
Features of Retinal Neurogenesis as a Key Factor of Age-Related Neurodegeneration: Myth or Reality?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
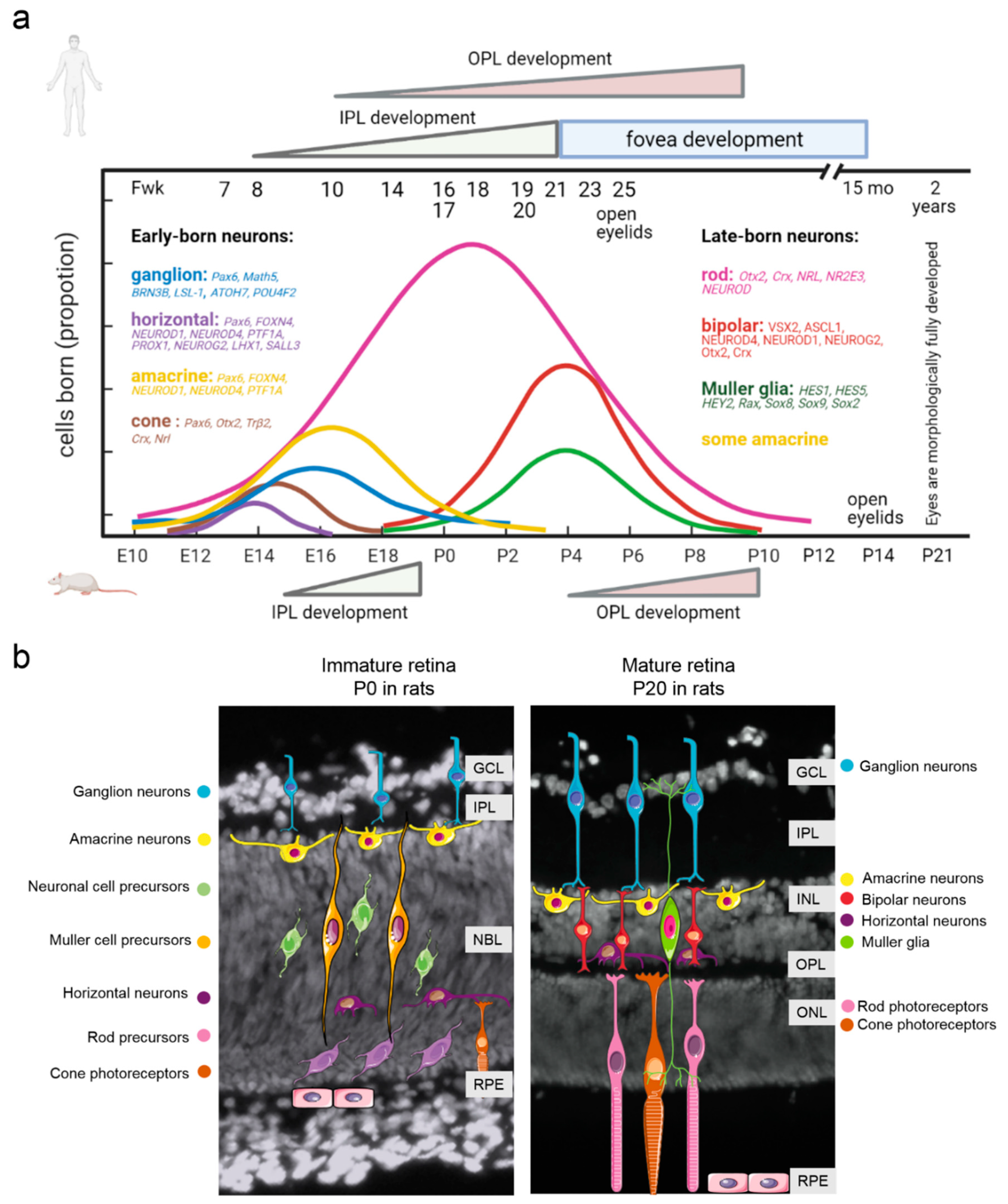
2. Retina Development: Similarities and Differences between Humans and Laboratory Animals

3. The Transcriptional Network Regulating Retinal Neurogenesis
4. The Role of Transcription Factors during Retinal Aging and Age-Related Degeneration
5. Stimulation of Neurogenesis as a New Strategy for the Treatment of Degenerative Diseases of the Mammalian Retina
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ardeljan, D.; Chan, C.-C. Aging is not a disease: Distinguishing age-related macular degeneration from aging. Prog. Retin. Eye Res. 2013, 37, 68–89. [Google Scholar] [CrossRef]
- Jun, S.; Datta, S.; Wang, L.; Pegany, R.; Cano, M.; Handa, J.T. The impact of lipids, lipid oxidation, and inflammation on AMD, and the potential role of miRNAs on lipid metabolism in the RPE. Exp. Eye Res. 2019, 181, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Telegina, D.; Kozhevnikova, O.; Kolosova, N.G. Changes in Retinal Glial Cells with Age and during Development of Age-Related Macular Degeneration. Biochemistry 2018, 83, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 2020, 77, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell. Mol. Life Sci. 2019, 76, 1901–1918. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between Autophagy and the Ubiquitin-Proteasome System and Its Role in the Pathogenesis of Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Garcia, T.B.; Hollborn, M.; Bringmann, A. Expression and signaling of NGF in the healthy and injured retina. Cytokine Growth Factor Rev. 2017, 34, 43–57. [Google Scholar] [CrossRef]
- Tekin, M.I.; Sekeroglu, M.A.; Demirtas, C.; Tekin, K.; Doguizi, S.; Bayraktar, S.; Yilmaz, C. Brain-Derived Neurotrophic Factor in Patients With Age-Related Macular Degeneration and Its Correlation With Retinal Layer Thicknesses. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2833–2840. [Google Scholar] [CrossRef]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 1–20. [Google Scholar] [CrossRef]
- Wu, J.; Sun, X. Complement system and age-related macular degeneration: Drugs and challenges. Drug Des. Dev. Ther. 2019, ume 13, 2413–2425. [Google Scholar] [CrossRef]
- Bandello, F.; Sacconi, R.; Querques, L.; Corbelli, E.; Cicinelli, M.V.; Querques, G. Recent advances in the management of dry age-related macular degeneration: A review. F1000Research 2017, 6, 245. [Google Scholar] [CrossRef]
- Bertolotti, E.; Neri, A.; Camparini, M.; Macaluso, C.; Marigo, V. Stem cells as source for retinal pigment epithelium transplantation. Prog. Retin. Eye Res. 2014, 42, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.K.; Cooley-Themm, C.A.; Barnett, J.D.; Bach, H.B.; Vainner, J.M.; Webster, S.E.; Linn, C.L. Evidence of BrdU-positive retinal neurons after application of an Alpha7 nicotinic acetylcholine receptor agonist. Neuroscience 2017, 346, 437–446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Otteson, D.C. Talkin’ about my (re)generation: The who of intrinsic retinal stem cells. Neuroscience 2017, 346, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.K.; Barnett, B.J.; Stanchfield, M.L.; Paris, J.R.; Webster, S.E.; Cooley-themm, C.A.; Levine, E.M.; Otteson, D.C.; Linn, C.L. Stimulation of Retinal Pigment Epithelium With an α7 nAChR Agonist Leads to Müller Glia Dependent Neurogenesis in the Adult Mammalian Retina. Investig. Ophthalmol. Vis. Sci. 2019, 60, 570–579. [Google Scholar] [CrossRef]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.; Jobling, A.; Greferath, U.; Mills, S.A.; Waugh, M.; Ho, T.; de Iongh, R.; Phipps, J.A.; Vessey, K.A. Studying Age-Related Macular Degeneration Using Animal Models. Optom. Vis. Sci. 2014, 91, 878–886. [Google Scholar] [CrossRef]
