-Trojanowska, N.; Stepniewski, J.; Dulak, J. Human iPSCs-Derived Endothelial Cells with Mutation in HNF1A as a Model of Maturity-Onset Diabetes of the Young. Cells 2019, 8, 1440. [Google Scholar] [CrossRef] [PubMed]
- Lehto, M.; Bitzén, P.O.; Isomaa, B.; Wipemo, C.; Wessman, Y.; Forsblom, C.; Tuomi, T.; Taskinen, M.R.; Groop, L. Mutation in the HNF-4alpha gene affects insulin secretion and triglyceride metabolism. Diabetes 1999, 48, 423–425. [Google Scholar] [CrossRef]
- Gonzalez, F.J. Regulation of hepatocyte nuclear factor 4 alpha-mediated transcription. Drug Metab. Pharm. 2008, 23, 2–7. [Google Scholar] [CrossRef]
- Dubois, V.; Staels, B.; Lefebvre, P.; Verzi, M.P.; Eeckhoute, J. Control of Cell Identity by the Nuclear Receptor HNF4 in Organ Pathophysiology. Cells 2020, 9, 2185. [Google Scholar] [CrossRef] [PubMed]
- Marchesin, V.; Pérez-Martí, A.; Le Meur, G.; Pichler, R.; Grand, K.; Klootwijk, E.D.; Kesselheim, A.; Kleta, R.; Lienkamp, S.; Simons, M. Molecular Basis for Autosomal-Dominant Renal Fanconi Syndrome Caused by HNF4A. Cell Rep. 2019, 29, 4407.e5–4421.e5. [Google Scholar] [CrossRef] [PubMed]
- Cattin, A.-L.; Le Beyec, J.; Barreau, F.; Saint-Just, S.; Houllier, A.; Gonzalez, F.J.; Robine, S.; Pinçon-Raymond, M.; Cardot, P.; Lacasa, M.; et al. Hepatocyte nuclear factor 4alpha, a key factor for homeostasis, cell architecture, and barrier function of the adult intestinal epithelium. Mol. Cell. Biol. 2009, 29, 6294–6308. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Mane-Padros, D.; Bolotin, E.; Jiang, T.; Sladek, F.M. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res. 2012, 40, 5343–5356. [Google Scholar] [CrossRef] [PubMed]
- Fajans, S.S.; Cloutier, M.C.; Crowther, R.L. Clinical and Etiologic Heterogeneity of Idiopathic Diabetes Mellitus. Diabetes 1978, 27, 1112–1125. [Google Scholar] [CrossRef]
- Fajans, S.S.; Bell, G.I. MODY. Diabetes Care 2011, 34, 1878–1884. [Google Scholar] [CrossRef]
- Colclough, K.; Bellanne-Chantelot, C.; Saint-Martin, C.; Flanagan, S.E.; Ellard, S. Mutations in the Genes Encoding the Transcription Factors Hepatocyte Nuclear Factor 1 Alpha and 4 Alpha in Maturity-Onset Diabetes of the Young and Hyperinsulinemic Hypoglycemia. Hum. Mutat. 2013, 34, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Arya, V.B.; Rahman, S.; Senniappan, S.; Flanagan, S.E.; Ellard, S.; Hussain, K. HNF4A mutation: Switch from hyperinsulinaemic hypoglycaemia to maturity-onset diabetes of the young, and incretin response. Diabet. Med. 2014, 31, e11–e15. [Google Scholar] [CrossRef] [PubMed]
- Bacon, S.; Kyithar, M.P.; Condron, E.M.; Vizzard, N.; Burke, M.; Byrne, M.M. Prolonged episodes of hypoglycaemia in HNF4A-MODY mutation carriers with IGT. Evidence of persistent hyperinsulinism into early adulthood. Acta Diabetol. 2016, 53, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.; Vargas, A.; Ariceta, G.; Martínez, R.; Campos, A.; Yeste, D. Hyperinsulinaemic hypoglycaemia, renal Fanconi syndrome and liver disease due to a mutation in the HNF4A gene. Endocrinol. Diabetes Metab. Case Rep. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Boj, S.F.; Steele, A.M.; Barrett, T.; Stals, K.; Shield, J.P.; Ellard, S.; Ferrer, J.; Hattersley, A.T. Macrosomia and Hyperinsulinaemic Hypoglycaemia in Patients with Heterozygous Mutations in the HNF4A Gene. PLoS Med. 2007, 4, e118. [Google Scholar] [CrossRef]
- Barroso, I.; Luan, J.; Middelberg, R.P.S.; Harding, A.-H.; Franks, P.W.; Jakes, R.W.; Clayton, D.; Schafer, A.J.; O’Rahilly, S.; Wareham, N.J. Candidate gene association study in type 2 diabetes indicates a role for genes involved in beta-cell function as well as insulin action. PLoS Biol. 2003, 1, E20. [Google Scholar]
- Miura, A.; Yamagata, K.; Kakei, M.; Hatakeyama, H.; Takahashi, N.; Fukui, K.; Nammo, T.; Yoneda, K.; Inoue, Y.; Sladek, F.M.; et al. Hepatocyte nuclear factor-4alpha is essential for glucose-stimulated insulin secretion by pancreatic beta-cells. J. Biol. Chem. 2006, 281, 5246–5257. [Google Scholar] [CrossRef]
