
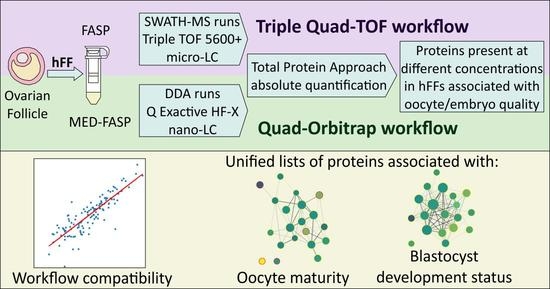
Compatibility of Distinct Label-Free Proteomic Workflows in Absolute Quantification of Proteins Linked to the Oocyte Quality in Human Follicular Fluid

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results
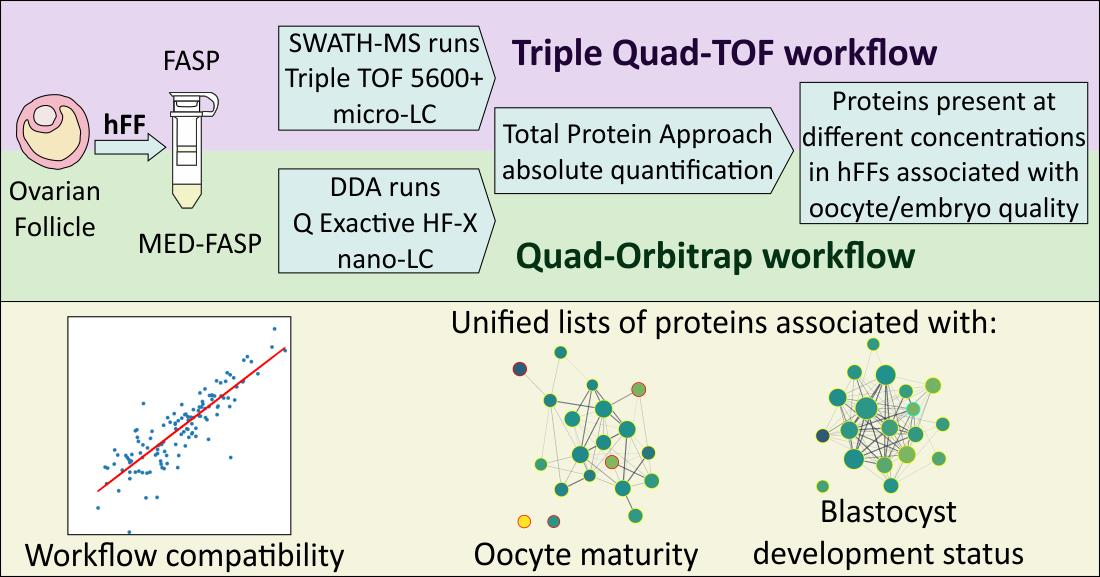
2.1. Experimental Design
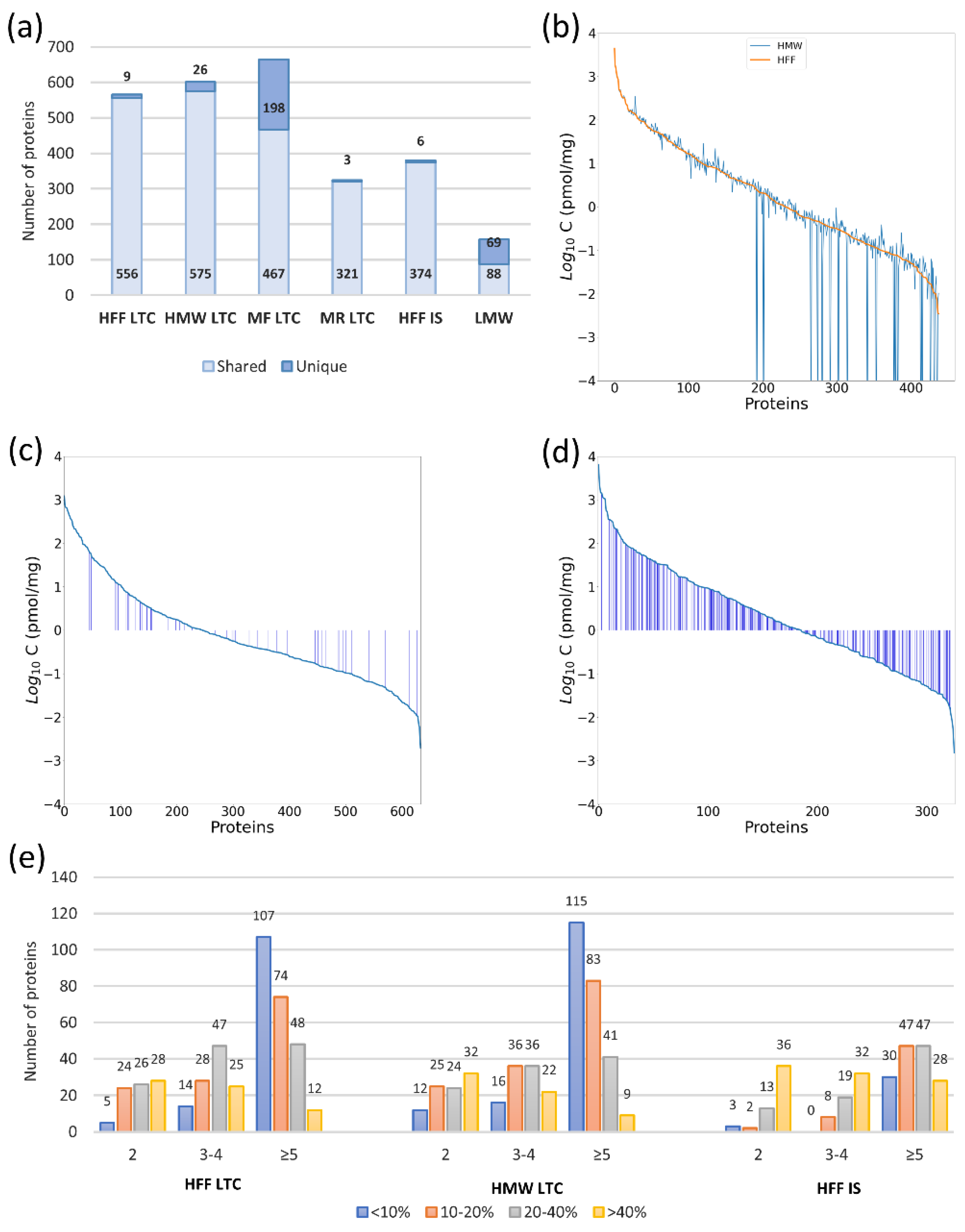
2.2. The Quad-Orbitrap Workflow
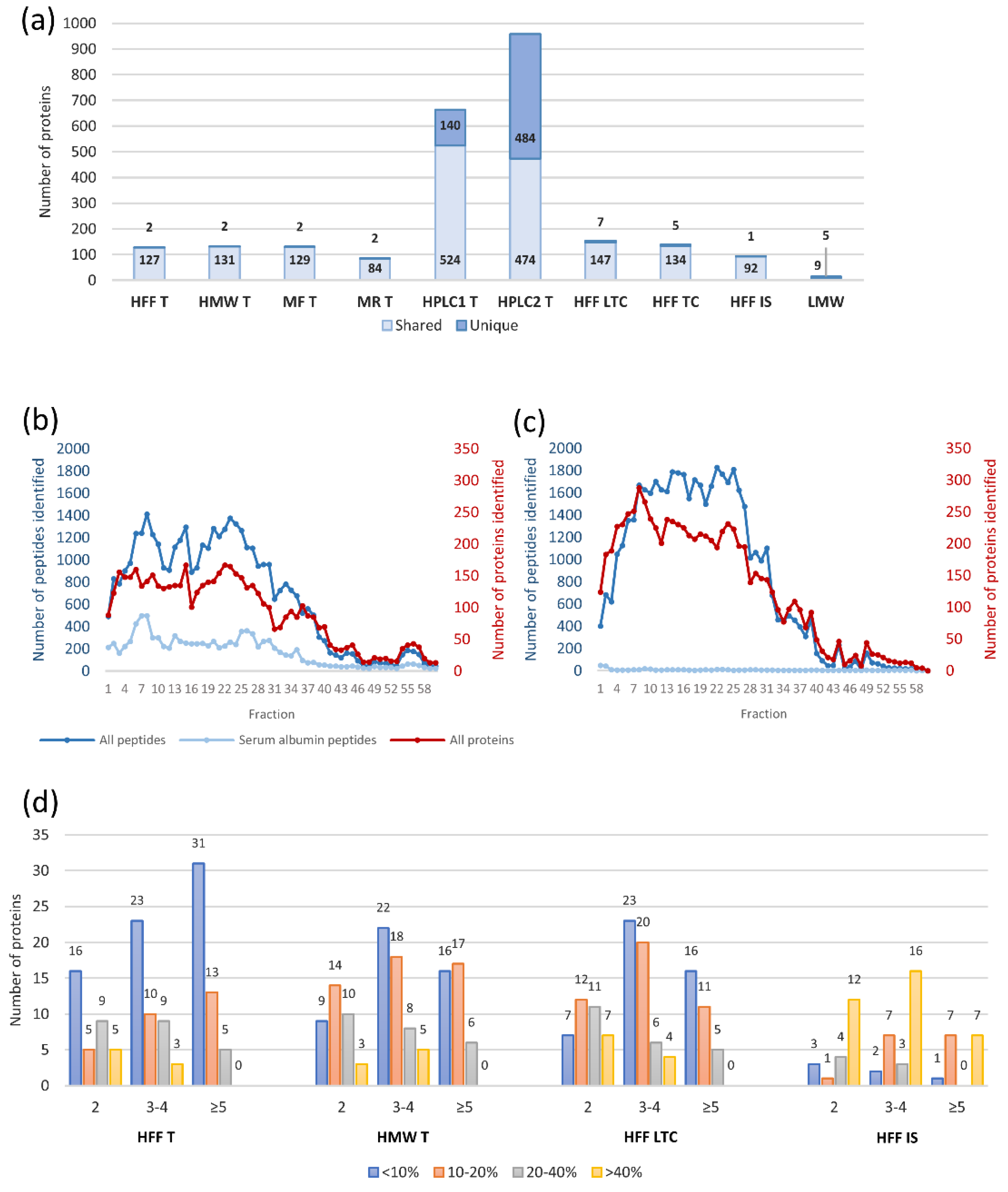
2.2.1. Workflow Optimization
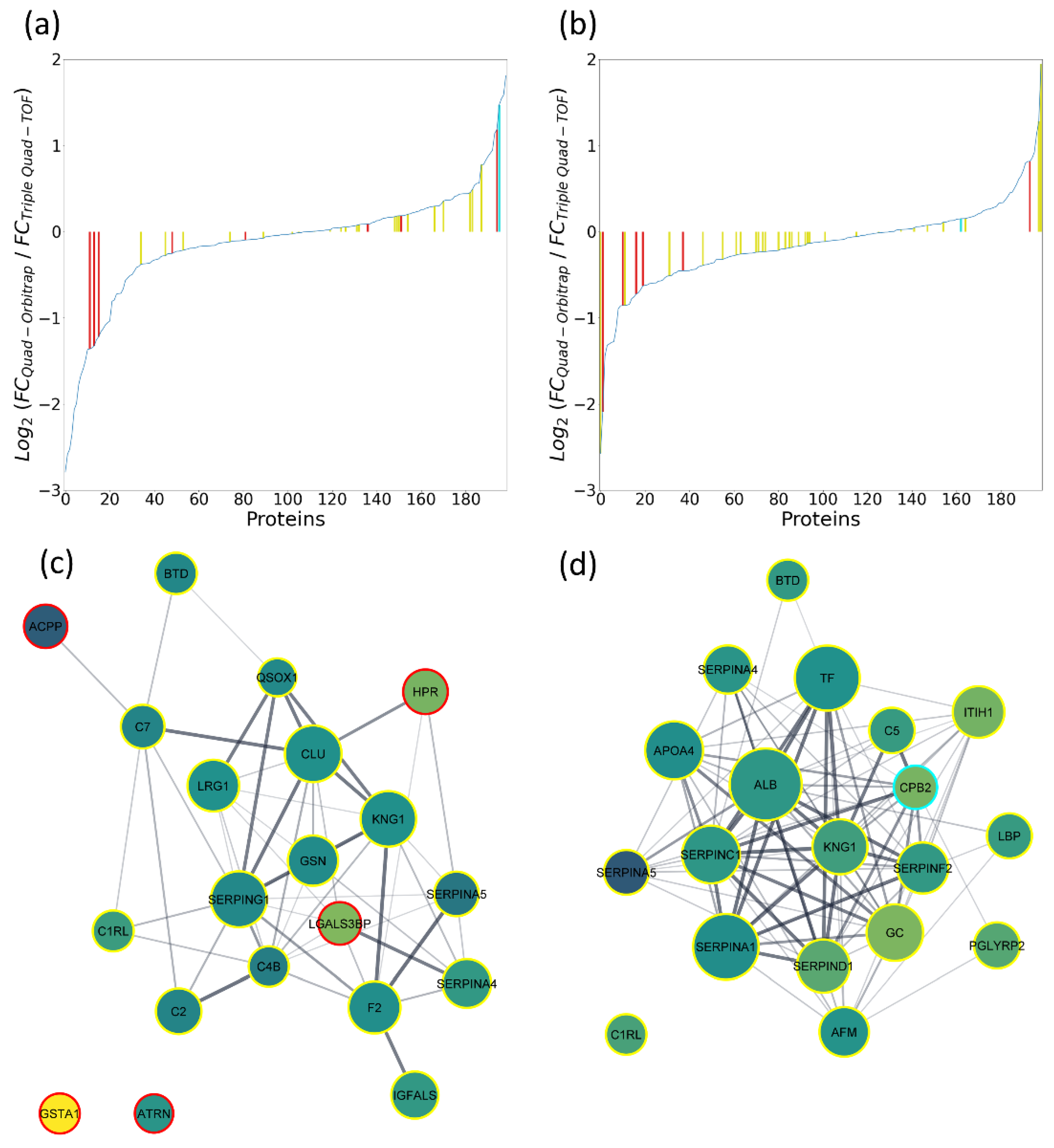
2.2.2. Functional Enrichment Analysis on Interaction Networks Created in STRING Database
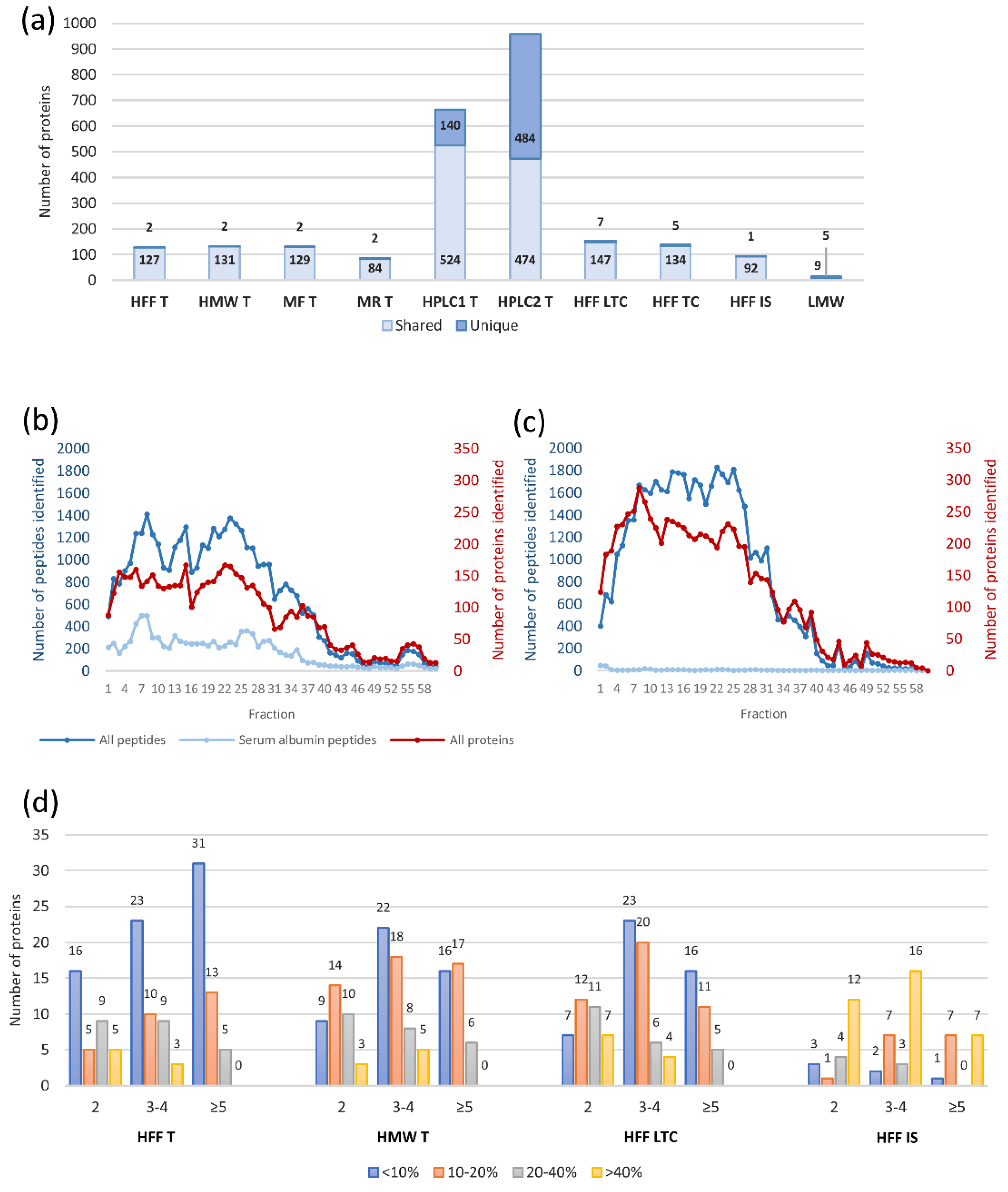
2.3. The Triple Quad-TOF Workflow
2.4. Literature Comparison to Proteomic Studies of hFF and Related Biological Materials
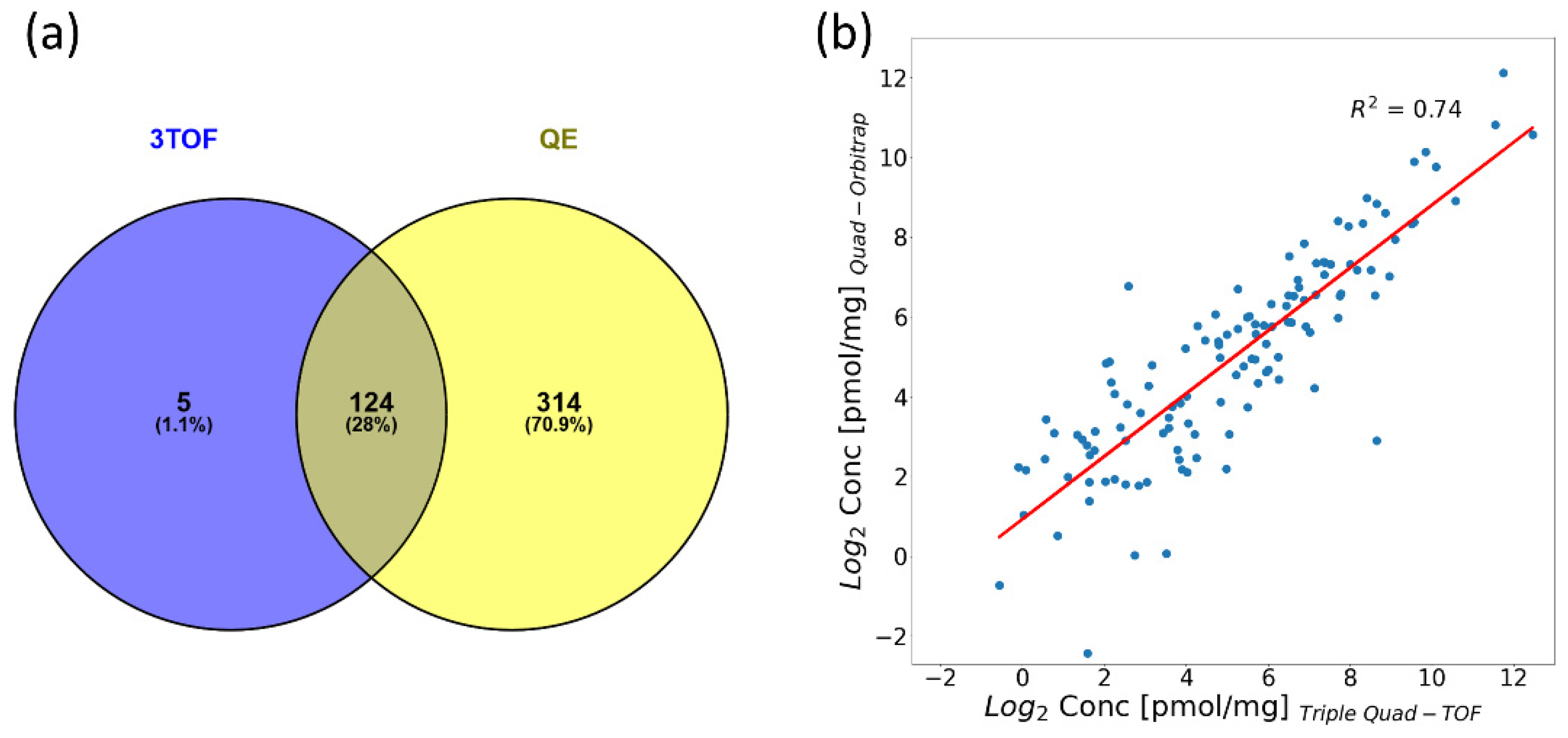
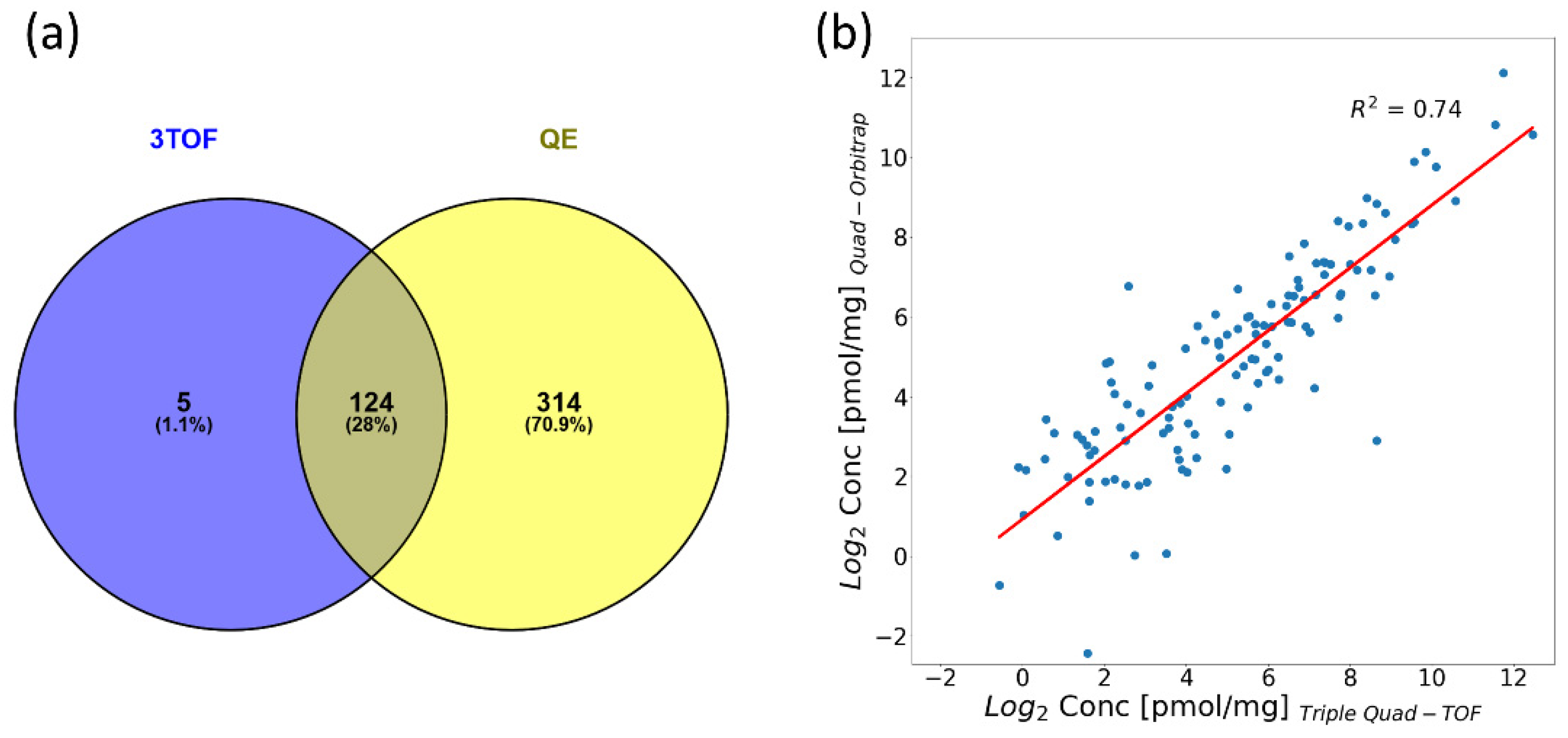
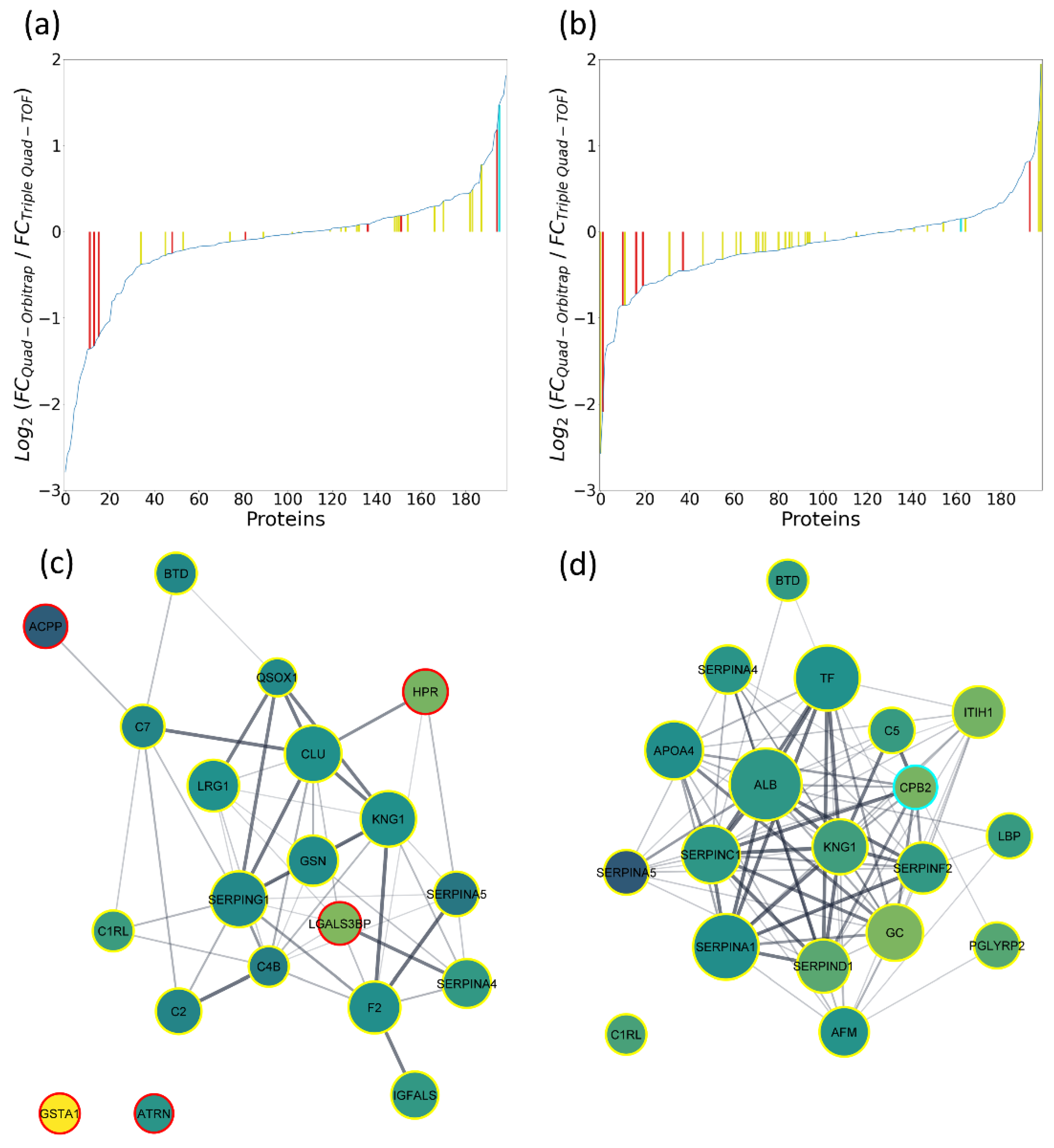
2.5. Compatibility of the Quad-Orbitrap and Triple Quad-TOF Workflows
2.5.1. Quantification Capability in Pooled Material
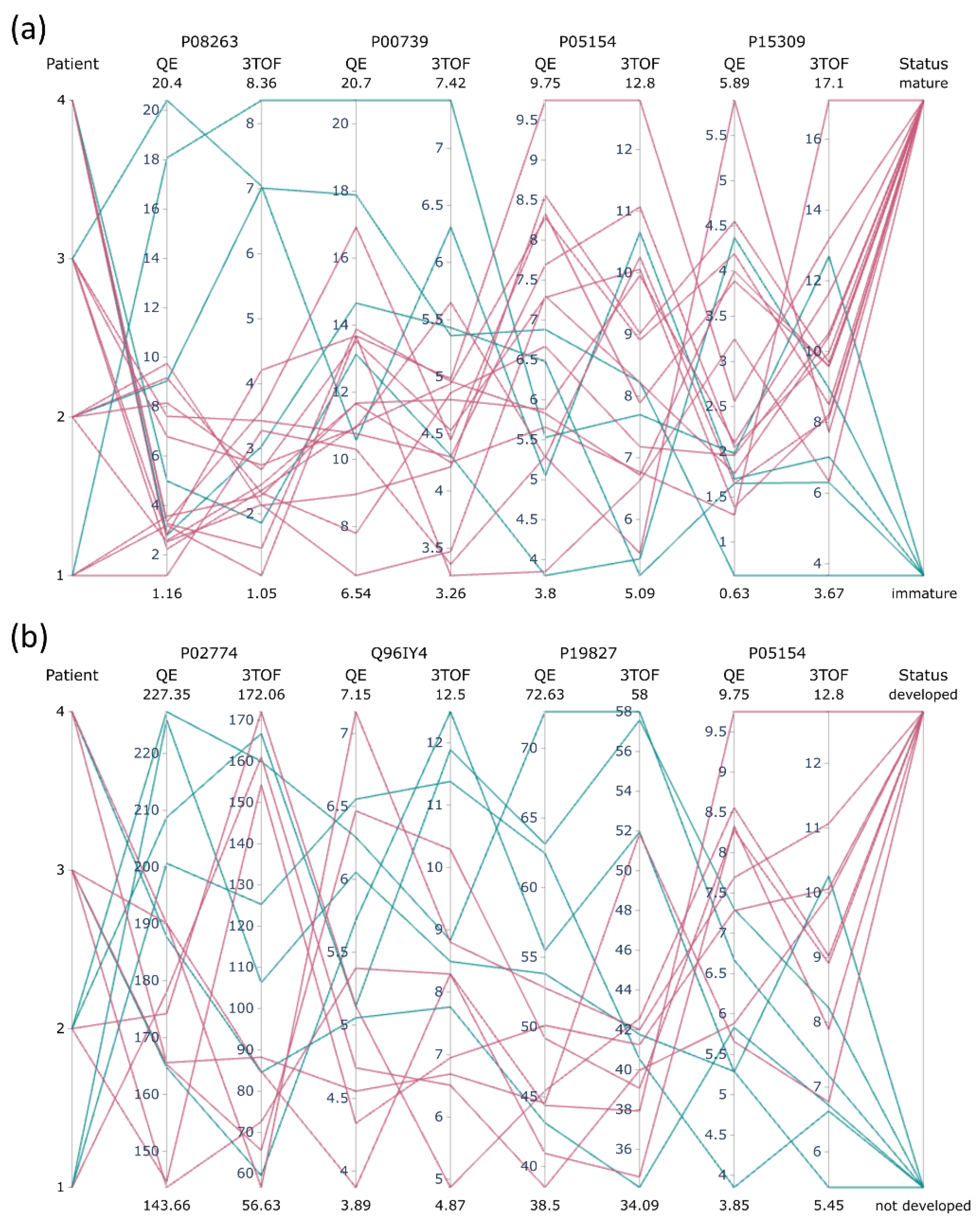
2.5.2. Pilot Study on Clinical Samples
3. Discussion
4. Materials and Methods
4.1. Research Approval
4.2. Collection of Samples
4.3. Experimental Design
4.4. Sample Preparation
4.4.1. Protein Fractionation
4.4.2. Protein Digestion
4.4.3. Peptide Fractionation by High pH RP-HPLC
4.5. LC-MS/MS Measurements and Quantitative Data Processing
4.5.1. Triple Quad-TOF Workflow
4.5.2. Quad-Orbitrap Workflow
4.6. Data Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virant-Klun, I.; Leicht, S.; Hughes, C.; Krijgsveld, J. Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell. Proteomics 2016, 15, 2616–2627. [Google Scholar] [CrossRef] [Green Version]