- Peynshaert, K.; Devoldere, J.; Minnaert, A.-K.; De Smedt, S.C.; Remaut, K. Morphology and Composition of the Inner Limiting Membrane: Species-Specific Variations and Relevance toward Drug Delivery Research. Curr. Eye Res. 2019, 44, 465–475. [Google Scholar] [CrossRef]
- Ramkumar, H.L.; Zhang, J.; Chan, C.-C. Retinal ultrastructure of murine models of dry age-related macular degeneration (AMD). Prog. Retin. Eye Res. 2010, 29, 169–190. [Google Scholar] [CrossRef]
- Narayan, D.S.; Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations Into Bioenergetic Neuroprotection of Cone Photoreceptors: Relevance to Retinitis Pigmentosa. Front. Neurosci. 2019, 13, 1234. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.O.; Nadal-Nicolás, F.M.; Jiménez-López, M.; Béjar, J.J.A.; Nieto-López, L.; García-Ayuso, D.; Villegas-Pérez, M.P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Number and Distribution of Mouse Retinal Cone Photoreceptors: Differences between an Albino (Swiss) and a Pigmented (C57/BL6) Strain. PLoS ONE 2014, 9, e102392. [Google Scholar] [CrossRef]
- Shah, M.; Cabrera-Ghayouri, S.; Christie, L.-A.; Held, K.S.; Viswanath, V. Translational Preclinical Pharmacologic Disease Models for Ophthalmic Drug Development. Pharm. Res. 2019, 36, 1–34. [Google Scholar] [CrossRef]
- Vessey, K.A.; Gu, B.J.; Jobling, A.; Phipps, J.A.; Greferath, U.; Tran, M.X.; Dixon, M.A.; Baird, P.N.; Guymer, R.; Wiley, J.; et al. Loss of Function of P2X7 Receptor Scavenger Activity in Aging Mice. Am. J. Pathol. 2017, 187, 1670–1685. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Wang, K.; Zheng, M.; Lester, K.L.; Han, Z. Light-induced Nrf2−/− mice as atrophic age-related macular degeneration model and treatment with nanoceria laden injectable hydrogel. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Im, S.; Jun, H.O.; Lee, K.; Park, Y.-J.; Kim, J.H.; Park, W.J.; Lee, Y.-H.; Kim, J.H. Dry age-related macular degeneration like pathology in aged 5XFAD mice: Ultrastructure and microarray analysis. Oncotarget 2017, 8, 40006–40018. [Google Scholar] [CrossRef]
- Bora, P.; Hu, Z.; Tezel, T.H.; Sohn, J.-H.; Kang, S.G.; Cruz, J.M.C.; Bora, N.S.; Garen, A.; Kaplan, H.J. Immunotherapy for choroidal neovascularization in a laser-induced mouse model simulating exudative (wet) macular degeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 2679–2684. [Google Scholar] [CrossRef]
- Telegina, D.; Kozhevnikova, O.; Bayborodin, S.I.; Kolosova, N.G. Contributions of age-related alterations of the retinal pigment epithelium and of glia to the AMD-like pathology in OXYS rats. Sci. Rep. 2017, 7, srep41533. [Google Scholar] [CrossRef]
- Feng, L.; Wang, F. Detecting A-beta deposition and RPE cell senescence in the retinas of SAMP8 mice. Discov. Med. 2016, 21, 149–158. [Google Scholar]
- Schnichels, S.; Paquet-Durand, F.; Löscher, M.; Tsai, T.; Hurst, J.; Joachim, S.C.; Klettner, A. Retina in a dish: Cell cultures, retinal explants and animal models for common diseases of the retina. Prog. Retin. Eye Res. 2021, 81, 100880. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Ahmad, I. Unlocking the Neurogenic Potential of Mammalian Müller Glia. Int. J. Stem Cells 2016, 9, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Crump, C. An overview of adult health outcomes after preterm birth. Early Hum. Dev. 2020, 150, 105187. [Google Scholar] [CrossRef]
