
Upregulation of Anti-Oxidative Stress Response Improves Metabolic Changes in L-Selectin-Deficient Mice but Does Not Prevent NAFLD Progression or Fecal Microbiota Shifts

 ,
,
Abstract
:1. Introduction
2. Results
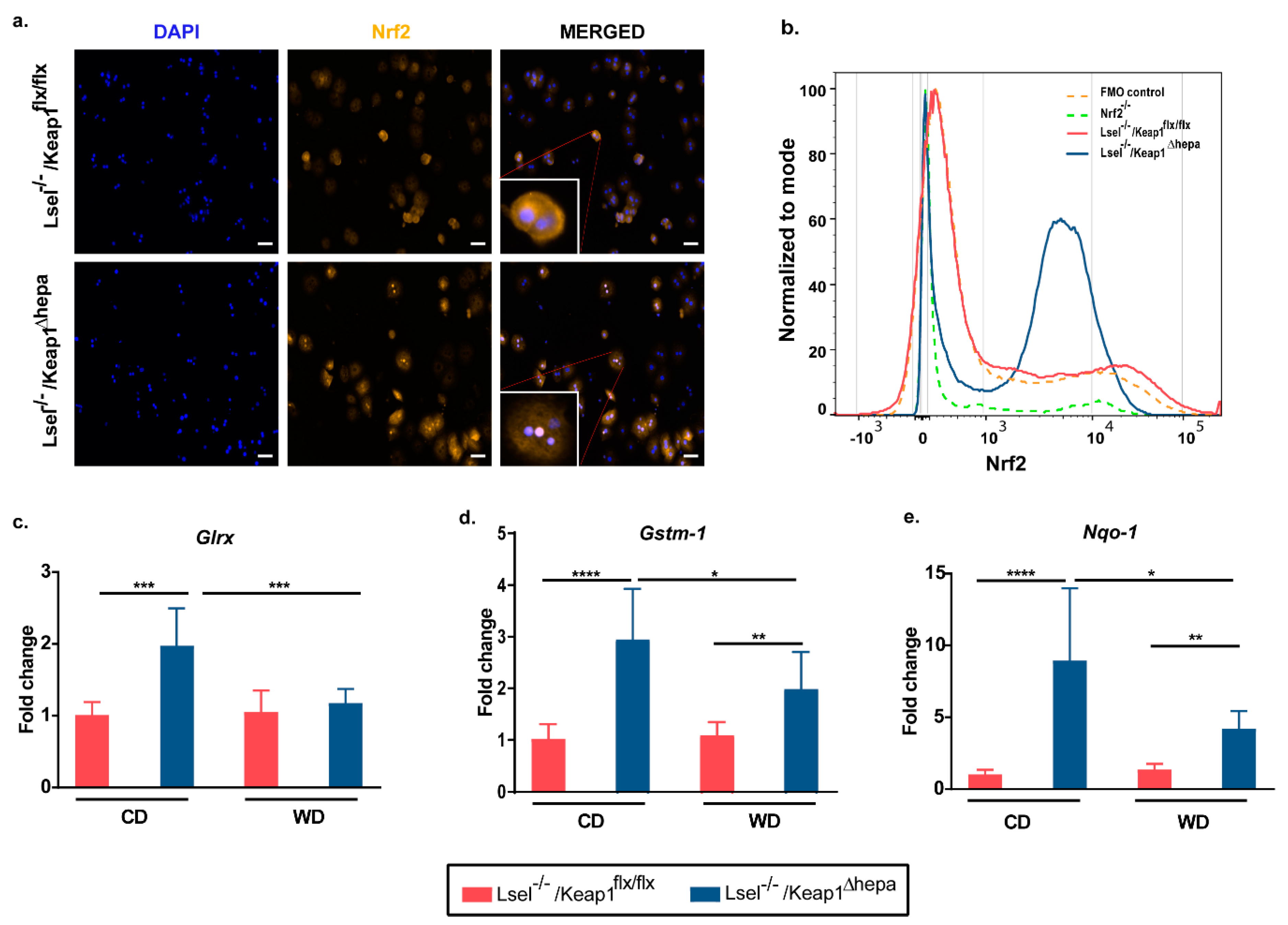
2.1. Nrf2 Activation in Hepatocytes of L-Selectin-Deficient Mice