- Revelli, A.; Piane, L.D.; Casano, S.; Molinari, E.; Massobrio, M.; Rinaudo, P. Follicular Fluid Content and Oocyte Quality: From Single Biochemical Markers to Metabolomics. Reprod. Biol. Endocrinol. 2009, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Vargas, P.; Muñoz, M.; Domínguez, F. Identifying Biomarkers for Predicting Successful Embryo Implantation: Applying Single to Multi-OMICs to Improve Reproductive Outcomes. Hum. Reprod. Update 2020, 26, 264–301. [Google Scholar] [CrossRef] [PubMed]
- La Cour Poulsen, L.; Pla, I.; Sanchez, A.; Grøndahl, M.L.; Marko-Varga, G.; Yding Andersen, C.; Englund, A.L.M.; Malm, J. Progressive Changes in Human Follicular Fluid Composition over the Course of Ovulation: Quantitative Proteomic Analyses. Mol. Cell. Endocrinol. 2019, 495, 110522. [Google Scholar] [CrossRef]
- Bianchi, L.; Gagliardi, A.; Landi, C.; Focarelli, R.; Leo, V.D.; Luddi, A.; Bini, L.; Piomboni, P. Protein Pathways Working in Human Follicular Fluid: The Future for Tailored IVF? Expert Rev. Mol. Med. 2016, 18. [Google Scholar] [CrossRef] [PubMed]
- Zamah, A.M.; Hassis, M.E.; Albertolle, M.E.; Williams, K.E. Proteomic Analysis of Human Follicular Fluid from Fertile Women. Clin. Proteomics 2015, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.W.; Kim, S.K.; Cho, K.-C.; Kim, M.-S.; Suh, C.S.; Lee, J.R.; Kim, K.P. Proteomic Analysis of Human Follicular Fluid in Poor Ovarian Responders during in Vitro Fertilization. Proteomics 2017, 17, 1600333. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, R.; Schmidt, A.; Pastuschek, J.; Müller, M.M.; Fritzsche, A.; Dieterle, S.; Greb, R.R.; Markert, U.R.; Slevogt, H. Comparison of Sample Preparation Techniques and Data Analysis for the LC-MS/MS-Based Identification of Proteins in Human Follicular Fluid. Am. J. Reprod. Immunol. 2018, 80, e12994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, X.; Li, P.; Zhou, F.; Kong, L.; Qiu, J.; Yuan, Z.; Tan, J. TMT Based Proteomic Analysis of Human Follicular Fluid from Overweight/Obese and Normal-Weight Patients With Polycystic Ovary Syndrome. Front. Endocrinol. 2019, 10, 821. [Google Scholar] [CrossRef] [PubMed]
- Pla, I.; Sanchez, A.; Pors, S.E.; Pawlowski, K.; Appelqvist, R.; Sahlin, K.B.; Poulsen, L.L.C.; Marko-Varga, G.; Andersen, C.Y.; Malm, J. Proteome of Fluid from Human Ovarian Small Antral Follicles Reveals Insights in Folliculogenesis and Oocyte Maturation. Hum. Reprod. 2021, 36, 756–770. [Google Scholar] [CrossRef]
- Kushnir, M.M.; Naessén, T.; Wanggren, K.; Rockwood, A.L.; Crockett, D.K.; Bergquist, J. Protein and Steroid Profiles in Follicular Fluid after Ovarian Hyperstimulation as Potential Biomarkers of IVF Outcome. J. Proteome Res. 2012, 11, 5090–5100. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.W.; Bolbot, T.; Koulova, A.; Sneeringer, R.; Humm, K.; Dagon, Y.; Usheva, A. Complement Factors Are Secreted in Human Follicular Fluid by Granulosa Cells and Are Possible Oocyte Maturation Factors. J. Obstet. Gynaecol. Res. 2013, 39, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Bayasula; Iwase, A.; Kobayashi, H.; Goto, M.; Nakahara, T.; Nakamura, T.; Kondo, M.; Nagatomo, Y.; Kotani, T.; Kikkawa, F. A Proteomic Analysis of Human Follicular Fluid: Comparison between Fertilized Oocytes and Non-Fertilized Oocytes in the Same Patient. J. Assist. Reprod. Genet. 2013, 30, 1231–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowska, A.E.; Macur, K.; Czaplewska, P.; Liss, J.; Łukaszuk, K.; Ołdziej, S. Qualitative and Quantitative Analysis of Proteome and Peptidome of Human Follicular Fluid Using Multiple Samples from Single Donor with LC–MS and SWATH Methodology. J. Proteome Res. 2017, 16, 3053–3067. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Rakus, D. Multi-Enzyme Digestion FASP and the ‘Total Protein Approach’-Based Absolute Quantification of the Escherichia Coli Proteome. J. Proteomics 2014, 109, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11, O111.016717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Shi, J.; Wang, X.; Jiang, H.; Zhu, H.-J. Label-Free Absolute Protein Quantification with Data-Independent Acquisition. J. Proteomics 2019, 200, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Ahmad Khan, A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a Resource for Proteomics Research: 2014 Update. Nucleic Acids Res. 2014, 42, D959–D965. [Google Scholar] [CrossRef] [Green Version]
- Bagnjuk, K.; Mayerhofer, A. Human Luteinized Granulosa Cells—A Cellular Model for the Human Corpus Luteum. Front. Endocrinol. 2019, 10, 452. [Google Scholar] [CrossRef]