- Shah, P.K.; Prabhu, V.; Karandikar, S.S.; Ranjan, R.; Narendran, V.; Kalpana, N. Retinopathy of prematurity: Past, present and future. World J. Clin. Pediatr. 2016, 5, 35–46. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Guo, X.; Zhang, Q. CRX variants in cone–rod dystrophy and mutation overview. Biochem. Biophys. Res. Commun. 2012, 426, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Fieβ, A.; Janz, J.; Schuster, A.K.; Kölb-Keerl, R.; Knuf, M.; Kirchhof, B.; Muether, P.S.; Bauer, J. Macular morphology in former preterm and full-term infants aged 4 to 10 years. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1433–1442. [Google Scholar] [CrossRef]
- E Yanni, S.; Wang, J.; Chan, M.; Carroll, J.; Farsiu, S.; Leffler, J.N.; Spencer, R.; E Birch, E. Foveal avascular zone and foveal pit formation after preterm birth. Br. J. Ophthalmol. 2012, 96, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-A.; Oh, S.Y. Analysis of Spectral-Domain Optical Coherence Tomography in Preterm Children: Retinal Layer Thickness and Choroidal Thickness Profiles. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7201–7207. [Google Scholar] [CrossRef]
- Bowl, W.; Stieger, K.; Bokun, M.; Schweinfurth, S.; Holve, K.; Andrassi-Darida, M.; Lorenz, B. OCT-Based Macular Structure–Function Correlation in Dependence on Birth Weight and Gestational Age—the Giessen Long-Term ROP Study. Investig. Ophthalmol. Vis. Sci. 2016, 57, OCT235–OCT241. [Google Scholar] [CrossRef]
- Fieβ, A.; Elbaz, H.; Korb, C.A.; Nickels, S.; Schulz, A.; Münzel, T.; Wild, P.S.; Beutel, M.E.; Schmidtmann, I.; Lackner, K.J.; et al. Low Birth Weight Is Linked to Age-Related Macular Degeneration: Results From the Population-Based Gutenberg Health Study (GHS). Investig. Ophthalmol. Vis. Sci. 2019, 60, 4943–4950. [Google Scholar] [CrossRef]
- Hall, N.F.; Gale, C.R.; Syddall, H.; Martyn, C.N.; Phillips, D.I.W. Relation between size at birth and risk of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3641–3645. [Google Scholar]
- Liew, G.; Wang, J.J.; Klein, R.; Duncan, B.B.; Brancati, F.; Yeh, H.-C.; Wong, T.Y. The Relationship between Birthweight and Early Age-Related Maculopathy: The Atherosclerosis Risk in Communities Study. Ophthalmic Epidemiol. 2008, 15, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.P. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Barker, D. Developmental origins of chronic disease. Public Health 2012, 126, 185–189. [Google Scholar] [CrossRef]
- Allegra, A.; Giarratana, R.M.; Scola, L.; Balistreri, C.R. The close link between the fetal programming imprinting and neurodegeneration in adulthood: The key role of “hemogenic endothelium” programming. Mech. Ageing Dev. 2021, 195, 111461. [Google Scholar] [CrossRef]
- Engerer, P.; Suzuki, S.C.; Yoshimatsu, T.; Chapouton, P.; Obeng, N.; Odermatt, B.; Williams, P.R.; Misgeld, T.; Godinho, L. Uncoupling of neurogenesis and differentiation during retinal development. EMBO J. 2017, 36, 1134–1146. [Google Scholar] [CrossRef]
- Xiang, M. Intrinsic control of mammalian retinogenesis. Cell. Mol. Life Sci. 2013, 70, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Ringuette, R.; Atkins, M.; Lagali, P.S.; Bassett, E.A.; Campbell, C.; Mazerolle, C.; Mears, A.J.; Picketts, D.J.; Wallace, V.A. A Notch-Gli2 axis sustains Hedgehog responsiveness of neural progenitors and Müller glia. Dev. Biol. 2016, 411, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Serb, J.; Greenlee, M.H.W. Mouse Retinal Development: A Dark Horse Model for Systems Biology Research. Bioinform. Biol. Insights 2011, 5, BBI-S6930. [Google Scholar] [CrossRef]
- Cepko, C.L. The Determination of Rod and Cone Photoreceptor Fate. Annu. Rev. Vis. Sci. 2015, 1, 211–234. [Google Scholar] [CrossRef]
- Rapaport, D.H.; Wong, L.; Wood, E.D.; Yasumura, D.; Lavail, M.M. Timing and topography of cell genesis in the rat retina. J. Comp. Neurol. 2004, 474, 304–324. [Google Scholar] [CrossRef]
