
Role of miR-24 in Multiple Endocrine Neoplasia Type 1: A Potential Target for Molecular Therapy



Abstract
:1. Introduction
2. The Autoregulatory Network between miR-24, MEN1, and Menin: A Possible Effector of MEN1 Tumorigenesis
2.1. miR-24, MEN1 mRNA, and Menin in Parathyroid Glands
2.2. miR-24, MEN1 mRNA, and Menin in the Endocrine Pancreas
2.3. miR-24, MEN1 mRNA, and Menin in Non-MEN1 Tumors
3. Targeting miR-24: A Potential Therapeutic Tool for MEN1 Tumorigenesis
4. Future Research Needed in the Field of MEN1 Syndrome and miRNAs
5. Conclusions
- The absence of a genotype-phenotype correlation in MEN1 syndrome suggested a possible role of epigenetic factors in the development of the individual clinical phenotype in any single patient, even in presence of the same MEN1 mutation.
- miRNAs have shown increasing evidence of a direct role in human malignancies, both for sporadic and hereditary cancers. Several miRNAs resulted to be deregulated in the sporadic tumor counterparts of the neuroendocrine tissues commonly affected in MEN1 syndrome. A role of specific mi-RNA deregulation also in MEN1 tumorigenesis can be suspected.
- miR-24 has been demonstrated to negatively regulate menin expression in the parathyroids and the endocrine pancreas in MEN1 syndrome, and in other non-MEN1 sporadic tumors, suggesting it as initiator of menin loss-derived tumorigenesis.
- Targeting/silencing miR-24, during the hyperplastic phase of parathyroid and endocrine pancreas tumorigenesis and before the occurrence of the somatic MEN1 LOH, could by a promising tissue-specific RNA-based anti-cancer therapy, aimed to restore the correct expression of menin in pre-cancerous cells.
Author Contributions
Funding
Conflicts of Interest
References
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L. Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011. [Google Scholar] [CrossRef]
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170. [Google Scholar] [CrossRef]
- Geslot, A.; Vialon, M.; Caron, P.; Grunenwald, S.; Vezzosi, D. New therapies for patients with multiple endocrine neoplasia type 1. Ann. Endocrinol. 2021, 82, 112–120. [Google Scholar] [CrossRef]
- Al-Salameh, A.; Cadiot, G.; Calender, A.; Goudet, P.; Chanson, P. Clinical aspects of multiple endocrine neoplasia type 1. Nat. Rev. Endocrinol. 2021, 17, 207–224. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Pannett, A.A.; Thakker, R.V. Somatic mutations in MEN type 1 tumors, consistent with the Knudson “two-hit” hypothesis. J. Clin. Endocrinol. Metab. 2001, 86, 4371–4374. [Google Scholar] [CrossRef]