- Zakerkish, F.; Brännström, M.; Carlsohn, E.; Sihlbom, C.; van der Post, S.; Thoroddsen, A. Proteomic Analysis of Follicular Fluid during Human Ovulation. Acta Obstet. Gynecol. Scand. 2020, 99, 917–924. [Google Scholar] [CrossRef]
- Bianchi, L.; Gagliardi, A.; Campanella, G.; Landi, C.; Capaldo, A.; Carleo, A.; Armini, A.; De Leo, V.; Piomboni, P.; Focarelli, R.; et al. A Methodological and Functional Proteomic Approach of Human Follicular Fluid En Route for Oocyte Quality Evaluation. J. Proteomics 2013, 90, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.C.; Tatone, C.; Monache, S.D.; Marci, R.; Caserta, D.; Colonna, R.; Amicarelli, F. Antioxidant Enzymatic Defences in Human Follicular Fluid: Characterization and Age-dependent Changes. Mol. Hum. Reprod. 2003, 9, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, D.; Insler, V.; Leiberman, J.R.; Glezerman, M.; Albotiano, S.; Potashnik, G.; Meizner, I. Acid Phosphatase Levels in Follicular Fluids Following Induction of Ovulation in in Vitro Fertilization Patients. J. In Vitro Fertil. Embryo Transf. 1987, 4, 181–184. [Google Scholar] [CrossRef]
- Chen, F.; Spiessens, C.; D’Hooghe, T.; Peeraer, K.; Carpentier, S. Follicular Fluid Biomarkers for Human in Vitro Fertilization Outcome: Proof of Principle. Proteome Sci. 2016, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Estes, S.J.; Ye, B.; Qiu, W.; Cramer, D.; Hornstein, M.D.; Missmer, S.A. A Proteomic Analysis of IVF Follicular Fluid in Women ≤32 Years Old. Fertil. Steril. 