- Wu, P.; Deng, J.-B.; Fan, W.-J.; Li, X.; Yao, H.-L.; Deng, J.-X.; Liu, H.-L.; Cui, Z.-J.; Wang, Q. Neural differentiation and synaptogenesis in retinal development. Neural Regen. Res. 2016, 11, 312–318. [Google Scholar] [CrossRef]
- Meng, Q.; Mongan, M.; Carreira, V.; Kurita, H.; Liu, C.-Y.; Kao, W.W.-Y.; Xia, Y. Eyelid Closure in Embryogenesis Is Required for Ocular Adnexa Development. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7652–7661. [Google Scholar] [CrossRef]
- Nguyen-Ba-Charvet, K.T.; Chédotal, A. Development of retinal layers. Comptes Rendus Biol. 2014, 337, 153–159. [Google Scholar] [CrossRef]
- Hendrickson, A. Development of Retinal Layers in Prenatal Human Retina. Am. J. Ophthalmol. 2016, 161, 29–35.e1. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Lang, R.A. Retinal ganglion cell interactions shape the developing mammalian visual system. Development 2020, 147, 1–13. [Google Scholar] [CrossRef]
- Hoshino, A.; Ratnapriya, R.; Brooks, M.J.; Chaitankar, V.; Wilken, M.S.; Zhang, C.; Starostik, M.; Gieser, L.; La Torre, A.; Nishio, M.; et al. Molecular Anatomy of the Developing Human Retina. Dev. Cell 2017, 43, 763–779.e4. [Google Scholar] [CrossRef]
- Van Cruchten, S.; Vrolyk, V.; Lepage, M.-F.P.; Baudon, M.; Voute, H.; Schoofs, S.; Haruna, J.; Benoit-Biancamano, M.-O.; Ruot, B.; Allegaert, K. Pre- and Postnatal Development of the Eye: A Species Comparison. Birth Defects Res. 2017, 109, 1540–1567. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.; Wijnholds, J. Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective. Genes 2019, 10, 987. [Google Scholar] [CrossRef] [PubMed]
- Grünert, U.; Martin, P.R. Cell types and cell circuits in human and non-human primate retina. Prog. Retin. Eye Res. 2020, 78, 100844. [Google Scholar] [CrossRef]
- Bringmann, A.; Syrbe, S.; Görner, K.; Kacza, J.; Francke, M.; Wiedemann, P.; Reichenbach, A. The primate fovea: Structure, function and development. Prog. Retin. Eye Res. 2018, 66, 49–84. [Google Scholar] [CrossRef]
- Hendrickson, A.; Possin, D.; Vajzovic, L.; Toth, C.A. Histologic Development of the Human Fovea from Midgestation to Maturity. Am. J. Ophthalmol. 2012, 154, 767–778.e2. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Lindsey, D.T. Contrast Insensitivity: The Critical Immaturity in Infant Visual Performance. Optom. Vis. Sci. 2009, 86, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Aldiri, I.; Xu, B.; Wang, L.; Chen, X.; Hiler, D.; Griffiths, L.; Valentine, M.; Shirinifard, A.; Thiagarajan, S.; Sablauer, A.; et al. The Dynamic Epigenetic Landscape of the Retina During Development, Reprogramming, and Tumorigenesis. Neuron 2017, 94, 550–568.e10. [Google Scholar] [CrossRef] [PubMed]
- Braekevelt, C.R.; Hollenberg, M.J. The development of the retina of the albino rat. Am. J. Anat. 1970, 127, 281–301. [Google Scholar] [CrossRef]
- Huang, X.; Ledesma, H.A.; Wei, W. Synapse formation in the developing vertebrate retina. Synap. Dev. Maturation 2020, 213–234. [Google Scholar] [CrossRef]
- Wilk, M.A.; McAllister, J.T.; Cooper, R.F.; Dubis, A.M.; Patitucci, T.N.; Summerfelt, P.; Anderson, J.L.; Stepien, K.E.; Costakos, D.M.; Connor, T.B.; et al. Relationship Between Foveal Cone Specialization and Pit Morphology in Albinism. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4186–4198. [Google Scholar] [CrossRef]
- Bhansali, P.; Rayport, I.; Rebsam, A.; Mason, C. Delayed neurogenesis leads to altered specification of ventrotemporal retinal ganglion cells in albino mice. Neural Dev. 2014, 11, 1–15. [Google Scholar] [CrossRef]
