
Butyrate Feeding Reverses CypD-Related Mitoflash Phenotypes in Mouse Myofibers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
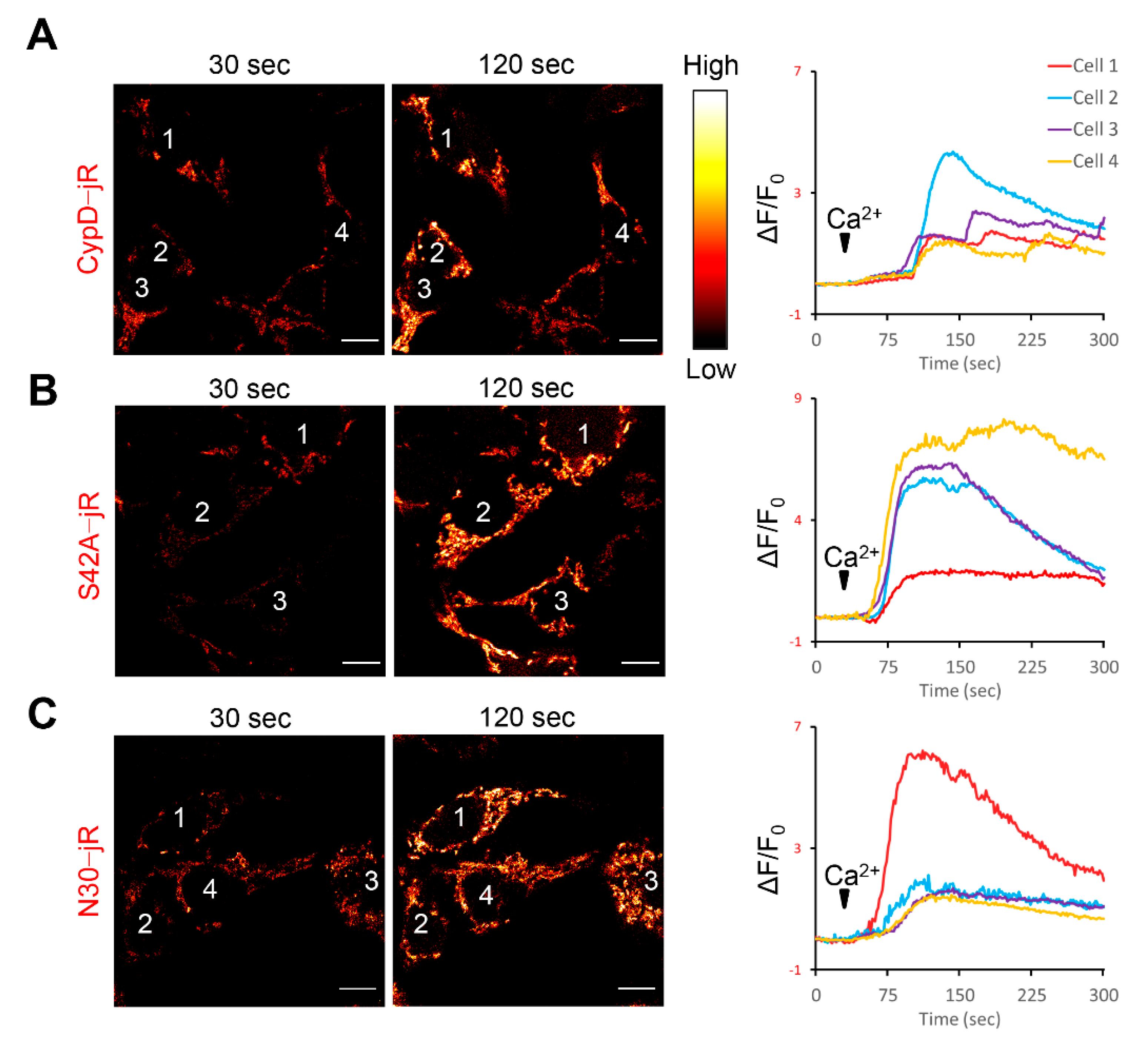
2.1. Fusion Constructs of CypD Derived Peptide and jRCaMP1b Exhibit Mitochondrial Localization and Respond to Ca2+ Stimulation
2.2. Observation of Mitoflash Associated Mitochondrial Ca2+ Efflux in the Perinuclear Region of Myofibers
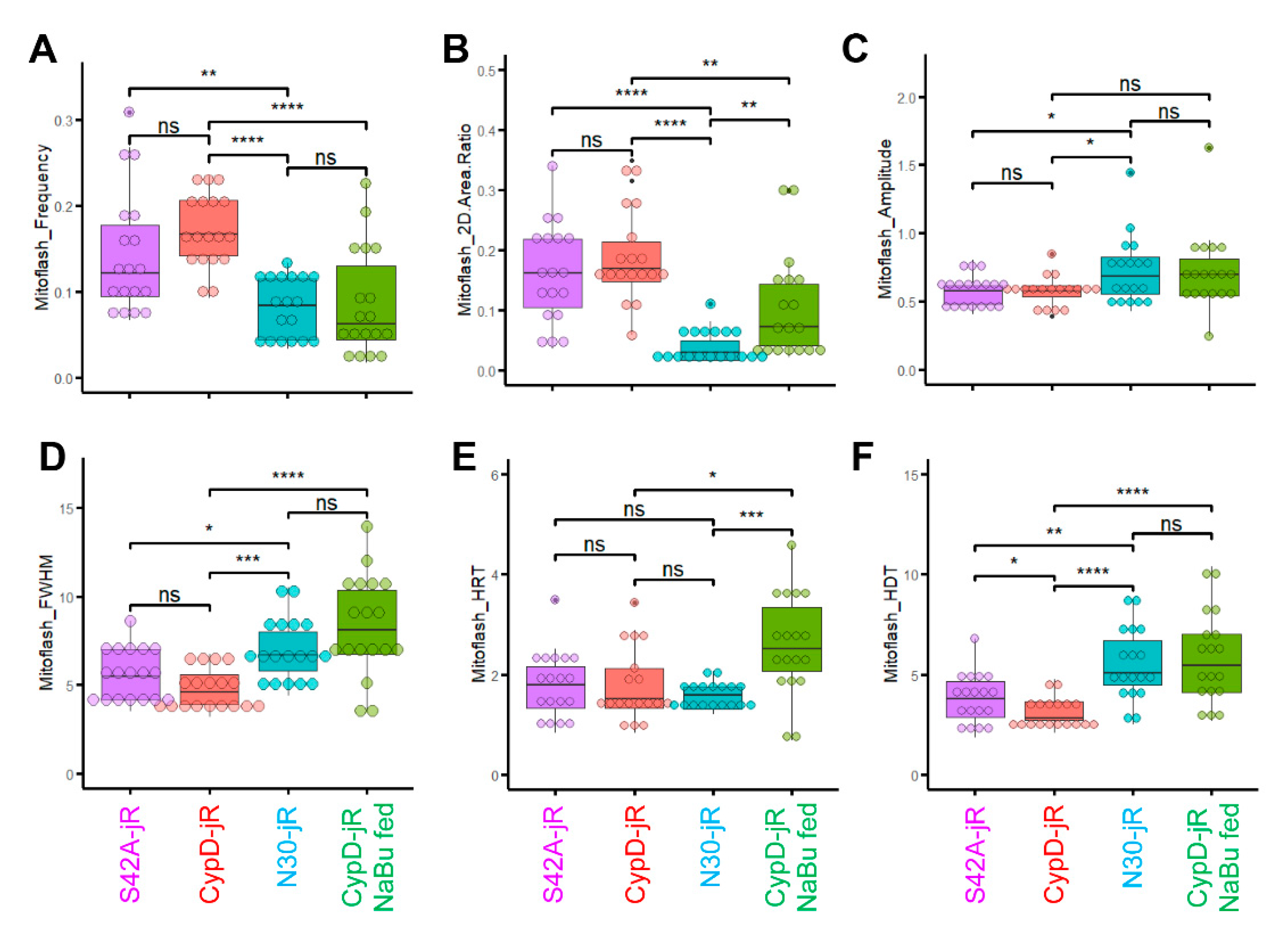
2.3. Reversal of CypD-Related Mitoflash Phenotypes in Myofibers by Butyrate Treatment
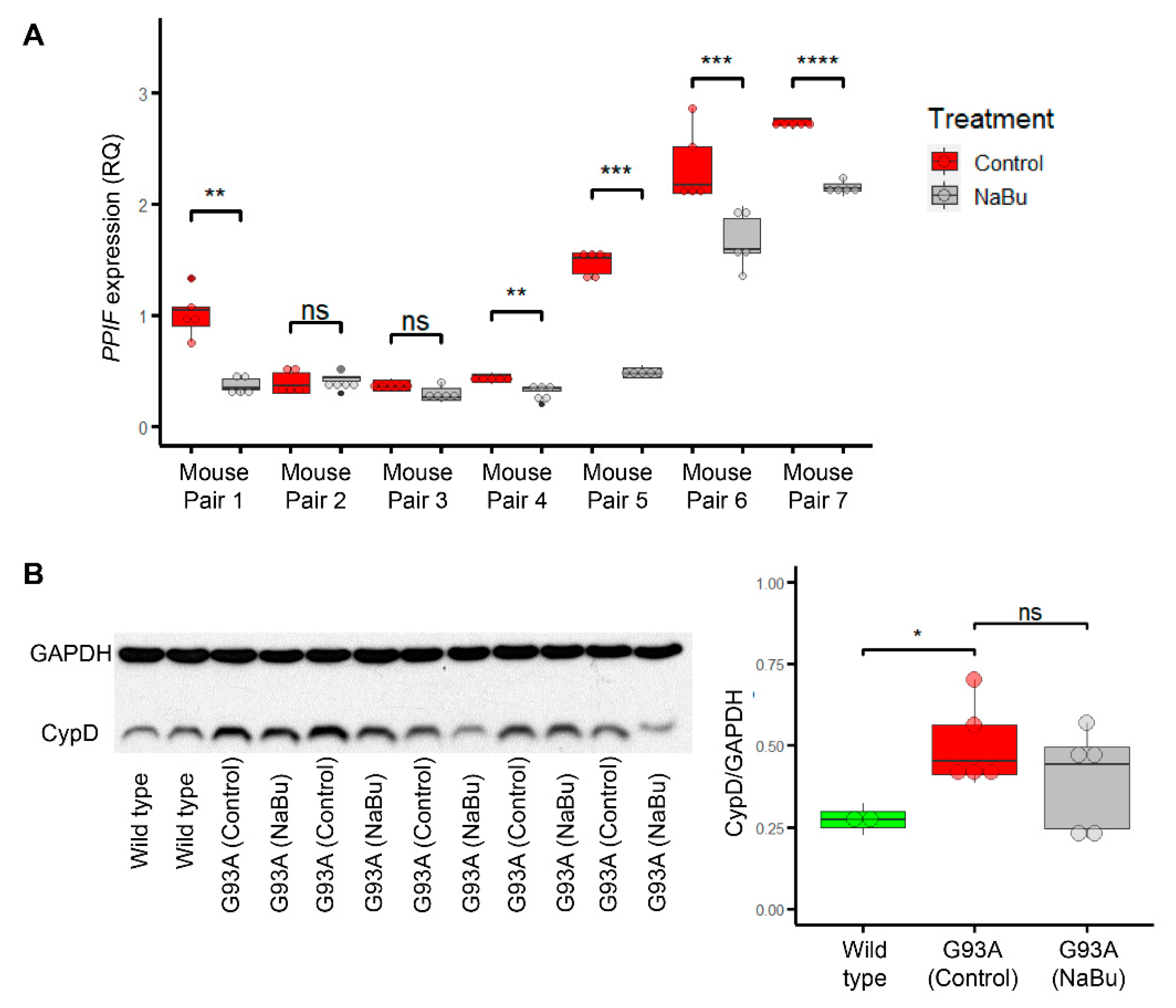
2.4. Butyrate Feeding Protects against Ectopic Upregulation of CypD in Myofibers from hSOD1G93A Mice
3. Discussion
4. Materials and Methods
4.1. Animal
4.2. Plasmid Construction
4.3. Time-lapse and Static Imaging of Cultured Cells
4.4. Electroporation, Dissection, and Time-lapse Imaging of Myofibers
4.5. SICT Based Analysis of Mitoflash Properties
4.6. RNA Extraction, Reverse Transcription, and qPCR
4.7. Western Blot
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M. Superoxide flashes in single mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef]
- Wei, L.; Salahura, G.; Boncompagni, S.; Kasischke, K.A.; Protasi, F.; Sheu, S.S.; Dirksen, R.T. Mitochondrial superoxide flashes: Metabolic biomarkers of skeletal muscle activity and disease. FASEB J. 2011, 25, 3068–3078. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S. Superoxide flashes in mouse skeletal muscle are produced by discrete arrays of active mitochondria operating coherently. PLoS ONE 2010, 5, e13035. [Google Scholar] [CrossRef] [PubMed]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H.; et al. Absence of physiological Ca2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, H.; Cheng, H. Mitochondrial flashes: From indicator characterization to in vivo imaging. Methods 2016, 109, 12–20. [Google Scholar] [CrossRef][Green Version]