2009, 92, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.A.; Kim, P.D.; Patel, B.B.; Kelsen, S.G.; Braverman, A.; Swinton, D.J.; Gafken, P.R.; Jones, L.A.; Lane, W.S.; Neveu, J.M.; et al. Immunodepletion Plasma Proteomics by TripleTOF 5600 and Orbitrap Elite/LTQ-Orbitrap Velos/Q Exactive Mass Spectrometers. J. Proteome Res. 2013, 12, 4351–4365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Zheng, R.; Bayer, F.P.; Wong, C.; Chang, Y.-C.; Meng, C.; Zolg, D.P.; Reinecke, M.; Zecha, J.; Wiechmann, S.; et al. Robust, Reproducible and Quantitative Analysis of Thousands of Proteomes by Micro-Flow LC–MS/MS. Nat. Commun. 2020, 11, 157. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Hunter, C.; Chen, C.; Ge, W.; Morrice, N.; Liang, S.; Zhu, T.; Yuan, C.; Ruan, G.; Zhang, Q.; et al. Accelerated Protein Biomarker Discovery from FFPE Tissue Samples Using Single-Shot, Short Gradient Microflow SWATH MS. J. Proteome Res. 2020, 19, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R. Filter-Aided Sample Preparation for Proteome Analysis. In Microbial Proteomics: Methods and Protocols; Becher, D., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 3–10. ISBN 9781493986958. [Google Scholar]
- Carvalho, L.B.; Capelo-Martínez, J.-L.; Lodeiro, C.; Wiśniewski, J.R.; Santos, H.M. Ultrasonic-Based Filter Aided Sample Preparation as the General Method to Sample Preparation in Proteomics. Anal. Chem. 2020, 92, 9164–9171. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skowrońska, P.; Kunicki, M.; Pastuszek, E.; Konieczna, L.; Bączek, T.; Łukaszuk, K. Follicular Fat-Soluble Vitamins as Markers of Oocyte Competency. Syst. Biol. Reprod. Med. 2020, 66, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Alpha Scientists in Reproductive Medicine and ESHRE