- Begum, R.; Powner, M.B.; Hudson, N.; Hogg, C.; Jeffery, G. Treatment with 670 nm Light Up Regulates Cytochrome C Oxidase Expression and Reduces Inflammation in an Age-Related Macular Degeneration Model. PLoS ONE 2013, 8, e57828. [Google Scholar] [CrossRef]
- Savaskan, E.; Wirz-Justice, A.; Olivieri, G.; Pache, M.; Kräuchi, K.; Brydon, L.; Jockers, R.; Müller-Spahn, F.; Meyer, P. Distribution of Melatonin MT1 Receptor Immunoreactivity in Human Retina. J. Histochem. Cytochem. 2002, 50, 519–525. [Google Scholar] [CrossRef]
- Seo, J.H.; Yu, Y.S.; Kim, J.H.; Choung, H.K.; Heo, J.W.; Kim, S.-J. Correlation of Visual Acuity with Foveal Hypoplasia Grading by Optical Coherence Tomography in Albinism. Ophthalmology 2007, 114, 1547–1551. [Google Scholar] [CrossRef]
- Zagozewski, J.; Zhang, Q.; Eisenstat, D. Genetic regulation of vertebrate eye development. Clin. Genet. 2014, 86, 453–460. [Google Scholar] [CrossRef]
- Jin, K. Transitional Progenitors during Vertebrate Retinogenesis. Mol. Neurobiol. 2017, 54, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Monsoro-Burq, A.H. PAX transcription factors in neural crest development. Semin. Cell Dev. Biol. 2015, 44, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, S.; Basu, B.; Surya, S.; Meera, V.; Riya, P.A.; Parvathy, S.; Das, A.V.; Sivakumar, K.C.; Nelson-Sathi, S.; James, J. Pax6 modulates intra-retinal axon guidance and fasciculation of retinal ganglion cells during retinogenesis. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Manuel, M.; Pratt, T.; Liu, M.; Jeffery, G.; Price, D.J. Overexpression of Pax6 results in microphthalmia, retinal dysplasia and defective retinal ganglion cell axon guidance. BMC Dev. Biol. 2008, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Fu, X.; Sun, H.; Beremand, P.D.; Thomas, T.L.; Klein, W.H. A gene network downstream of transcription factor Math5 regulates retinal progenitor cell competence and ganglion cell fate. Dev. Biol. 2005, 280, 467–481. [Google Scholar] [CrossRef]
- Andreazzoli, M. Molecular regulation of vertebrate retina cell fate. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 284–295. [Google Scholar] [CrossRef]
- Le, T.T.; Wroblewski, E.; Patel, S.; Riesenberg, A.N.; Brown, N.L. Math5 is required for both early retinal neuron differentiation and cell cycle progression. Dev. Biol. 2006, 295, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Casarosa, S.; Murenu, E. Stem Cells and the Retina–Challenges for Regenerative Medicine. In Embryonic Stem Cells-Recent Advances in Pluripotent Stem Cell-Based Regenerative Medicine; IntechOpen: London, UK, 2011; pp. 211–236. [Google Scholar]
- Beby, F.; Lamonerie, T. The homeobox gene Otx2 in development and disease. Exp. Eye Res. 2013, 111, 9–16. [Google Scholar] [CrossRef]
- Roger, J.E.; Chang, B.; Swaroop, A.; Roger, J.E.; Hiriyanna, A.; Gotoh, N.; Hao, H.; Cheng, D.F.; Ratnapriya, R.; Kautzmann, M.I.; et al. Congenital blindness OTX2 loss causes rod differentiation defect in CRX-associated congenital blindness. J. Clin. Investig. 2014, 124, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Nakhai, H.; Sel, S.; Favor, J.; Mendoza-Torres, L.; Paulsen, F.; Duncker, G.I.W.; Schmid, R.M. Ptf1a is essential for the differentiation of GABAergic and glycinergic amacrine cells and horizontal cells in the mouse retina. Development 2007, 134, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mo, Z.; Yang, X.; Price, S.M.; Shen, M.; Xiang, M. Foxn4 Controls the Genesis of Amacrine and Horizontal Cells by Retinal Progenitors. Neuron 2004, 43, 795–807. [Google Scholar] [CrossRef]
- Cepko, C. Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci. 2014, 15, 615–627. [Google Scholar] [CrossRef]