- Schwarzländer, M.; Murphy, M.P.; Duchen, M.R.; Logan, D.C.; Fricker, M.D.; Halestrap, A.P.; Müller, F.L.; Rizzuto, R.; Dick, T.P.; Meyer, A.J. Mitochondrial ‘flashes’: A radical concept repHined. Trends Cell Biol. 2012, 22, 503–508. [Google Scholar] [CrossRef]
- Demaurex, N.; Schwarzländer, M. Mitochondrial flashes: Dump superoxide and dance with protons now. Antioxid. Redox Signal. 2016, 25, 550–551. [Google Scholar] [CrossRef]
- Ichas, F.; Mazat, J.-P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low-to high-conductance state. Biochim. Biophys. Acta Bioenerg. 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 2020, 11, 1370. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Muller, F.; Crofts, A.R.; Kramer, D.M. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc 1 complex. Biochemistry 2002, 41, 7866–7874. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Roberts, A.G.; Bowman, M.K.; Kramer, D.M. Architecture of the Qo site of the cytochrome bc 1 complex probed by superoxide production. Biochemistry 2003, 42, 6493–6499. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria: I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria: II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria: III. Transitional Ca2+ release. Arch. Biochem. Biophys. 1979, 195, 468–477. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Antonenko, Y.N.; Zorov, D.B. Modulation of inner mitochondrial membrane channel activity. J. Bioenerg. Biomembr. 1992, 24, 99–110. [Google Scholar] [CrossRef]
- Szabó, I.; Zoratti, M. The mitochondrial megachannel is the permeability transition pore. J. Bioenerg. Biomembr. 1992, 24, 111–117. [Google Scholar] [CrossRef]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef]
- Giorgio, V.; Von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.-A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca2+ binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F 1 FO ATP synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Jouaville, L.S.; Mazat, J.-P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef]