- Valdes, N.; Alvarez, V.; Diaz-Cadorniga, F.; Aller, J.; Villazon, F.; Garcia, I.; Herrero, A.; Coto, E. Multiple endocrine neoplasia type 1 (MEN1): LOH studies in an affected family and in sporadic cases. Anticancer Res. 1998, 18, 2685–2689. [Google Scholar] [PubMed]
- Dreijerink, K.M.A.; Timmers, H.T.M.; Brown, M. Twenty years of menin: Emerging opportunities for restoration of transcriptional regulation in MEN1. Endocr. Relat. Cancer 2017, 24, T135–T145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Ma, J.; Hua, X. Epigenetic regulation by the menin pathway. Endocr. Relat. Cancer 2017, 24, T147–T159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, F.; Giusti, F.; Tonelli, F.; Brandi, M.L. Pancreatic Neuroendocrine Neoplasms in Multiple Endocrine Neoplasia Type 1. Int. J. Mol. Sci. 2021, 22, 4041. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Shukla, G.C.; Singh, J.; Barik, S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol. Cell Pharmacol. 2011, 3, 83–92. [Google Scholar]
- Tüfekci, K.U.; Meuwissen, R.L.J.; Genç, S. The role of microRNAs in biological processes. Methods Mol. Biol. 2014, 1107, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015, 25, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-C.; Tso-Hsiao Chen, T.-H.; Huang, Y.-M.; Wei, P.-L.; Lin, J.-C. Involvement of microRNA in Solid Cancer: Role and Regulatory Mechanisms. Biomedicines 2021, 9, 343. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Liu, B.; Qu, S.; Liang, G.; Luo, W.; Gong, C. MicroRNAs and cancer: Key paradigms in molecular therapy. Oncol. Lett. 2018, 15, 2735–2742. [Google Scholar] [CrossRef] [Green Version]
- Bouyssou, J.M.; Manier, S.; Huynh, D.; Issa, S.; Roccaro, A.M.; Ghobrial, I.M. Regulation of microRNAs in cancer metastasis. Biochim. Biophys. Acta 2014, 1845, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.A.; Al-Qahtani, M.H.; Shay, J.W. Role of miRNAs in human cancer metastasis: Implications for therapeutic intervention. Semin. Cancer Biol. 2017, 44, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Szabó, P.M.; Grolmusz, V.K.; Perge, P.; Igaz, I.; Patócs, A.; Igaz, P. MEN1 and microRNAs: The link between sporadic pituitary, parathyroid and adrenocortical tumors? Med. Hypotheses 2017, 99, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Grolmusz, V.K.; Borka, K.; Kövesdi, A.; Németh, K.; Balogh, K.; Dékány, C.; Kiss, A.; Szentpéteri, A.; Sármán, B.; Somogyi, A.; et al. MEN1 mutations and potentially MEN1-targeting miRNAs are responsible for menin deficiency in sporadic and MEN1 syndrome-associated primary hyperparathyroidism. Virchows Arch. 2017, 471, 401–411. [Google Scholar] [CrossRef]