- de Melo, J.; Peng, G.-H.; Chen, S.; Blackshaw, S. The Spalt family transcription factor Sall3 regulates the development of cone photoreceptors and retinal horizontal interneurons. Development 2011, 138, 2325–2336. [Google Scholar] [CrossRef]
- Warre-Cornish, K.; Barber, A.C.; Sowden, J.C.; Ali, R.; Pearson, R.A. Migration, Integration and Maturation of Photoreceptor Precursors Following Transplantation in the Mouse Retina. Stem Cells Dev. 2014, 23, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Brzezinski, J.A.; Reh, T.A. Photoreceptor cell fate specification in vertebrates. Development 2015, 142, 3263–3273. [Google Scholar] [CrossRef]
- Swaroop, A.; Kim, D.; Forrest, D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 2010, 11, 563–576. [Google Scholar] [CrossRef]
- Del Debbio, C.B.; Mir, Q.; Parameswaran, S.; Mathews, S.; Xia, X.; Zheng, L.; Neville, A.J.; Ahmad, I. Notch Signaling Activates Stem Cell Properties of Müller Glia through Transcriptional Regulation and Skp2-mediated Degradation of p27Kip1. PLoS ONE 2016, 11, e0152025. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.R.; Ueki, Y.; Reardon, S.; Karl, M.; Georgi, S.; Hartman, B.H.; Lamba, D.A.; Reh, T.A. Genome-Wide Analysis of Müller Glial Differentiation Reveals a Requirement for Notch Signaling in Postmitotic Cells to Maintain the Glial Fate. PLoS ONE 2011, 6, e22817. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Mukherjee, S.; Bao, Z.; Morrow, E.M.; Cepko, C.L. rax, Hes1, and notch1 Promote the Formation of Müller Glia by Postnatal Retinal Southwestern Medical Center at Dallas. Neuron 2000, 26, 383–394. [Google Scholar] [CrossRef]
- Faigle, R.; Song, H. Signaling mechanisms regulating adult neural stem cells and neurogenesis. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 2435–2448. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Ji, Z.; Cepko, C.L.; Wang, S.; Wang, S. Cell type- and stage-specific expression of Otx2 is coordinated by a cohort of transcription factors and multiple cis-regulatory modules in the retina. Development 2019. [Google Scholar] [CrossRef]
- Hennig, A.K.; Peng, G.-H.; Chen, S. Regulation of photoreceptor gene expression by Crx-associated transcription factor network. Brain Res. 2008, 1192, 114–133. [Google Scholar] [CrossRef]
- Vergara, M.N.; Gutierrez, C.; O’Brien, D.R.; Canto-Soler, M.V. Ex vivo electroporation of retinal cells: A novel, high efficiency method for functional studies in primary retinal cultures. Exp. Eye Res. 2013, 109, 40–50. [Google Scholar] [CrossRef]
- Kautzmann, M.-A.I.; Kim, D.; Felder-Schmittbuhl, M.-P.; Swaroop, A. Combinatorial Regulation of Photoreceptor Differentiation Factor, Neural Retina Leucine Zipper Gene Nrl, Revealed by in Vivo Promoter Analysis. J. Biol. Chem. 2011, 286, 28247–28255. [Google Scholar] [CrossRef]
- Dna-, T.; Ser, T. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat. Genet. 1999, 21, 355–356. [Google Scholar]
- Martinez-gimeno, M.; Maseras, M.; Beneito, M. Mutations P51L and G122E in Retinal Transcription Factor NRL Associated With Autosomal Dominant and Sporadic Retinitis Pigmentosa. Hum. Mutat. 2001, 17, 520. [Google Scholar] [CrossRef]
- Laranjeiro, R.; Whitmore, D. Transcription factors involved in retinogenesis are co-opted by the circadian clock following photoreceptor differentiation. Development 2014, 141, 2644–2656. [Google Scholar] [CrossRef]
- Liu, H.; Etter, P.; Hayes, S.; Jones, I.; Nelson, B.; Hartman, B.; Forrest, D.; Reh, T.A. NeuroD1 Regulates Expression of Thyroid Hormone Receptor 2 and Cone Opsins in the Developing Mouse Retina. J. Neurosci. 2008, 28, 749–756. [Google Scholar] [CrossRef]