- Selivanov, V.A.; Ichas, F.; Holmuhamedov, E.L.; Jouaville, L.S.; Evtodienko, Y.V.; Mazat, J.-P. A model of mitochondrial Ca2+-induced Ca2+ release simulating the Ca2+ oscillations and spikes generated by mitochondria. Biophys. Chem. 1998, 72, 111–121. [Google Scholar] [CrossRef]
- Batandier, C.; Leverve, X.; Fontaine, E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 2004, 279, 17197–17204. [Google Scholar] [CrossRef]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 2016, 5, e12727. [Google Scholar] [CrossRef]
- Linard, D.; Kandlbinder, A.; Degand, H.; Morsomme, P.; Dietz, K.-J.; Knoops, B. Redox characterization of human cyclophilin D: Identification of a new mammalian mitochondrial redox sensor? Arch. Biochem. Biophys. 2009, 491, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Sciacovelli, M.; Chiara, F.; Pantic, B.; Brusilow, W.S.; Bernardi, P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. USA 2010, 107, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Varela, A.T.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin and NAD+ prevent mitochondrial ischaemia/reperfusion damage in fatty livers. Eur. J. Clin. Investig. 2018, 48, e12932. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Siegelin, M.D.; Vaira, V.; Faversani, A.; Tavecchio, M.; Chae, Y.C.; Lisanti, S.; Rampini, P.; Giroda, M.; Caino, M.C.; et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J. Natl. Cancer Inst. 2015, 107, dju502. [Google Scholar] [CrossRef]
- Parks, R.J.; Menazza, S.; Holmström, K.M.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc. Res. 2019, 115, 385–394. [Google Scholar] [CrossRef]
- Tong, H.; Chen, W.; Steenbergen, C.; Murphy, E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ. Res. 2000, 87, 309–315. [Google Scholar] [CrossRef]
- Tong, H.; Imahashi, K.; Steenbergen, C.; Murphy, E. Phosphorylation of glycogen synthase kinase-3β during preconditioning through a phosphatidylinositol-3-kinase–dependent pathway is cardioprotective. Circ. Res. 2002, 90, 377–379. [Google Scholar] [CrossRef]