- Luzi, E.; Pandolfini, L.; Ciuffi, S.; Marini, F.; Cremisi, F.; Nesi, G.; Brandi, M.L. MicroRNAs regulatory networks governing the epigenetic landscape of MEN1 gastro-entero-pancreatic neuroendocrine tumor: A case report. Clin. Transl. Med. 2021, 11, e351. [Google Scholar] [CrossRef]
- Donati, S.; Ciuffi, S.; Marini, F.; Palmini, G.; Miglietta, F.; Aurilia, C.; Brandi, M.L. Multiple Endocrine Neoplasia Type 1: The Potential Role of microRNAs in the Management of the Syndrome. Int. J. Mol. Sci. 2020, 21, 7592. [Google Scholar] [CrossRef]
- Lal, A.; Navarro, F.; Maher, C.A.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofmann, O.; et al. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3’UTR microRNA recognition elements. Mol. Cell 2009, 35, 610–625. [Google Scholar] [CrossRef] [Green Version]
- Luzi, E.; Marini, F.; Giusti, F.; Galli, G.; Cavalli, L.; Brandi, M.L. The negative feedback-loop between the oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by mimicking the “Knudson’s second hit”. PLoS ONE 2012, 7, e39767. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.M.; Ishizuka, J.; Townsend, C.M., Jr.; Thompson, J.C. The human carcinoid cell line, BON. A model system for the study of carcinoid tumors. Ann. N. Y. Acad. Sci. 1994, 733, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Luzi, E.; Marini, F.; Ciuffi, S.; Galli, G.; Brandi, M.L. An autoregulatory network between menin and pri-miR-24-1 is required for the processing of its specific modulator miR-24-1 in BON1 cells. Mol. Biosyst. 2016, 12, 1922–1928. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, M.; Liu, R.-F.; Zhang, J.-S.; Xu, M. Cardiac Hypertrophy is Positively Regulated by MicroRNA-24 in Rats. Chin. Med. J. 2018, 131, 1333–1341. [Google Scholar] [CrossRef]
- Hall, C.; Ehrlich, L.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Lairmore, T.C.; Alpini, G.; Glaser, S. Inhibition of microRNA-24 increases liver fibrosis by enhanced menin expression in Mdr2(−/−) mice. J. Surg. Res. 2017, 217, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiaoqiao, C.; Li, H.; Liu, X.; Yan, Z.; Zhao, M.; Xu, Z.; Wang, Z.; Shi, K. MiR-24-3p regulates cell proliferation and milk protein synthesis of mammary epithelial cells through menin in dairy cows. J. Cell Physiol. 2019, 234, 1522–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrezk, R.; Hannah-Shmouni, F.; Stratakis, C.A. MEN4 and CDKN1B mutations: The latest of the MEN syndromes. Endocr. Relat. Cancer. 2017, 24, T195–T208. [Google Scholar] [CrossRef] [Green Version]
- Luzi, E.; Ciuffi, S.; Marini, F.; Mavilia, C.; Galli, G.; Brandi, M.L. Analysis of differentially expressed microRNAs in MEN1 parathyroid adenomas. Am. J. Transl Res. 2017, 9, 1743–1753. [Google Scholar] [PubMed]