- Zagozewski, J.L.; Zhang, Q.; Pinto, V.I.; Wigle, J.; Eisenstat, D.D. The role of homeobox genes in retinal development and disease. Dev. Biol. 2014, 393, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kon, T.; Omori, Y.; Furukawa, T. Functional and Evolutionary Diversification of Otx2 and Crx in Vertebrate Retinal Photoreceptor and Bipolar Cell Development. Cell Rep. 2020, 30, 658–671.e5. [Google Scholar] [CrossRef] [PubMed]
- Bernard, C.; Kim, H.-T.; Ibad, R.T.; Lee, E.J.; Simonutti, M.; Picaud, S.; Acampora, D.; Simeone, A.; Di Nardo, A.A.; Prochiantz, A.; et al. Graded Otx2 activities demonstrate dose-sensitive eye and retina phenotypes. Hum. Mol. Genet. 2013, 23, 1742–1753. [Google Scholar] [CrossRef]
- Béby, F.; Housset, M.; Fossat, N.; Le Greneur, C.; Flamant, F.; Godement, P.; Lamonerie, T. Otx2 Gene Deletion in Adult Mouse Retina Induces Rapid RPE Dystrophy and Slow Photoreceptor Degeneration. PLoS ONE 2010, 5, e11673. [Google Scholar] [CrossRef]
- Di Nardo, A.A.; Fuchs, J.; Joshi, R.L.; Moya, K.L.; Prochiantz, A. The Physiology of Homeoprotein Transduction. Physiol. Rev. 2018, 98, 1943–1982. [Google Scholar] [CrossRef]
- Assawachananont, J.; Kim, S.-Y.; Kaya, K.D.; Fariss, R.; E Roger, J.; Swaroop, A. Cone-rod homeobox CRX controls presynaptic active zone formation in photoreceptors of mammalian retina. Hum. Mol. Genet. 2018, 27, 3555–3567. [Google Scholar] [CrossRef]
- Whitaker, D.T.; Mondal, A.K.; Fann, H.; Hargrove, P.; Brooks, M.J.; Chaitankar, V.; Yu, W.; Wu, Z.; Kim, S.-Y.; Swaroop, A. NRL- and CRX-guided gene network modulates photoreceptor presynapse size and positioning during retinal development. Biorxiv 2019, 753012. [Google Scholar] [CrossRef]
- Omori, Y.; Katoh, K.; Sato, S.; Muranishi, Y.; Chaya, T.; Onishi, A.; Minami, T.; Fujikado, T.; Furukawa, T. Analysis of Transcriptional Regulatory Pathways of Photoreceptor Genes by Expression Profiling of the Otx2-Deficient Retina. PLoS ONE 2011, 6, e19685. [Google Scholar] [CrossRef] [PubMed]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.; Xing, Y.; Brown, L.N.; Samuvel, D.J.; Panganiban, C.H.; Havens, L.T.; Balasubramanian, S.; Wegner, M.; Krug, E.; Barth, J.L. Neural stem/progenitor cell properties of glial cells in the adult mouse auditory nerve. Sci. Rep. 2015, 5, 13383. [Google Scholar] [CrossRef]
- Bachleda, A.R.; Pevny, L.H.; Weiss, E.R. Sox2-Deficient Müller Glia Disrupt the Structural and Functional Maturation of the Mammalian Retina. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cugnon, L.; Anasagasti, A.; Ezquerra-Inchausti, M.; Izeta, A.; De La Villa, P.; Ruiz-Ederra, J.; Matheu, A. SOX2 haploinsufficiency promotes impaired vision at advanced age. Oncotarget 2018, 9, 36684–36692. [Google Scholar] [CrossRef][Green Version]
- Surzenko, N.; Crowl, T.; Bachleda, A.; Langer, L.; Pevny, L. SOX2 maintains the quiescent progenitor cell state of postnatal retinal Müller glia. Development 2013, 140, 1445–1456. [Google Scholar] [CrossRef]
- Kautzman, A.G.; Keeley, P.W.; Nahmou, M.M.; Luna, G.; Fisher, S.K.; Reese, B.E. Sox2 regulates astrocytic and vascular development in the retina. Glia 2018, 66, 623–636. [Google Scholar] [CrossRef]
- Taranova, O.V.; Magness, S.T.; Fagan, B.M.; Wu, Y.; Surzenko, N.; Hutton, S.R.; Pevny, L.H. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006, 20, 1187–1202. [Google Scholar] [CrossRef]
- Stanescu, D.; Iseli, H.P.; Schwerdtfeger, K.; Ittner, L.; E Remé, C.; Hafezi, F. Continuous expression of the homeobox gene Pax6 in the ageing human retina. Eye 2005, 21, 90–93. [Google Scholar] [CrossRef]
- Stanescu-Segall, D.; Birke, K.; Wenzel, A.; Grimm, C.; Orgul, S.; Fischer, J.A.; Born, W.; Hafezi, F. PAX6 Expression and Retinal Cell Death in a Transgenic Mouse Model for Acute Angle-Closure Glaucoma. J. Glaucoma 2015, 24, 426–432. [Google Scholar] [CrossRef]