Results in Increased Expression of Nrf2 Target Genes
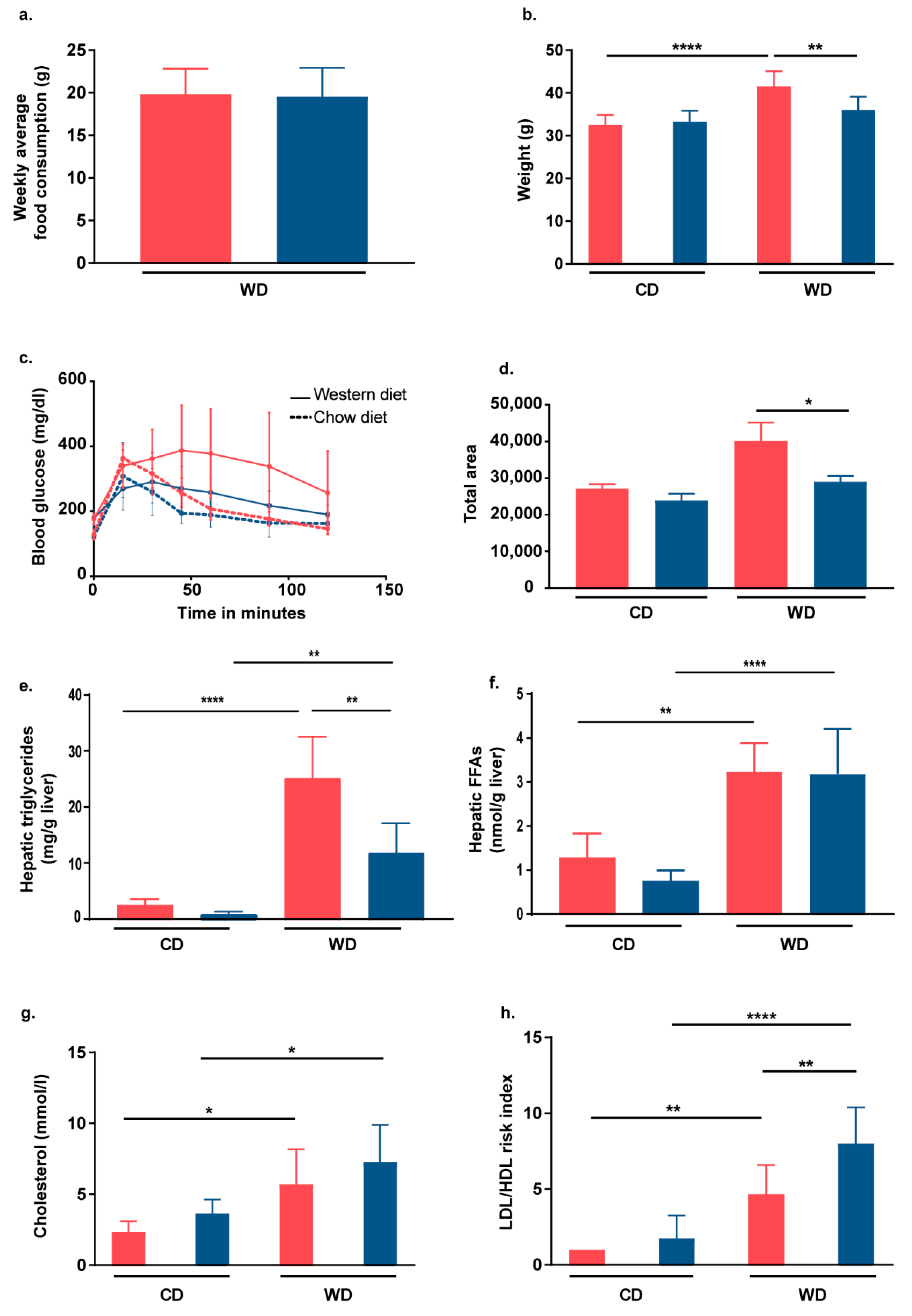
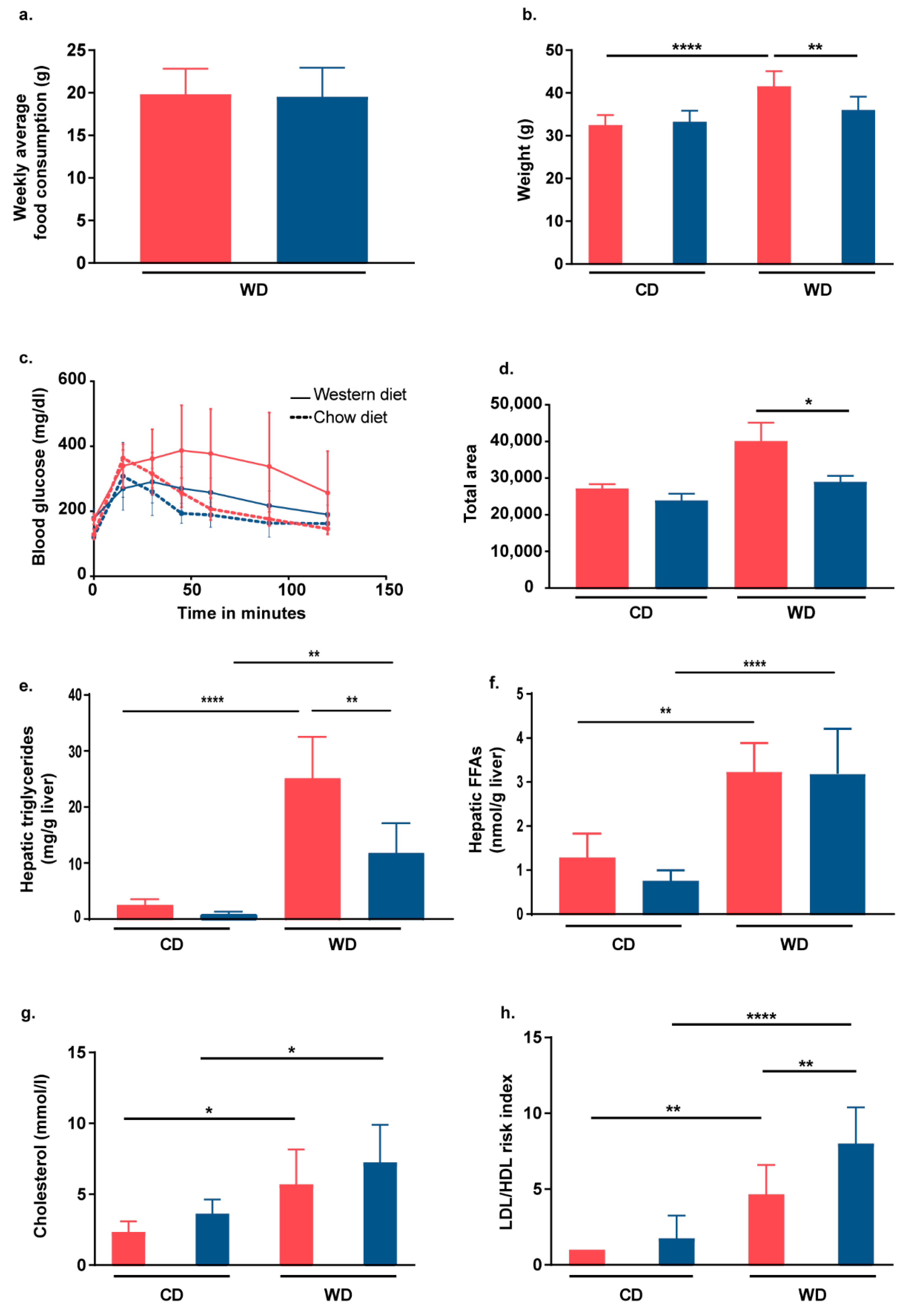
2.2. Nrf2 Activation in