- Falchetti, A.; Marin, F.; Luzi, E.; Giusti, F.; Cavalli, L.; Cavalli, T.; Brandi, M.L. Multiple endocrine neoplasia type 1 (MEN1): Not only inherited endocrine tumors. Genet. Med. 2009, 11, 825–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayaraghavan, J.; Maggi, E.C.; Crabtree, J.S. miR-24 regulates menin in the endocrine pancreas. Am. J. Physiol Endocrinol. Metab. 2014, 307, E84–E92. [Google Scholar] [CrossRef]
- Ehrlich, L.; Hall, C.; Venter, J.; Dostal, D.; Bernuzzi, F.; Invernizzi, P.; Meng, F.; Trzeciakowski, J.P.; Zhou, T.; Standeford, H.; et al. miR-24 Inhibition Increases Menin Expression and Decreases Cholangiocarcinoma Proliferation. Am. J. Pathol. 2017, 187, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, H.; Ma, D.; Ji, Z.; Luo, L.; Cao, F.; Huang, F.; Liu, Y.; Dong, Y.; Chen, Y. miR-24 may be a negative regulator of menin in lung cancer. Oncol. Rep. 2018, 39, 2342–2350. [Google Scholar] [CrossRef] [Green Version]
- Montero, C.; Sanjuán, P.; del Mar Fernández, M.; Vidal, I.; Verea, H.; Cordido, F. Bronchial carcinoid and type 1 multiple endocrine neoplasia syndrome. A case report. Arch. Bronconeumol. 2010, 46, 559–561. (In Spanish) [Google Scholar] [CrossRef]
- Gang, D.; Hongwei, H.; Hedai, L.; Ming, Z.; Qian, H.; Zhijun, L. The tumor suppressor protein menin inhibits NF-κB-mediated transactivation through recruitment of Sirt1 in hepatocellular carcinoma. Mol. Biol. Rep. 2013, 40, 2461–2466. [Google Scholar] [CrossRef]
- Gallo, A.; Agnese, S.; Esposito, I.; Galgani, M.; Avvedimento, V.E. Menin stimulates homology-directed DNA repair. FEBS Lett. 2010, 584, 4531–4536. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Mao, H.; Schnepp, R.W.; Sykes, S.M.; Silva, A.C.; D’Andrea, A.D.; Hua, X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003, 63, 4204–4210. [Google Scholar]
- Gambari, R.; Brognara, E.; Spandidos, D.A.; Fabbri, E. Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: Νew trends in the development of miRNA therapeutic strategies in oncology (Review). Int. J. Oncol. 2016, 49, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Mattes, J.; Yang, M.; Foster, P.S. Regulation of microRNA by antagomirs: A new class of pharmacological antagonists for the specific regulation of gene function? Am. J. Respir. Cell Mol. Biol. 2007, 36, 8–12. [Google Scholar] [CrossRef]
- Vester, B.; Wengel, J. LNA (locked nucleic acid): High-affinity targeting of complementary RNA and DNA. Biochemistry 2004, 43, 13233–13241. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Danquah, M.; Chaudhary, A.K.; Mahato, R.I. Small molecules targeting microRNA for cancer therapy: Promises and obstacles. J. Control. Release 2015, 219, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z. The principles of MiRNA-masking antisense oligonucleotides technology. Methods Mol. Biol. 2011, 676, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Preethi, K.A.; Lakshmanan, G.; Sekar, D. Antagomir technology in the treatment of different types of cancer. Epigenomics 2021, 13, 481–484. [Google Scholar] [CrossRef]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Effects of AntagomiRs on Different Lung Diseases in Human, Cellular, and Animal Models. Int. J. Mol. Sci. 2019, 20, 3938. [Google Scholar] [CrossRef] [Green Version]
- Innao, V.; Allegra, A.; Pulvirenti, N.; Allegra, A.G.; Musolino, C. Therapeutic potential of antagomiRs in haematological and oncological neoplasms. Eur. J. Cancer Care 2020, 29, e13208. [Google Scholar] [CrossRef]
- Krutzfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007, 35, 2885–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oli-gonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Chen, J.; Huang, Z. Recent progress in microRNA-based delivery systems for the treatment of human disease. ExRNA 2019, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, I.; Anushila Chatterjee, A. Recent Advances in miRNA Delivery Systems. Methods Protoc. 2021, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Muthana, M.; Scott, S.D.; Farrow, N.; Morrow, F.; Murdoch, C.; Grubb, S.; Brown, N.; Dobson, J.; Lewis, C.E. A novel magnetic approach to enhance the efficacy of cell-based gene therapies. Gene Ther. 2008, 15, 902–910. [Google Scholar] [CrossRef]
- Revia, R.A.; Stephen, Z.R.; Zhang, M. Theranostic Nanoparticles for RNA-Based Cancer Treatment. Acc. Chem Res. 2019, 52, 1496–1506. [Google Scholar] [CrossRef]
- Hofmann, A.; Wenzel, D.; Becher, U.M.; Freitag, D.F.; Klein, A.M.; Eberbeck, D.; Schulte, M.; Zimmermann, K.; Bergemann, C.; Gleich, B.; et al. Combined targeting of lentiviral vectors and positioning of transduced cells by magnetic nanoparticles. Proc. Natl. Acad. Sci. USA 2009, 106, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Carpten, J.D.; Robbins, C.M.; Villablanca, A.; Forsberg, L.; Presciuttini, S.; Bailey-Wilson, J.; Simonds, W.F.; Gillanders, E.M.; Kennedy, A.M.; Chen, J.D.; et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat. Genet. 2002, 32, 676–680. [Google Scholar] [CrossRef]
- Shattuck, T.M.; Välimäki, S.; Obara, T.; Gaz, R.D.; Clark, O.H.; Shoback, D.; Wierman, M.E.; Tojo, K.; Robbins, C.M.; Carpten, J.D.; et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N. Engl. J. Med. 2003, 349, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, M.; Mörz, M.; Pinzer, T.; Schackert, H.K.; Schackert, G. Frequent loss of the CDKN2C (p18INK4c) gene product in pituitary adenomas. Genes Chromosomes Cancer 2009, 48, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Fei, X.-Q.; Yang, S.-F.; Xu, B.-K.; Li, Y.-Y. Glucose-induced microRNA-17 promotes pancreatic beta cell proliferation through down-regulation of Menin. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 624–629. [Google Scholar] [PubMed]