- Kohr, M.J.; Aponte, A.M.; Sun, J.; Wang, G.; Murphy, E.; Gucek, M.; Steenbergen, C. Characterization of potential S-nitrosylation sites in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1327–H1335. [Google Scholar] [CrossRef] [PubMed]
- Kohr, M.J.; Sun, J.; Aponte, A.; Wang, G.; Gucek, M.; Murphy, E.; Steenbergen, C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ. Res. 2011, 108, 418–426. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011, 286, 40184–40192. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, G.; Fernández, C.; Montecinos, L.; Domenech, R.J.; Donoso, P. Preconditioning tachycardia decreases the activity of the mitochondrial permeability transition pore in the dog heart. Biochem. Biophys. Res. Commun. 2011, 410, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.O.; Woodley, K.T.; Choudhary, J.S. Global, site-specific analysis of neuronal protein S-acylation. Sci. Rep. 2017, 7, 4683. [Google Scholar] [CrossRef]
- Amanakis, G.; Sun, J.; Fergusson, M.M.; McGinty, S.; Liu, C.; Molkentin, J.D.; Murphy, E. Cysteine 202 of cyclophilin D is a site of multiple post-translational modifications and plays a role in cardioprotection. Cardiovasc. Res. 2021, 117, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Artiga, D.J.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA 2016, 113, E1872–E1880. [Google Scholar] [CrossRef] [PubMed]
- Schlatter, D.; Thoma, R.; Küng, E.; Stihle, M.; Müller, F.; Borroni, E.; Cesura, A.; Hennig, M. Crystal engineering yields crystals of cyclophilin D diffracting to 1.7 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 513–519. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 2013, CJ-13-0321. [Google Scholar] [CrossRef]