- Stoffel, M.; Duncan, S.A. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc. Natl. Acad. Sci. USA 1997, 94, 13209–13214. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maechler, P.; Antinozzi, P.A.; Hagenfeldt, K.A.; Wollheim, C.B. Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic beta -cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J. Biol. Chem. 2000, 275, 35953–35959. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Lu, H. Novel Mechanisms of Regulation of the Expression and Transcriptional Activity of HNF4α. J. Cell. Biochem. 2019, 120, 519–532. [Google Scholar] [CrossRef]
- Jafar-Mohammadi, B.; Groves, C.J.; Gjesing, A.P.; Herrera, B.M.; Winckler, W.; Stringham, H.M.; Morris, A.P.; Lauritzen, T.; Doney, A.S.F.; Morris, A.D.; et al. A role for coding functional variants in HNF4A in type 2 diabetes susceptibility. Diabetologia 2011, 54, 111–119. [Google Scholar] [CrossRef]
- Andrulionytė, L.; Laukkanen, O.; Chiasson, J.-L.; Laakso, M. STOP-NIDDM Study Group Single nucleotide polymorphisms of the HNF4α gene are associated with the conversion to type 2 diabetes mellitus: The STOP-NIDDM trial. J. Mol. Med. 2006, 84, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Granados-Silvestre, M.A.; Ortiz-López, M.G.; Granados, J.; Canizales-Quinteros, S.; Peñaloza-Espinosa, R.I.; Lechuga, C.; Acuña-Alonzo, V.; Sánchez-Pozos, K.; Menjivar, M. Susceptibility background for type 2 diabetes in eleven Mexican Indigenous populations: HNF4A gene analysis. Mol. Genet. Genom. 2017, 292, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Azizi, S.M.; Sarhangi, N.; Afshari, M.; Abbasi, D.; Aghaei Meybodi, H.R.; Hasanzad, M. Association Analysis of the HNF4A Common Genetic Variants with Type 2 Diabetes Mellitus Risk. Int. J. Mol. Cell. Med. 2019, 8, 56–62. [Google Scholar]
- Matschinsky, F.M. Regulation of pancreatic beta-cell glucokinase: From basics to therapeutics. Diabetes 2002, 51 (Suppl. 3), S394–S404. [Google Scholar] [CrossRef]
- Wang, Z.; Diao, C.; Liu, Y.; Li, M.; Zheng, J.; Zhang, Q.; Yu, M.; Zhang, H.; Ping, F.; Li, M.; et al. Identification and functional analysis of GCK gene mutations in 12 Chinese families with hyperglycemia. J. Diabetes Investig. 2019, 10, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Chakera, A.J.; Steele, A.M.; Gloyn, A.L.; Shepherd, M.H.; Shields, B.; Ellard, S.; Hattersley, A.T. Recognition and Management of Individuals With Hyperglycemia Because of a Heterozygous Glucokinase Mutation. Diabetes Care 2015, 38, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Hulín, J.; Škopková, M.; Valkovičová, T.; Mikulajová, S.; Rosoľanková, M.; Papcun, P.; Gašperíková, D.; Staník, J. Clinical implications of the glucokinase impaired function-GCK MODY today. Physiol. Res. 2020, 69, 995–1011. [Google Scholar] [CrossRef]
- Rudland, V.L. Diagnosis and management of glucokinase monogenic diabetes in pregnancy: Current perspectives. Diabetes Metab. Syndr. Obes. 2019, 12, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.M.; Shields, B.M.; Wensley, K.J.; Colclough, K.; Ellard, S.; Hattersley, A.T. Prevalence of Vascular Complications Among Patients With Glucokinase Mutations and Prolonged, Mild Hyperglycemia. JAMA 2014, 311, 279–286. [Google Scholar] [CrossRef]
- Solar, M.; Cardalda, C.; Houbracken, I.; Martín, M.; Maestro, M.A.; De Medts, N.; Xu, X.; Grau, V.; Heimberg, H.; Bouwens, L.; et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev. Cell 2009, 17, 849–860. [Google Scholar] [CrossRef] [PubMed]