Hepatocytes of L-Selectin-Deficient Mice Partially Protects Mice from WD-Induced Metabolic Dysfunction
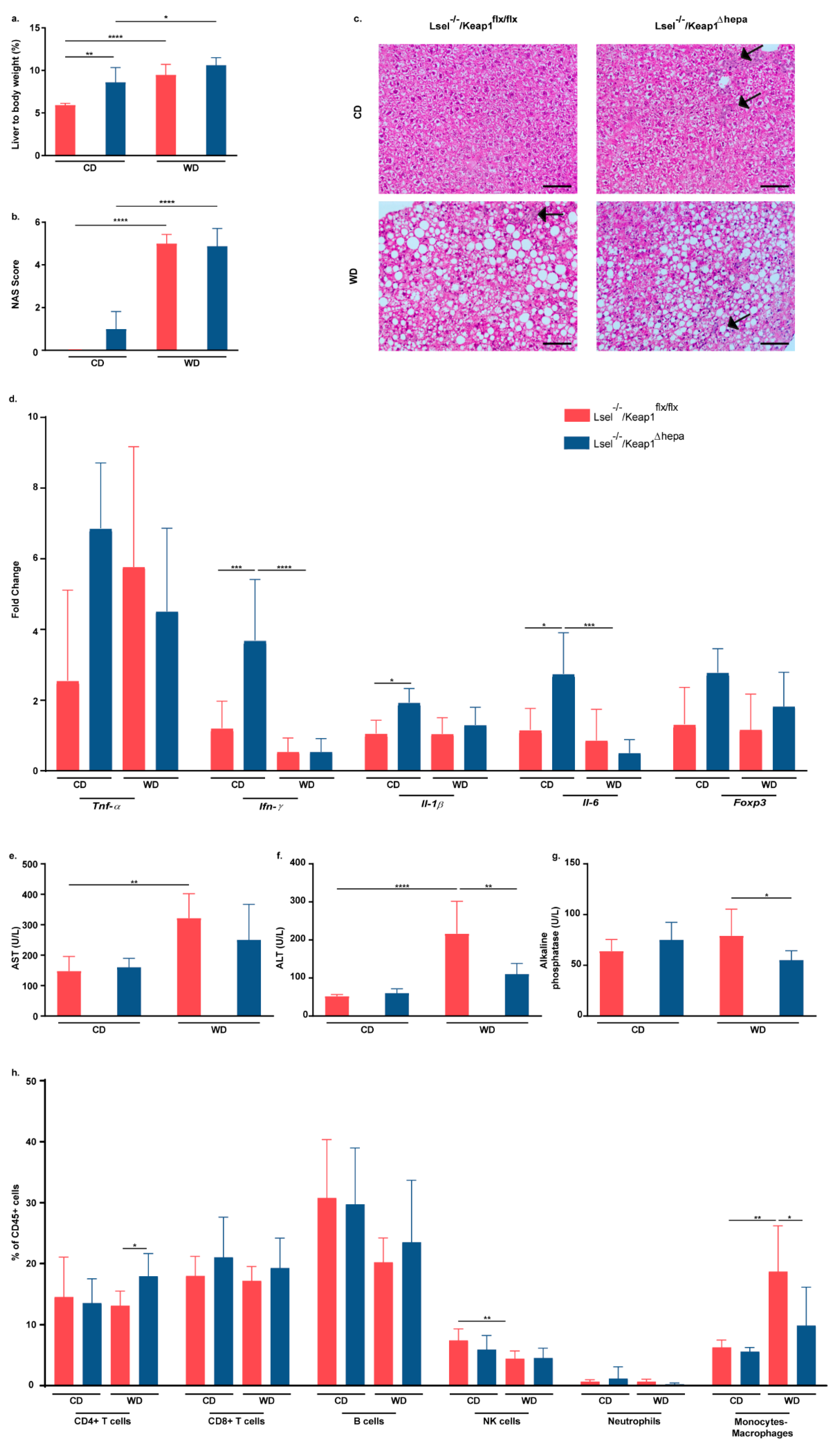