- Joly, S.; Pernet, V.; Samardzija, M.; Grimm, C. Pax6-positive müller glia cells express cell cycle markers but do not proliferate after photoreceptor injury in the mouse retina. Glia 2011, 59, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Pirmardan, E.R.; Soheili, Z.-S.; Samiei, S.; Ahmadieh, H.; Mowla, S.J.; Naseri, M.; Daftarian, N. In Vivo Evaluation of PAX6 Overexpression and NMDA Cytotoxicity to Stimulate Proliferation in the Mouse Retina. Sci. Rep. 2018, 8, 17700. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, E.N. Potential Endogenous Cell Sources for Retinal Regeneration in Vertebrates and Humans: Progenitor Traits and Specialization. Biomedicines 2020, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin. Eye Res. 2020, 74, 100771. [Google Scholar] [CrossRef]
- Hoang, T.; Wang, J.; Boyd, P.; Wang, F.; Santiago, C.; Jiang, L.; Yoo, S.; Lahne, M.; Todd, L.J.; Jia, M.; et al. Gene regulatory networks controlling vertebrate retinal regeneration. Science 2020, 370, eabb8598. [Google Scholar] [CrossRef]
- Wilken, M.S.; A Reh, T. Retinal regeneration in birds and mice. Curr. Opin. Genet. Dev. 2016, 40, 57–64. [Google Scholar] [CrossRef]
- Karl, M.; Hayes, S.; Nelson, B.R.; Tan, K.; Buckingham, B.; Reh, T.A. Stimulation of neural regeneration in the mouse retina. Proc. Natl. Acad. Sci. USA 2008, 105, 19508–19513. [Google Scholar] [CrossRef]
- Ohsawa, R.; Kageyama, R. Regulation of retinal cell fate specification by multiple transcription factors. Brain Res. 2008, 1192, 90–98. [Google Scholar] [CrossRef]
- Ueki, Y.; Wilken, M.S.; Cox, K.E.; Chipman, L.; Jorstad, N.L.; Sternhagen, K.; Simic, M.; Ullom, K.; Nakafuku, M.; Reh, T.A. Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13717–13722. [Google Scholar] [CrossRef]
- Jorstad, N.L.; Wilken, M.S.; Grimes, W.N.; Wohl, S.G.; VandenBosch, L.S.; Yoshimatsu, T.; Wong, R.O.; Rieke, F.; Reh, T.A. Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 2017, 548, 103–107. [Google Scholar] [CrossRef] [PubMed]
- VandenBosch, L.S.; Wohl, S.G.; Wilken, M.S.; Hooper, M.; Finkbeiner, C.; Cox, K.; Chipman, L.; Reh, T.A. Developmental changes in the accessible chromatin, transcriptome and Ascl1-binding correlate with the loss in Müller Glial regenerative potential. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 2020, 181, 590–603.e16. [Google Scholar] [CrossRef] [PubMed]
- Van Lieshout, R.J.; McGowan, P.O.; de Vega, W.C.; Savoy, C.D.; Morrison, K.M.; Saigal, S.; Mathewson, K.J.; Schmidt, L.A. Extremely Low Birth Weight and Accelerated Biological Aging. Pediatrics 2021, 147. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telegina, D.V.; Kozhevnikova, O.S.; Antonenko, A.K.; Kolosova, N.G. Features of Retinal Neurogenesis as a Key Factor of Age-Related Neurodegeneration: Myth or Reality? Int. J. Mol. Sci. 2021, 22, 7373. https://doi.org/10.3390/ijms22147373
Telegina DV, Kozhevnikova OS, Antonenko AK, Kolosova NG. Features of Retinal Neurogenesis as a Key Factor of Age-Related Neurodegeneration: Myth or Reality? International Journal of Molecular Sciences. 2021; 22(14):7373. https://doi.org/10.3390/ijms22147373
Chicago/Turabian StyleTelegina, Darya V., Oyuna S. Kozhevnikova, Anna K. Antonenko, and Nataliya G. Kolosova. 2021. "Features of Retinal Neurogenesis as a Key Factor of Age-Related Neurodegeneration: Myth or Reality?" International Journal of Molecular Sciences 22, no. 14: 7373. https://doi.org/10.3390/ijms22147373
APA StyleTelegina, D. V., Kozhevnikova, O. S., Antonenko, A. K., & Kolosova, N. G. (2021). Features of Retinal Neurogenesis as a Key Factor of Age-Related Neurodegeneration: Myth or Reality? International Journal of Molecular Sciences, 22(14), 7373. https://doi.org/10.3390/ijms22147373