- De Vas, M.G.; Kopp, J.L.; Heliot, C.; Sander, M.; Cereghini, S.; Haumaitre, C. Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development 2015, 142, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Bellanné-Chantelot, C.; Chauveau, D.; Gautier, J.-F.; Dubois-Laforgue, D.; Clauin, S.; Beaufils, S.; Wilhelm, J.-M.; Boitard, C.; Noël, L.-H.; Velho, G.; et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann. Intern. Med. 2004, 140, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Z.; Gao, Q.; Zhao, X.-Z.; Chen, Y.-Z.; Bennett, C.L.; Xiong, X.-S.; Mei, C.-L.; Shi, Y.-Q.; Chen, X.-M. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin. Med. J. 2010, 123, 3326–3333. [Google Scholar] [PubMed]
- Bockenhauer, D.; Jaureguiberry, G. HNF1B-associated clinical phenotypes: The kidney and beyond. Pediatr. Nephrol. 2016, 31, 707–714. [Google Scholar] [CrossRef]
- Kato, T.; Tanaka, D.; Muro, S.; Jambaljav, B.; Mori, E.; Yonemitsu, S.; Oki, S.; Inagaki, N. A Novel p.L145Q Mutation in the HNF1B Gene in a Case of Maturity-onset Diabetes of the Young Type 5 (MODY5). Intern. Med. 2018, 57, 2035–2039. [Google Scholar] [CrossRef]
- Fujita, Y.; Tanaka, D.; Tatsuoka, H.; Matsubara, M.; Hyo, T.; Hamamoto, Y.; Komiya, T.; Inagaki, N.; Seino, Y.; Yamazaki, Y. A novel splice-site mutation of the HNF1B gene in a family with maturity onset diabetes of the young type 5 (MODY5). Endocrinol. Diabetes Metab. Case Rep. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Yang, Y.; Hu, Z. A novel HNF1B mutation p.R177Q in autosomal dominant tubulointerstitial kidney disease and maturity-onset diabetes of the young type 5: A pedigree-based case report. Medicine 2020, 99, e21438. [Google Scholar] [CrossRef]
- Kim, E.K.; Lee, J.S.; Cheong, H.I.; Chung, S.S.; Kwak, S.H.; Park, K.S. Identification and Functional Characterization of P159L Mutation in HNF1B in a Family with Maturity-Onset Diabetes of the Young 5 (MODY5). Genom. Inform. 2014, 12, 240–246. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sztromwasser, P.; Michalak, A.; Małachowska, B.; Młudzik, P.; Antosik, K.; Hogendorf, A.; Zmysłowska, A.; Borowiec, M.; Młynarski, W.; Fendler, W. A cross-sectional study of patients referred for HNF1B-MODY genetic testing due to cystic kidneys and diabetes. Pediatr. Diabetes 2020, 21, 422–430. [Google Scholar] [CrossRef]
- Bustamante, C.; Sanchez, J.; Seeherunvong, T.; Ukarapong, S. Early Onset of Mody5 Due to Haploinsufficiency of HNF1B. AACE Clin. Case Rep. 2020, 6, e243–e246. [Google Scholar] [CrossRef]
- Motyka, R.; Kołbuc, M.; Wierzchołowski, W.; Beck, B.B.; Towpik, I.E.; Zaniew, M. Four Cases of Maturity Onset Diabetes of the Young (MODY) Type 5 Associated with Mutations in the Hepatocyte Nuclear Factor 1 Beta (HNF1B) Gene Presenting in a 13-Year-Old Boy and in Adult Men Aged 33, 34, and 35 Years in Poland. Am. J. Case Rep. 2021, 22, e928994. [Google Scholar] [PubMed]
- Dubois-Laforgue, D.; Cornu, E.; Saint-Martin, C.; Coste, J.; Bellanné-Chantelot, C.; Timsit, J. Monogenic Diabetes Study Group of the Société Francophone du Diabète Diabetes, Associated Clinical Spectrum, Long-term Prognosis, and Genotype/Phenotype Correlations in 201 Adult Patients With Hepatocyte Nuclear Factor 1B (HNF1B) Molecular Defects. Diabetes Care 2017, 40, 1436–1443. [Google Scholar] [CrossRef]
- Ivanoshchuk, D.E.; Shakhtshneider, E.V.; Rymar, O.D.; Ovsyannikova, A.K.; Mikhailova, S.V.; Orlov, P.S.; Ragino, Y.I.; Voevoda, M.I. Analysis of APPL1 Gene Polymorphisms in Patients with a Phenotype of Maturity Onset Diabetes of the Young. J. Pers. Med. 2020, 10, 100. [Google Scholar] [CrossRef]
- De Santana, L.S.; Caetano, L.A.; Costa-Riquetto, A.D.; Franco, P.C.; Dotto, R.P.; Reis, A.F.; Weinert, L.S.; Silveiro, S.P.; Vendramini, M.F.; do Prado, F.A.; et al. Targeted sequencing identifies novel variants in common and rare MODY genes. Mol. Genet. Genom. Med. 2019, 7, e962. [Google Scholar] [CrossRef]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P.J.; Ashcroft, F.M. New insights into KATP channel gene mutations and neonatal diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Beltrand, J.; Busiah, K.; Vaivre-Douret, L.; Fauret, A.L.; Berdugo, M.; Cavé, H.; Polak, M. Neonatal Diabetes Mellitus. Front. Pediatr. 2020, 8, 540718. [Google Scholar] [CrossRef] [PubMed]