Special Interest Group of Embryology; Balaban, B.; Brison, D.R.; Calderón, G.; Catt, J.; Conaghan, J.; Cowan, L.; Ebner, T.; Gardner, D.; Hardarson, T.; et al. The Istanbul consensus workshop on embryo assessment: Proceedings of an expert meeting. Hum. Reprod. 2011, 26, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Tirumalai, R.S.; Chan, K.C.; Prieto, D.A.; Issaq, H.J.; Conrads, T.P.; Veenstra, T.D. Characterization of the Low Molecular Weight Human Serum Proteome. Mol. Cell. Proteomics 2003, 2, 1096–1103. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [Green Version]
- Lewandowska, A.E.; Macur, K.; Czaplewska, P.; Liss, J.; Łukaszuk, K.; Ołdziej, S. Human Follicular Fluid Proteomic and Peptidomic Composition Quantitative Studies by SWATH-MS Methodology. Applicability of High PH RP-HPLC Fractionation. J. Proteomics 2019, 191, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A.; Eyk, J.E.V. Preparation of Proteins and Peptides for Mass Spectrometry Analysis in a Bottom-Up Proteomics Workflow. Curr. Protoc. Mol. Biol. 2010, 90, 10–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC–SWATH–MS Acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef]
- Fel, A.; Lewandowska, A.E.; Petrides, P.E.; Wiśniewski, J.R. Comparison of Proteome Composition of Serum Enriched in Extracellular Vesicles Isolated from Polycythemia Vera Patients and Healthy Controls. Proteomes 2019, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. (2007–2015). Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://Bioinfogp.Cnb.Csic.Es/Tools/Venny/Index.Html (accessed on 14 June 2021).