- Zhang, Y.-G.; Wu, S.; Yi, J.; Xia, Y.; Jin, D.; Zhou, J.; Sun, J. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin. Ther. 2017, 39, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.J.; Jones, P.P.; Wang, R.; Chen, W.; Bolstad, J.; Chen, K.; Shimoni, Y.; Chen, S.R.W. K201 (JTV519) suppresses spontaneous Ca2+ release and [3H] ryanodine binding to RyR2 irrespective of FKBP12. 6 association. Biochem. J. 2007, 404, 431–438. [Google Scholar] [CrossRef]
- Zhou, X.; Park, K.H.; Yamazaki, D.; Lin, P.H.; Nishi, M.; Ma, Z.; Qiu, L.; Murayama, T.; Zou, X.; Takeshima, H.; et al. TRIC-A Channel Maintains Store Calcium Handling by Interacting With Type 2 Ryanodine Receptor in Cardiac Muscle. Circ. Res. 2020, 126, 417–435. [Google Scholar] [CrossRef]
- Egawa, T.; Hanaoka, K.; Koide, Y.; Ujita, S.; Takahashi, N.; Ikegaya, Y.; Matsuki, N.; Terai, T.; Ueno, T.; Komatsu, T.; et al. Development of a far-red to near-infrared fluorescence probe for calcium ion and its application to multicolor neuronal imaging. J. Am. Chem. Soc. 2011, 133, 14157–14159. [Google Scholar] [CrossRef]
- Mancini, R.; van der Bijl, T.; Bourgeois-Jaarsma, Q.; Lasabuda, R.; Groffen, A.J. SICT: Automated detection and supervised inspection of fast Ca2+ transients. Sci. Rep. 2018, 8, 15523. [Google Scholar] [CrossRef]
- Hong, J.; Jia, Y.; Pan, S.; Jia, L.; Li, H.; Han, Z.; Cai, D.; Zhao, R. Butyrate alleviates high fat diet-induced obesity through activation of adiponectin-mediated pathway and stimulation of mitochondrial function in the skeletal muscle of mice. Oncotarget 2016, 7, 56071–56082. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef]
- Canani, R.B.; Di Costanzo, M.; Leone, L. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin. Epigenetics 2012, 4, 1–7. [Google Scholar]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Yaniv, Y.; Nuss, H.B.; Wang, S.; Sollott, S.J. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc. Res. 2009, 83, 213–225. [Google Scholar] [CrossRef]