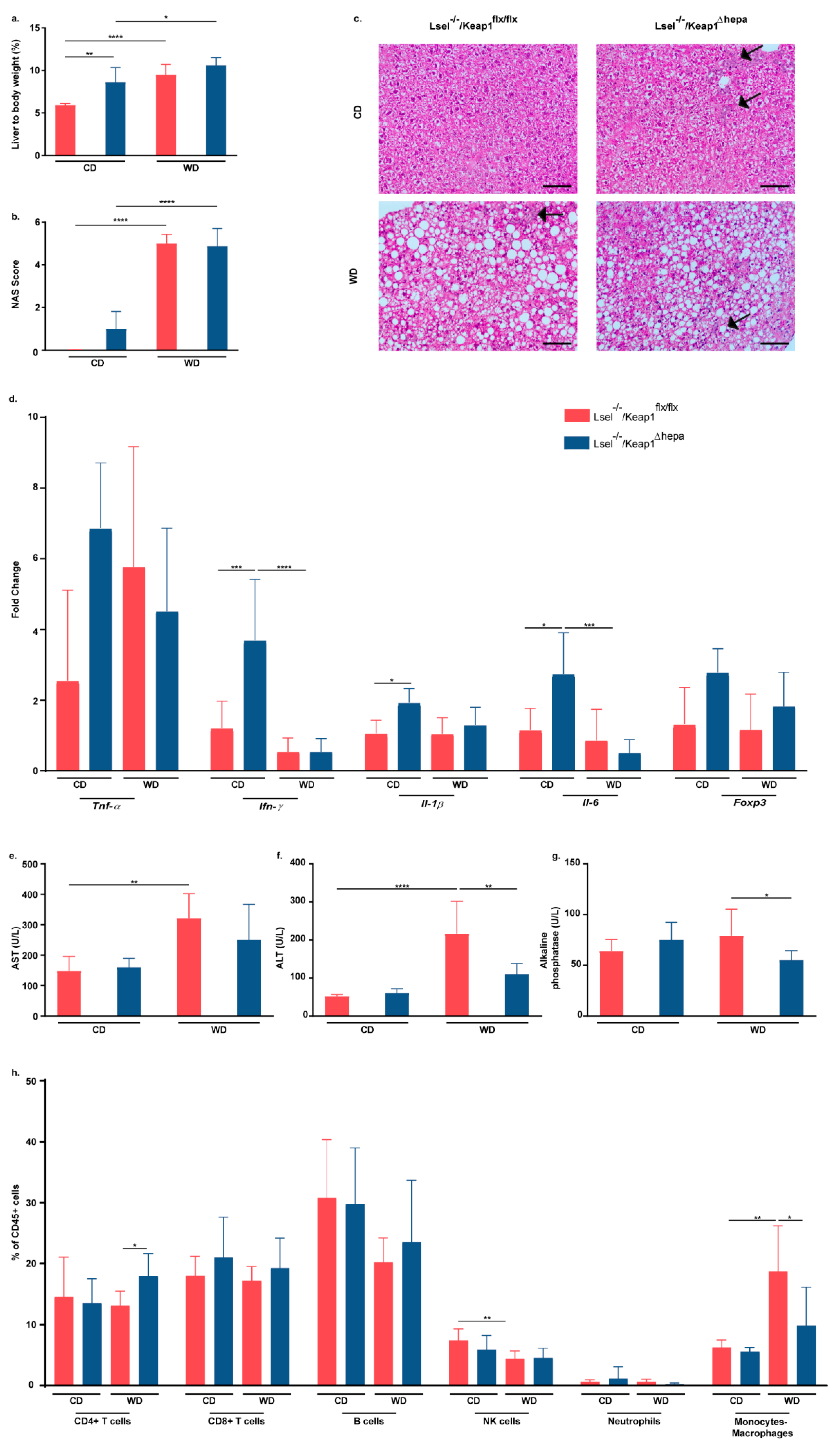
2.3. Nrf2 Activation in Hepatocytes of L-Selectin-Deficient Mice Has No Major Influence on WD-Induced NASH Progression