- Aarthy, R.; Aston-Mourney, K.; Mikocka-Walus, A.; Radha, V.; Amutha, A.; Anjana, R.M.; Unnikrishnan, R.; Mohan, V. Clinical features, complications and treatment of rarer forms of maturity-onset diabetes of the young (MODY)—A review. J. Diabetes Its Complicat. 2020, 35, 107640. [Google Scholar] [CrossRef]
- Kapoor, R.R.; Flanagan, S.E.; Arya, V.B.; Shield, J.P.; Ellard, S.; Hussain, K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur. J. Endocrinol. 2013, 168, 557–564. [Google Scholar] [CrossRef]
- Snider, K.E.; Becker, S.; Boyajian, L.; Shyng, S.-L.; MacMullen, C.; Hughes, N.; Ganapathy, K.; Bhatti, T.; Stanley, C.A.; Ganguly, A. Genotype and Phenotype Correlations in 417 Children With Congenital Hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, E355–E363. [Google Scholar] [CrossRef]
- Rozenkova, K.; Malikova, J.; Nessa, A.; Dusatkova, L.; Bjørkhaug, L.; Obermannova, B.; Dusatkova, P.; Kytnarova, J.; Aukrust, I.; Najmi, L.A.; et al. High Incidence of Heterozygous ABCC8 and HNF1A Mutations in Czech Patients with Congenital Hyperinsulinism. J. Clin. Endocrinol. Metab. 2015, 100, E1540–E1549. [Google Scholar] [CrossRef]
- Stanik, J.; Dusatkova, P.; Cinek, O.; Valentinova, L.; Huckova, M.; Skopkova, M.; Dusatkova, L.; Stanikova, D.; Pura, M.; Klimes, I.; et al. De novo mutations of GCK, HNF1A and HNF4A may be more frequent in MODY than previously assumed. Diabetologia 2014, 57, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Besser, R.E.J.; Shepherd, M.H.; McDonald, T.J.; Shields, B.M.; Knight, B.A.; Ellard, S.; Hattersley, A.T. Urinary C-peptide creatinine ratio is a practical outpatient tool for identifying hepatocyte nuclear factor 1-α/hepatocyte nuclear factor 4-α maturity-onset diabetes of the young from long-duration type 1 diabetes. Diabetes Care 2011, 34, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.R.; Thanabalasingham, G.; James, T.J.; Karpe, F.; Farmer, A.J.; McCarthy, M.I.; Gloyn, A.L. Assessment of high-sensitivity C-reactive protein levels as diagnostic discriminator of maturity-onset diabetes of the young due to HNF1A mutations. Diabetes Care 2010, 33, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.J.; Shields, B.M.; Lawry, J.; Owen, K.R.; Gloyn, A.L.; Ellard, S.; Hattersley, A.T. High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care 2011, 34, 1860–1862. [Google Scholar] [CrossRef] [PubMed]
- Thanabalasingham, G.; Shah, N.; Vaxillaire, M.; Hansen, T.; Tuomi, T.; Gašperíková, D.; Szopa, M.; Tjora, E.; James, T.J.; Kokko, P.; et al. A large multi-centre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia 2011, 54, 2801–2810. [Google Scholar] [CrossRef]
- Warncke, K.; Kummer, S.; Raile, K.; Grulich-Henn, J.; Woelfle, J.; Steichen, E.; Prinz, N.; Holl, R.W. Frequency and Characteristics of MODY 1 (HNF4A Mutation) and MODY 5 (HNF1B Mutation): Analysis From the DPV Database. J. Clin. Endocrinol. Metab. 2019, 104, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Rajadurai, V.S.; Hoi, W.H.; Flanagan, S.E.; Hussain, K.; Yap, F. A Novel HNF4A Mutation Causing Three Phenotypic Forms of Glucose Dysregulation in a Family. Front. Pediatr 2020, 8, 320. [Google Scholar] [CrossRef]
- Fu, J.; Wang, T.; Liu, J.; Wang, X.; Zhang, Q.; Li, M.; Xiao, X. Using Clinical Indices to Distinguish MODY2 (GCK Mutation) and MODY3 (HNF1A Mutation) from Type 1 Diabetes in a Young Chinese Population. Diabetes Ther. 2019, 10, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.M.; Wensley, K.J.; Ellard, S.; Murphy, R.; Shepherd, M.; Colclough, K.; Hattersley, A.T.; Shields, B.M. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: Observational case control studies. PLoS ONE 2013, 8, e65326. [Google Scholar] [CrossRef]