- Gurung, B.; Katona, B.W.; Hua, X. Menin-mediated regulation of miRNA biogenesis uncovers the IRS2 pathway as a target for regulating pancreatic beta cells. Oncoscience 2014, 1, 562–566. [Google Scholar] [CrossRef]
- Roldo, C.; Missiaglia, E.; Hagan, J.P.; Falconi, M.; Capelli, P.; Bersani, S.; Calin, G.A.; Volinia, S.; Liu, C.G.; Scarpa, A.; et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol. 2006, 24, 4677–4684. [Google Scholar] [CrossRef]
- Bottoni, A.; Piccin, D.; Tagliati, F.; Luchin, A.; Zatelli, M.C.; degli Uberti, E.C. miR-15a and miR-16-1 down-regulation in pituitary adenomas. J. Cell Physiol. 2005, 204, 280–285. [Google Scholar] [CrossRef]
- Lines, K.E.; Newey, P.J.; Yates, C.J.; Stevenson, M.; Dyar, R.; Walls, G.V.; Bowl, M.R.; Thakker, R.V. miR-15a/miR-16-1 expression inversely correlates with cyclin D1 levels in Men1 pituitary NETs. J. Endocrinol. 2018, 240, 41–50. [Google Scholar] [CrossRef] [Green Version]
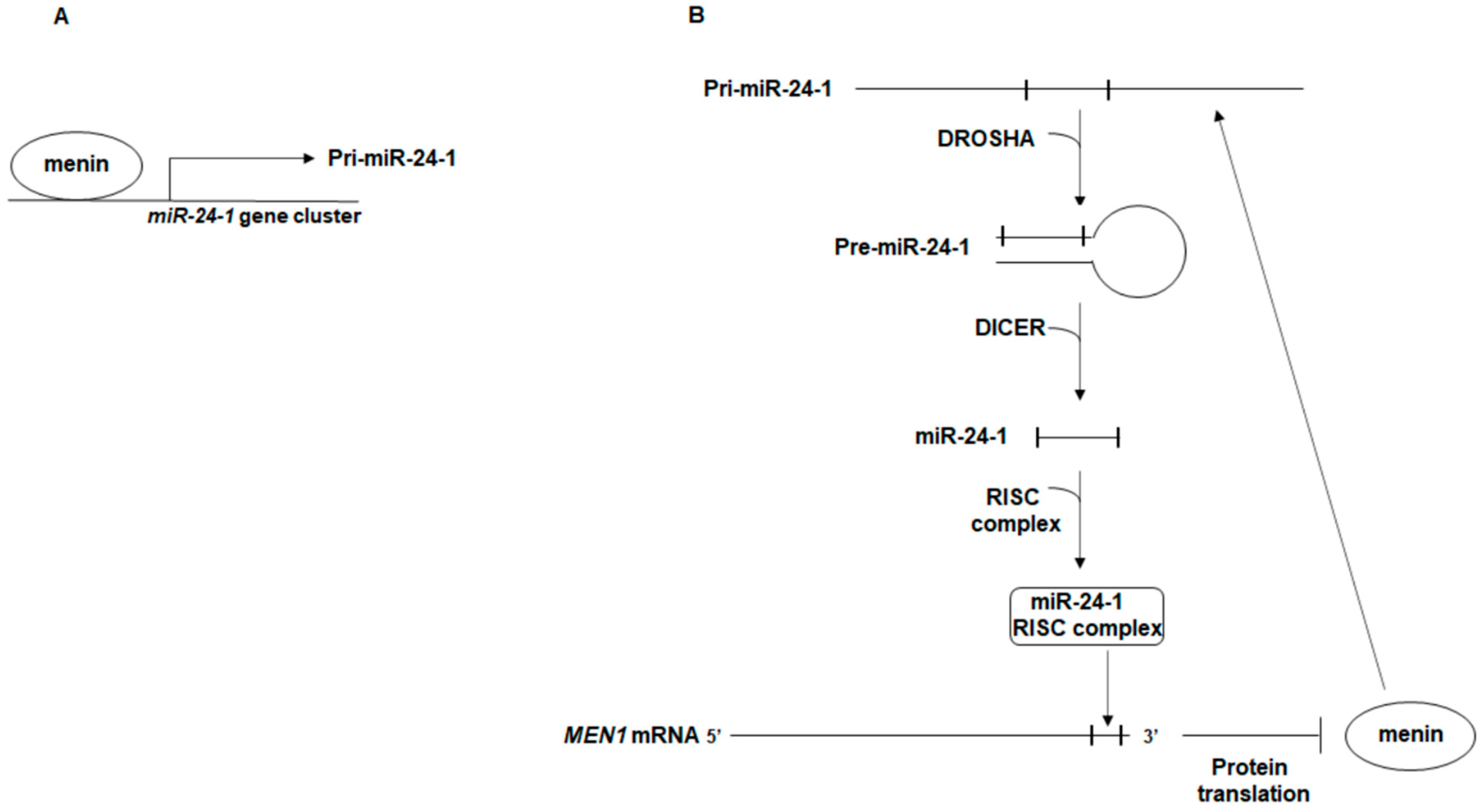

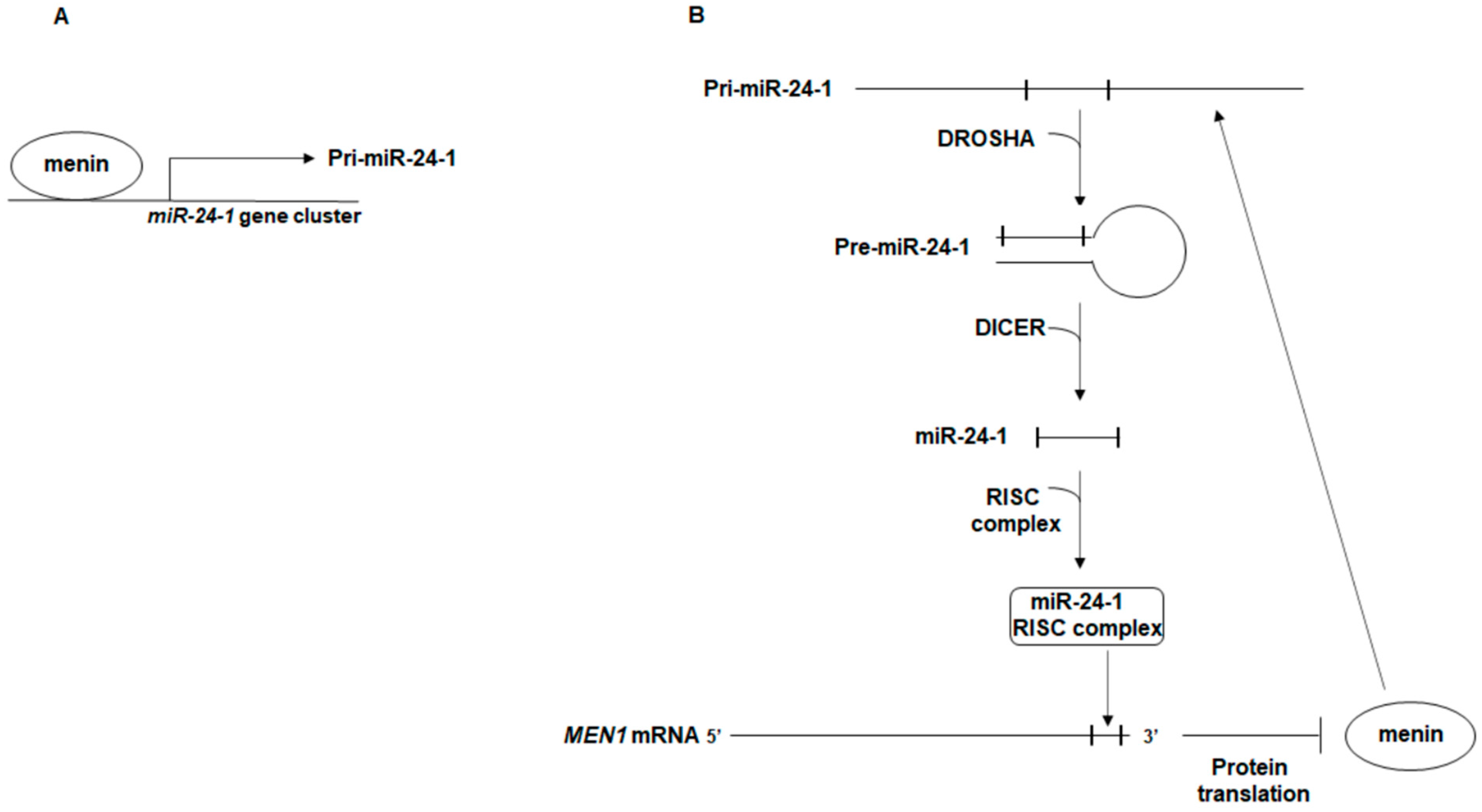{kind=link}
| miR-24 Target | Effect | Possible Role in MEN1 Tumorigenesis | Reference |
|---|---|---|---|
| Parathyroid glands | |||
| MEN1 | No effect on MEN1 mRNA expression. Loss of menin protein expression. | Uncontrolled cell proliferation | [27] |
| Endocrine pancreas | |||
| MEN1 | Reduction of both MEN1 mRNA and menin expression. | Uncontrolled cell proliferation | [36] |
| CDKN1B | Reduction of expression of both CDKN1B mRNA and of p27kip1 protein in a mouse insulinoma cell line (MIN6). | Enhanced proliferation of beta-cells and hyperplasia of pancreas islets | [36] |
| CDKN1B | Reduction of expression of CDKN1B mRNA in an immortalized human pancreas beta cell line (Blox5). No data on expression of p27kip1 protein. | Enhanced proliferation of beta-cells and hyperplasia of pancreas islets | [36] |
| CDKN2C | No effect on expression of both CDKN2C mRNA and of p18Ink4c protein in a mouse insulinoma cell line (MIN6). | Non applicable | [36] |
| CDKN2C | Reduction of expression of CDKN2C mRNA in an immortalized human pancreas beta cell line (Blox5). No data on expression of p18Ink4c protein. | Enhanced proliferation of beta-cells and hyperplasia of pancreas islets | [36] |
| Therapeutic Tool | Description | Mechanism of Action on the Target oncomiR | Reference |
|---|---|---|---|