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange Consortium in 2017: Supporting the Cultural Change in Proteomics Public Data Deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Workflow Abbreviation | Protein Fractionation | Fraction Analyzed | Digestion | Peptide Fractionation | Fraction Analyzed | Instrument |
|---|---|---|---|---|---|---|
| 3TOF HFF T | None | - | FASP with trypsin | None | - | Triple TOF 5600+ |
| 3TOF HMW T | Ultrafiltration | Retentate (>10 kDa) | FASP with trypsin | None | - | Triple TOF 5600+ |
| 3TOF MF T | Immunodepletion | Low abundant proteins (MARS-14) | FASP with trypsin | None | - | Triple TOF 5600+ |
| 3TOF MR T | Immunodepletion | High abundant proteins (MARS-14) | FASP with trypsin | None | - | Triple TOF 5600+ |
| 3TOF HPLC1 | None | - | FASP with trypsin | High pH RP-HPLC | 60 separate fractions | Triple TOF 5600+ |
| 3TOF HPLC2 | Immunodepletion | Low abundant proteins (MARS-14) | FASP with trypsin | High pH RP-HPLC | 60 separate fractions | Triple TOF 5600+ |
| 3TOF HFF LTC | None | MED-FASP (LysC, trypsin, and chymotrypsin) | None | - | Triple TOF 5600+ | |
| 3TOF HFF TC | None | - | MED-FASP (trypsin and chymotrypsin) | None | - | Triple TOF 5600+ |
| 3TOF HFF IS | None | - | In solution with trypsin | None | - | Triple TOF 5600+ |
| 3TOF LMW | Ultrafiltration | Filtrate (<10 kDa) | None | None | - | Triple TOF 5600+ |
| QE HFF LTC | None | - | MED-FASP (LysC, trypsin, and chymotrypsin) | None | - | Q Exactive HF-X |
| QE HMW LTC | Ultrafiltration | Retentate (>10 kDa) | MED-FASP (LysC, trypsin, and chymotrypsin) | None | - | Q Exactive HF-X |
| QE MF LTC | Immunodepletion | Low abundant proteins (MARS-14) | MED-FASP (LysC, trypsin, and chymotrypsin) | None | - | Q Exactive HF-X |
| QE MR LTC | Immunodepletion | High abundant proteins (MARS-14) | MED-FASP (LysC, trypsin, and chymotrypsin) | None | - | Q Exactive HF-X |
| QE HFF IS | None | - | In solution with trypsin | None | - | Q Exactive HF-X |
| QE LMW | Ultrafiltration | Filtrate (<10 kDa) | None | None | - | Q Exactive HF-X |
| All Identified Proteins | All Proteins Identified in HMW Fraction | Proteins Identified only in HMW Fraction | All Proteins Identified in LMW Fraction | Proteins Identified only in LMW Fraction | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Resource | QE (942) | 3TOF (1182) | QE (873) | 3TOF (1177) | QE (785) | 3TOF (1168) | QE (157) | 3TOF (14) | QE (69) | 3TOF (5) |
| Plasma Proteome Database [21] | 773 | 975 | 723 | 975 | 644 | 966 | 129 | 9 | 50 | 0 |
| Human oocyte [1] (oocyte specific) | 226 (22) | 301 (18) | 211 (22) | 301 (18) | 183 (18) | 294 (17) | 43 (4) | 7 (1) | 15 | 0 |
| Human granulosa cell [22] | 436 | 599 | 390 | 599 | 346 | 591 | 90 | 8 | 46 | 0 |
| HFF (Zamah et al., 2015) [6] | 545 | 610 | 542 | 610 | 470 | 602 | 75 | 8 | 3 | 0 |
| HFF (Bianchi et al., 2016) [5] | 357 | 368 | 352 | 368 | 284 | 360 | 73 | 8 | 5 | 0 |
| HFF (Oh et al., 2017) [7] | 521 | 534 | 518 | 534 | 445 | 525 | 76 | 9 | 3 | 0 |
| HFF (Poulsen et al., 2019) [4] | 336 | 330 | 333 | 330 | 269 | 321 | 67 | 9 | 3 | 0 |
| HFF (Zhang et al., 2019) [9] | 567 | 647 | 540 | 647 | 463 | 640 | 104 | 7 | 27 | 0 |
| HFF from hSAF (Pla et al., 2020) [10] | 829 | 987 | 794 | 987 | 708 | 978 | 121 | 9 | 35 | 0 |
| Unique | 28 | 40 | 20 | 35 | 20 | 35 | 8 | 5 | 8 | 5 |
| Method 1 (Quad-Orbitrap Workflow) | Method 2 (Triple Quad-TOF Workflow) | |
|---|---|---|
| Instrument | Q Exactive HF-X | TripleTOF 5600+ |
| LC (flowrate) | nanoflow (0.3 µL/min) | microflow (5 µL/min) |
| Digestion method (enzymes) | MED-FASP (Lys-C, Trypsin, Chymotrypsin) | FASP (Trypsin) |
| Quantification method | DDA-TPA | DIA-TPA (SWATH-MS based) |
| Single LC-MS/MS analysis time (full in triplicate) | 95 min (285 min) | 30 min (90 min) |
| Number of proteins identified in the unfractionated sample using the described digestion method | 565/380 (MED-FASP/IS) | 129/154/93 (FASP/MED-FASP/IS) |
| Number of proteins identified in the LMW fraction | 157 | 14 |
| Number of proteins quantified in the pool sample | 438 | 129 |
| Number of proteins quantified in the pool sample with CV < 10%/< 20% | 126/252 | 70/98 |
| Number of proteins quantified in the clinical samples | 455 | 215 |
| Number of proteins linked to the patient factor (inter-patient differences) at 1%/5% FDR | 68/101 | 64/96 |
| Number of proteins linked to the oocyte status (inter-follicle differences) at 1%/5% FDR | 11/49 | 1/10 |
| Number of proteins linked to the blastocyst status (inter-follicle differences) at 1%/5% FDR | 13/45 | 2/7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewandowska, A.E.; Fel, A.; Thiel, M.; Czaplewska, P.; Łukaszuk, K.; Wiśniewski, J.R.; Ołdziej, S. Compatibility of Distinct Label-Free Proteomic Workflows in Absolute Quantification of Proteins Linked to the Oocyte Quality in Human Follicular Fluid. Int. J. Mol. Sci. 2021, 22, 7415. https://doi.org/10.3390/ijms22147415
Lewandowska AE, Fel A, Thiel M, Czaplewska P, Łukaszuk K, Wiśniewski JR, Ołdziej S. Compatibility of Distinct Label-Free Proteomic Workflows in Absolute Quantification of Proteins Linked to the Oocyte Quality in Human Follicular Fluid. International Journal of Molecular Sciences. 2021; 22(14):7415. https://doi.org/10.3390/ijms22147415
Chicago/Turabian StyleLewandowska, Aleksandra E., Anna Fel, Marcel Thiel, Paulina Czaplewska, Krzysztof Łukaszuk, Jacek R. Wiśniewski, and Stanisław Ołdziej. 2021. "Compatibility of Distinct Label-Free Proteomic Workflows in Absolute Quantification of Proteins Linked to the Oocyte Quality in Human Follicular Fluid" International Journal of Molecular Sciences 22, no. 14: 7415. https://doi.org/10.3390/ijms22147415
APA StyleLewandowska, A. E., Fel, A., Thiel, M., Czaplewska, P., Łukaszuk, K., Wiśniewski, J. R., & Ołdziej, S. (2021). Compatibility of Distinct Label-Free Proteomic Workflows in Absolute Quantification of Proteins Linked to the Oocyte Quality in Human Follicular Fluid. International Journal of Molecular Sciences, 22(14), 7415. https://doi.org/10.3390/ijms22147415