- Shields, B.M.; Shepherd, M.; Hudson, M.; McDonald, T.J.; Colclough, K.; Peters, J.; Knight, B.; Hyde, C.; Ellard, S.; Pearson, E.R.; et al. Population-Based Assessment of a Biomarker-Based Screening Pathway to Aid Diagnosis of Monogenic Diabetes in Young-Onset Patients. Diabetes Care 2017, 40, 1017–1025. [Google Scholar] [CrossRef]
- Shepherd, M.; Shields, B.; Hammersley, S.; Hudson, M.; McDonald, T.J.; Colclough, K.; Oram, R.A.; Knight, B.; Hyde, C.; Cox, J.; et al. Systematic Population Screening, Using Biomarkers and Genetic Testing, Identifies 2.5% of the U.K. Pediatric Diabetes Population With Monogenic Diabetes. Diabetes Care 2016, 39, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, Y.; Cai, X.; Chen, L.; Zhou, L.; Ma, Y.; Gong, S.; Han, X.; Ji, L. Clinical Implications of Urinary C-Peptide Creatinine Ratio in Patients with Different Types of Diabetes. J. Diabetes Res. 2019, 2019, 1747684. [Google Scholar] [CrossRef]
- Naylor, R.; Knight Johnson, A.; del Gaudio, D. Maturity-Onset Diabetes of the Young Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gardner, D.S.; Tai, E.S. Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabetes Metab. Syndr. Obes. 2012, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Pruhova, S.; Tack, C.J.; Johansen, A.; Castleden, H.A.J.; Lumb, P.J.; Wierzbicki, A.S.; Clark, P.M.; Lebl, J.; Pedersen, O.; et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4α mutations in a large European collection. Diabetologia 2005, 48, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Španinger, E.; Potočnik, U.; Bren, U. Molecular Dynamics Simulations Predict that rSNP Located in the HNF-1α Gene Promotor Region Linked with MODY3 and Hepatocellular Carcinoma Promotes Stronger Binding of the HNF-4α Transcription Factor. Biomolecules 2020, 10, 1700. [Google Scholar] [CrossRef]
- Spiro, A.J.; Vu, K.N.; Warnock, A.L. An Atypical HNF4A Mutation Which Does Not Conform to the Classic Presentation of HNF4A-MODY. Case Rep. Endocrinol. 2018, 2018, 1560472. [Google Scholar] [CrossRef]
- Shih, D.Q.; Dansky, H.M.; Fleisher, M.; Assmann, G.; Fajans, S.S.; Stoffel, M. Genotype/phenotype relationships in HNF-4alpha/MODY1: Haploinsufficiency is associated with reduced apolipoprotein (AII), apolipoprotein (CIII), lipoprotein(a), and triglyceride levels. Diabetes 2000, 49, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K. Roles of HNF1α and HNF4α in pancreatic β-cells: Lessons from a monogenic form of diabetes (MODY). Vitam. Horm. 2014, 95, 407–423. [Google Scholar]
- Carlsson, A.; Shepherd, M.; Ellard, S.; Weedon, M.; Lernmark, Å.; Forsander, G.; Colclough, K.; Brahimi, Q.; Valtonen-Andre, C.; Ivarsson, S.A.; et al. Absence of Islet Autoantibodies and Modestly Raised Glucose Values at Diabetes Diagnosis Should Lead to Testing for MODY: Lessons From a 5-Year Pediatric Swedish National Cohort Study. Diabetes Care 2020, 43, 82–89. [Google Scholar] [CrossRef]
- Marchand, L.; Li, M.; Leblicq, C.; Rafique, I.; Alarcon-Martinez, T.; Lange, C.; Rendon, L.; Tam, E.; Courville-Le Bouyonnec, A.; Polychronakos, C. Monogenic Causes in the Type 1 Diabetes Genetics Consortium Cohort: Low Genetic Risk for Autoimmunity in Case Selection. J. Clin. Endocrinol. Metab. 2021, 106, 1804–1810. [Google Scholar] [CrossRef]
- Tatsi, E.B.; Kanaka-Gantenbein, C.; Scorilas, A.; Chrousos, G.P.; Sertedaki, A. Next generation sequencing targeted gene panel in Greek MODY patients increases diagnostic accuracy. Pediatr. Diabetes 2020, 21, 28–39. [Google Scholar] [CrossRef]