| Anti-miRNA oligonucleotides (AMOs) | Synthetic single-stranded RNA molecules complementary to the target miRNA | Competitive inhibition of the target mature miRNA by base pair | [44] |
| Modified AMOs | AMOs with a chemical modification of the 2′-OH into 2′-O′methyl- or 2′-O′methoxyethyl- groups, to increase intracellular stability | Competitive inhibition of the target mature miRNA by base pair | [44] |
| Antagomirs | AMOs with the 2′-O chemical modification and phosphorothioate bonds to increase intracellular stability and with a conjugated cholesterol tail at the 3′-end to favor cell membrane permeation | Competitive inhibition of the target mature miRNA by base pair | [45] |
| Locked nucleic acids (LNA)-based AMOs | AMOs containing an additional methylene link between the 2′-O atom and the 4′-C atom, that locks ribose into a more thermodynamically stable conformation | High-affinity base pair with their target mature miRNA. Inhibition of miRNA activity | [46] |
| Small molecules miRNA inhibitors | Small molecules (chemical compounds) that interfere with miRNA biogenesis and/or activity | Inhibition of a specific miRNA biogenesis and/or activity by chemical structure-based docking to miRNA precursor or to mature miRNA | [47] |
| miRNA sponges | RNA transcripts presenting multiple tandem repeats of the binding site (seed sequence) of the miRNA to be targeted | They stably interact with the endogenous target miRNA, preventing its interaction with its target mRNAs | [48] |
| miRNA masks | Single-stranded 2′-O′methyl-modified antisense oligonucleotides totally complementary to the miRNA binding sites in the 3′-UTR of the target mRNA | They “mask” the target mRNA from the endogenous miRNA, preventing the miRNA-driven suppression of protein translation | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marini, F.; Brandi, M.L. Role of miR-24 in Multiple Endocrine Neoplasia Type 1: A Potential Target for Molecular Therapy. Int. J. Mol. Sci. 2021, 22, 7352. https://doi.org/10.3390/ijms22147352
Marini F, Brandi ML. Role of miR-24 in Multiple Endocrine Neoplasia Type 1: A Potential Target for Molecular Therapy. International Journal of Molecular Sciences. 2021; 22(14):7352. https://doi.org/10.3390/ijms22147352
Chicago/Turabian StyleMarini, Francesca, and Maria Luisa Brandi. 2021. "Role of miR-24 in Multiple Endocrine Neoplasia Type 1: A Potential Target for Molecular Therapy" International Journal of Molecular Sciences 22, no. 14: 7352. https://doi.org/10.3390/ijms22147352
APA StyleMarini, F., & Brandi, M. L. (2021). Role of miR-24 in Multiple Endocrine Neoplasia Type 1: A Potential Target for Molecular Therapy. International Journal of Molecular Sciences, 22(14), 7352. https://doi.org/10.3390/ijms22147352