- Luciani, S.; Martini, N.; Santi, R. Effects of carboxyatractyloside a structural analogue of atractyloside on mitochondrial oxidative phosphorylation. Life Sci. 1971, 10, 961–968. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Klingenberg, M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 1996, 35, 8483–8488. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Tropschug, M.; Heimpel, S.; Heidkämper, D.; Klingenberg, M. A large Ca2+-dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore. Biochemistry 2002, 41, 11804–11811. [Google Scholar] [CrossRef]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; LaNoue, K.F.; Cheung, J.Y.; Scaduto, R.C. Regulation of citric acid cycle by calcium. J. Biol. Chem. 1989, 264, 13430–13439. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Das, A.M.; Harris, D.A. Control of mitochondrial ATP synthase in heart cells: Inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 1990, 24, 411–417. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Ma, C.; Li, Y.; Weisleder, N.; Ríos, E.; Ma, J.; Zhou, J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (EC) coupling. J. Biol. Chem. 2011, 286, 32436–32443. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, A.; Li, X.; Yi, J.; Ma, J.; Zhou, J. Butyrate Feeding Reverses CypD-Related Mitoflash Phenotypes in Mouse Myofibers. Int. J. Mol. Sci. 2021, 22, 7412. https://doi.org/10.3390/ijms22147412
Li A, Li X, Yi J, Ma J, Zhou J. Butyrate Feeding Reverses CypD-Related Mitoflash Phenotypes in Mouse Myofibers. International Journal of Molecular Sciences. 2021; 22(14):7412. https://doi.org/10.3390/ijms22147412
Chicago/Turabian StyleLi, Ang, Xuejun Li, Jianxun Yi, Jianjie Ma, and Jingsong Zhou. 2021. "Butyrate Feeding Reverses CypD-Related Mitoflash Phenotypes in Mouse Myofibers" International Journal of Molecular Sciences 22, no. 14: 7412. https://doi.org/10.3390/ijms22147412
APA StyleLi, A., Li, X., Yi, J., Ma, J., & Zhou, J. (2021). Butyrate Feeding Reverses CypD-Related Mitoflash Phenotypes in Mouse Myofibers. International Journal of Molecular Sciences, 22(14), 7412. https://doi.org/10.3390/ijms22147412