- Pihoker, C.; Gilliam, L.K.; Ellard, S.; Dabelea, D.; Davis, C.; Dolan, L.M.; Greenbaum, C.J.; Imperatore, G.; Lawrence, J.M.; Marcovina, S.M.; et al. Prevalence, Characteristics and Clinical Diagnosis of Maturity Onset Diabetes of the Young Due to Mutations in HNF1A, HNF4A, and Glucokinase: Results From the SEARCH for Diabetes in Youth. J. Clin. Endocrinol. Metab. 2013, 98, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.M.; Hicks, S.; Shepherd, M.H.; Colclough, K.; Hattersley, A.T.; Ellard, S. Maturity-onset diabetes of the young (MODY): How many cases are we missing? Diabetologia 2010, 53, 2504–2508. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Starkey, B.J.; Powell, R.J.; Gribble, F.M.; Clark, P.M.; Hattersley, A.T. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003, 362, 1275–1281. [Google Scholar] [CrossRef]
- Bacon, S.; Kyithar, M.P.; Rizvi, S.R.; Donnelly, E.; McCarthy, A.; Burke, M.; Colclough, K.; Ellard, S.; Byrne, M.M. Successful maintenance on sulphonylurea therapy and low diabetes complication rates in a HNF1A-MODY cohort. Diabet. Med. 2016, 33, 976–984. [Google Scholar] [CrossRef]
- Christensen, A.S.; Hædersdal, S.; Storgaard, H.; Rose, K.; Hansen, N.L.; Holst, J.J.; Hansen, T.; Knop, F.K.; Vilsbøll, T. GIP and GLP-1 Potentiate Sulfonylurea-Induced Insulin Secretion in Hepatocyte Nuclear Factor 1α Mutation Carriers. Diabetes 2020, 69, 1989–2002. [Google Scholar] [CrossRef]
- Tan, C.S.H.; Ang, S.F.; Lim, S.C. Response to multiple glucose-lowering agents in a sib-pair with a novel HNF1α (MODY3) variant. Eur. J. Hum. Genet. 2020, 28, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Sidelmann Christensen, A.; Storgaard, H.; Hædersdal, S.; Hansen, T.; Knop, F.K.; Vilsbøll, T. Glimepiride monotherapy versus combination of glimepiride and linagliptin therapy in patients with HNF1A-diabetes: A protocol for a randomised, double-blinded, placebo-controlled trial. BMJ Open 2018, 8, e022517. [Google Scholar] [CrossRef]
- Christensen, A.S.; Hædersdal, S.; Støy, J.; Storgaard, H.; Kampmann, U.; Forman, J.L.; Seghieri, M.; Holst, J.J.; Hansen, T.; Knop, F.K.; et al. Efficacy and Safety of Glimepiride with or without Linagliptin Treatment in Patients With HNF1A Diabetes (Maturity-Onset Diabetes of the Young Type 3): A Randomized, Double-Blinded, Placebo-Controlled, Crossover Trial (GLIMLINA). Diabetes Care 2020, 43, 2025–2033. [Google Scholar] [CrossRef] [PubMed]
- Østoft, S.H.; Bagger, J.I.; Hansen, T.; Pedersen, O.; Faber, J.; Holst, J.J.; Knop, F.K.; Vilsbøll, T. Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: A double-blind, randomized, crossover trial. Diabetes Care 2014, 37, 1797–1805. [Google Scholar] [CrossRef]
- Broome, D.T.; Tekin, Z.; Pantalone, K.M.; Mehta, A.E. Novel Use of GLP-1 Receptor Agonist Therapy in HNF4A-MODY. Diabetes Care 2020, 43, e65. [Google Scholar] [CrossRef]
- Broome, D.T.; Pantalone, K.M.; Kashyap, S.R.; Philipson, L.H. Approach to the Patient with MODY-Monogenic Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Sauer, B.; Gonzalez, F.J. Laron dwarfism and non-insulin-dependent diabetes mellitus in the Hnf-1alpha knockout mouse. Mol. Cell. Biol. 1998, 18, 3059–3068. [Google Scholar] [CrossRef]
- Hagenfeldt-Johansson, K.A.; Herrera, P.L.; Wang, H.; Gjinovci, A.; Ishihara, H.; Wollheim, C.B. Beta-cell-targeted expression of a dominant-negative hepatocyte nuclear factor-1 alpha induces a maturity-onset diabetes of the young (MODY)3-like phenotype in transgenic mice. Endocrinology 2001, 142, 5311–5320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shih, D.Q.; Heimesaat, M.; Kuwajima, S.; Stein, R.; Wright, C.V.E.; Stoffel, M. Profound defects in pancreatic β-cell function in mice with combined heterozygous mutations in Pdx-1, Hnf-1α, and Hnf-3β. Proc. Natl. Acad. Sci. USA 2002, 99, 3818–3823. [Google Scholar] [CrossRef]
- Quilichini, E.; Fabre, M.; Nord, C.; Dirami, T.; Le Marec, A.; Cereghini, S.; Pasek, R.C.; Gannon, M.; Ahlgren, U.; Haumaitre, C. Insights into the etiology and physiopathology of MODY5/HNF1B pancreatic phenotype with a mouse model of the human disease. J. Pathol. 2021, 254, 31–45. [Google Scholar]
- Saha, K.; Jaenisch, R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 2009, 5, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Models Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef]
- Balboa, D.; Saarimäki-Vire, J.; Borshagovski, D.; Survila, M.; Lindholm, P.; Galli, E.; Eurola, S.; Ustinov, J.; Grym, H.; Huopio, H.; et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife 2018, 7, e38519. [Google Scholar] [CrossRef] [PubMed]
- Stepniewski, J.; Kachamakova-Trojanowska, N.; Ogrocki, D.; Szopa, M.; Matlok, M.; Beilharz, M.; Dyduch, G.; Malecki, M.T.; Jozkowicz, A.; Dulak, J. Induced pluripotent stem cells as a model for diabetes investigation. Sci. Rep. 2015, 5, 8597. [Google Scholar] [CrossRef]
- Teo, A.K.K.; Gupta, M.K.; Doria, A.; Kulkarni, R.N. Dissecting diabetes/metabolic disease mechanisms using pluripotent stem cells and genome editing tools. Mol. Metab. 2015, 4, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Braverman-Gross, C.; Nudel, N.; Ronen, D.; Beer, N.L.; McCarthy, M.I.; Benvenisty, N. Derivation and molecular characterization of pancreatic differentiated MODY1-iPSCs. Stem Cell Res. 2018, 31, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.H.J.; Jasmen, J.B.; Lim, C.S.; Lau, H.H.; Krishnan, V.G.; Kadiwala, J.; Kulkarni, R.N.; Ræder, H.; Vallier, L.; Hoon, S.; et al. HNF4A Haploinsufficiency in MODY1 Abrogates Liver and Pancreas Differentiation from Patient-Derived Induced Pluripotent Stem Cells. iScience 2019, 16, 192–205. [Google Scholar] [CrossRef]
- Teo, A.K.K.; Lau, H.H.; Valdez, I.A.; Dirice, E.; Tjora, E.; Raeder, H.; Kulkarni, R.N. Early Developmental Perturbations in a Human Stem Cell Model of MODY5/HNF1B Pancreatic Hypoplasia. Stem Cell Rep. 2016, 6, 357–367. [Google Scholar] [CrossRef]
- Vethe, H.; Bjørlykke, Y.; Ghila, L.M.; Paulo, J.A.; Scholz, H.; Gygi, S.P.; Chera, S.; Ræder, H. Probing the missing mature β-cell proteomic landscape in differentiating patient iPSC-derived cells. Sci. Rep. 2017, 7, 4780. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Diaz, F.L.; Osorio-Quintero, C.; Diaz-Miranda, M.A.; Kishore, S.; Leavens, K.; Jobaliya, C.; Stanescu, D.; Ortiz-Gonzalez, X.; Yoon, C.; Chen, C.S.; et al. Modeling Monogenic Diabetes using Human ESCs Reveals Developmental and Metabolic Deficiencies Caused by Mutations in HNF1A. Cell Stem Cell 2019, 25, 273.e5–289.e5. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Guo, M.; Zhou, T.; Tan, L.; Chong, C.N.; Zhang, T.; Dong, X.; Xiang, J.Z.; Yu, A.S.; Yue, L.; et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell 2016, 19, 326–340. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef]
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 2018, 145, dev156166. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Z.; Song, E.; Xu, T. Islet organoid as a promising model for diabetes. Protein Cell 2021, 1–19. [Google Scholar]
- Balboa, D.; Iworima, D.G.; Kieffer, T.J. Human Pluripotent Stem Cells to Model Islet Defects in Diabetes. Front. Endocrinol. 2021, 12, 149. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Full Name | Gene Function | Clinical Manifestation | Diabetic Complications |
|---|---|---|---|---|
| HNF1A | Hepatocyte nuclear factor 1 alpha | transcription factor | progressive insulin secretory defect; diminished renal threshold for glycosuria; | common |
| GCK | Glucokinase | enzyme in the first step of glucose metabolism | stable, mild fasting hyperglycemia; | rare |
| HNF4A | Hepatocyte nuclear factor 4 alpha | transcription factor | transient neonatal diabetes; progressive insulin secretory defect | common |
| HNF1B | Hepatocyte nuclear factor 1 beta | transcription factor | renal abnormalities and insufficiency at young age; liver test abnormalities; exocrine pancreatic dysfunction; hyperuricemia | common |
| PDX1 | Pancreatic and duodenal homebox-1 | transcription factor | permanent neonatal diabetes in homozygote; pancreas agenesis | unknown |
| NEUROD1 | Neurogenic differentiation 1 | transcription factor | neonatal diabetes; pancreatic abnormalities; child or adult-onset diabetes neurological abnormalities | unknown |
| KLF11 | Krupell-like factor 11 | transcription factor | pancreatic malignancy; similar to T2DM | unknown |
| CEL | Carboxyl ester lipase | controls exocrine and endocrine functions of pancreas | exocrine pancreatic dysfunction; lipomatosis and fibrosis with posterior diabetes development | unknown |
| PAX4 | Paired box 4 | transcription factor | possible ketoacidosis | unknown |
| INS | Insulin | encode the insulin precursor | permanent neonatal diabetes | unknown |
| BLK | B-lymphoid tyrosine kinase | tyrosine kinase functions in signal transduction | overweight | unknown |
| ABCC8 | ATP-binding cassette C8 | regulating insulin release | permanent or transient neonatal diabetes | unknown |
| KCNJ11 | Inwardly rectifying potassium channel subfamily J member 11 | regulating insulin release | neonatal diabetes in homozygote | unknown |
| APPL1 | Adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 | insulin signaling pathway | insulin secretion defect; child or adult-onset diabetes | unknown |
| MODY Subtype | Pluripotent Cells | Differentiated Cell Type | Control Lines | Mechanism Revealed | Reference |
|---|---|---|---|---|---|
| HNF1B-MODY | Patient-derived hiPSCs | Pancreatic progenitors | Family non-diseased and non-family control individuals | Compensatory increase in PDX1 in mutant pancreatic progenitors. | [157] |
| HNF4A-MODY | Patient-derived hiPSCs | Insulin-producing beta-cells | Family non-diseased and non-family control individuals | No effect on expression of insulin genes, nor in the development of insulin-producing beta cells | [158] |
| HNF4A-MODY | Patient-derived hiPSCs | Hepatopancreatic progenitors (HPPs) | Family non-diseased and non-family control individuals | Alterations in hepatic and pancreatic beta-cell signatures and abnormal cytoplasmic localisation of HNF4A. | [156] |
| HNF1A-MODY | ESCs | Pancreatic beta-like cells | Isogenic control | Increase in alpha-cell gene expression markers, impaired insulin secretion, defect in glycolysis and mitochondrial respiration. | [159] |
| KCNJ11-MODY | ESCs (biallelic mutation introduced) | Pancreatic beta-like cells | Isogenic control | Impaired insulin secretion, defective glucose homeostasis | [160] |
| INS-MODY | Patient-derived hiPSCs | Pancreatic beta-like cells | Isogenic control | Increased expression of ER-stress associated genes, reduced proliferation in vitro, lower insulin secretion in vivo together with increased ER-stress markers. | [152] |
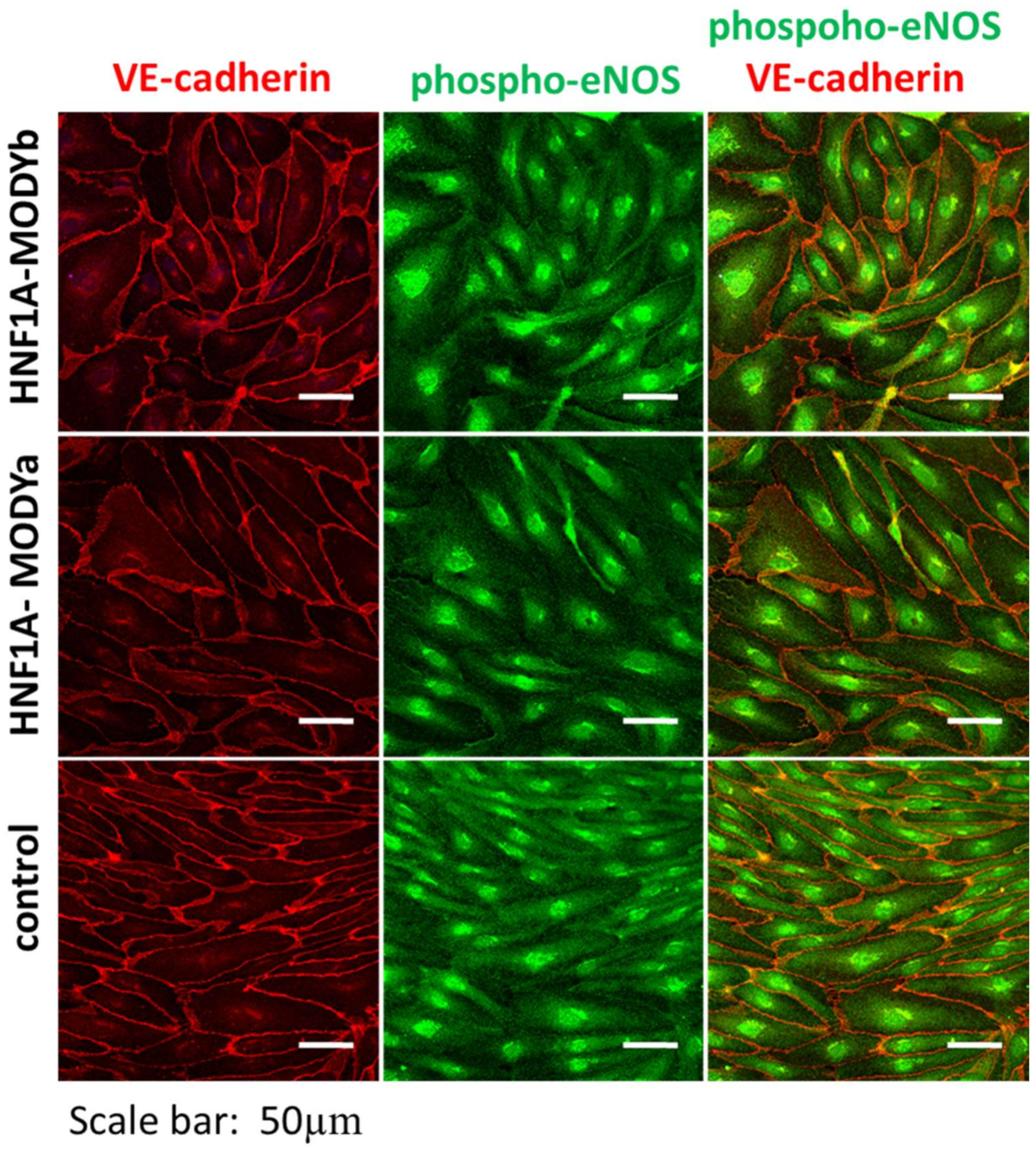
| HNF1A-MODY | hiPSCs | Endothelial cells | Isogenic control | Increased vascular permeability in response to pro-inflammatory cytokine, no difference in pro-angiogenic response. | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoczek, D.; Dulak, J.; Kachamakova-Trojanowska, N. Maturity Onset Diabetes of the Young—New Approaches for Disease Modelling. Int. J. Mol. Sci. 2021, 22, 7553. https://doi.org/10.3390/ijms22147553
Skoczek D, Dulak J, Kachamakova-Trojanowska N. Maturity Onset Diabetes of the Young—New Approaches for Disease Modelling. International Journal of Molecular Sciences. 2021; 22(14):7553. https://doi.org/10.3390/ijms22147553
Chicago/Turabian StyleSkoczek, Dawid, Józef Dulak, and Neli Kachamakova-Trojanowska. 2021. "Maturity Onset Diabetes of the Young—New Approaches for Disease Modelling" International Journal of Molecular Sciences 22, no. 14: 7553. https://doi.org/10.3390/ijms22147553
APA StyleSkoczek, D., Dulak, J., & Kachamakova-Trojanowska, N. (2021). Maturity Onset Diabetes of the Young—New Approaches for Disease Modelling. International Journal of Molecular Sciences, 22(14), 7553. https://doi.org/10.3390/ijms22147553