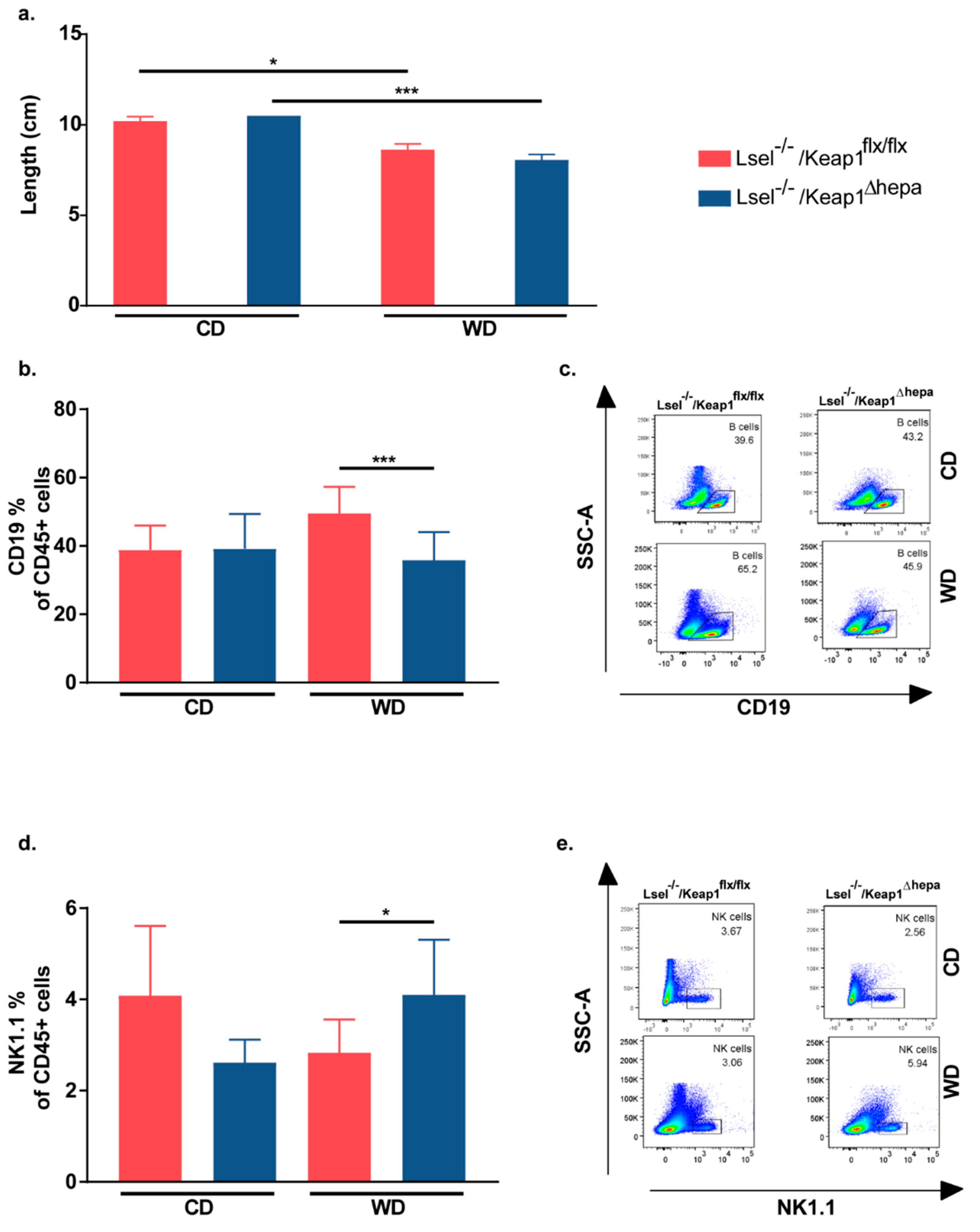
2.4. Nrf2 Activation in Hepatocytes of L-Selectin-Deficient Mice Results in a Decreased Epidydimal White Adipose Tissue/Body Weight Ratio
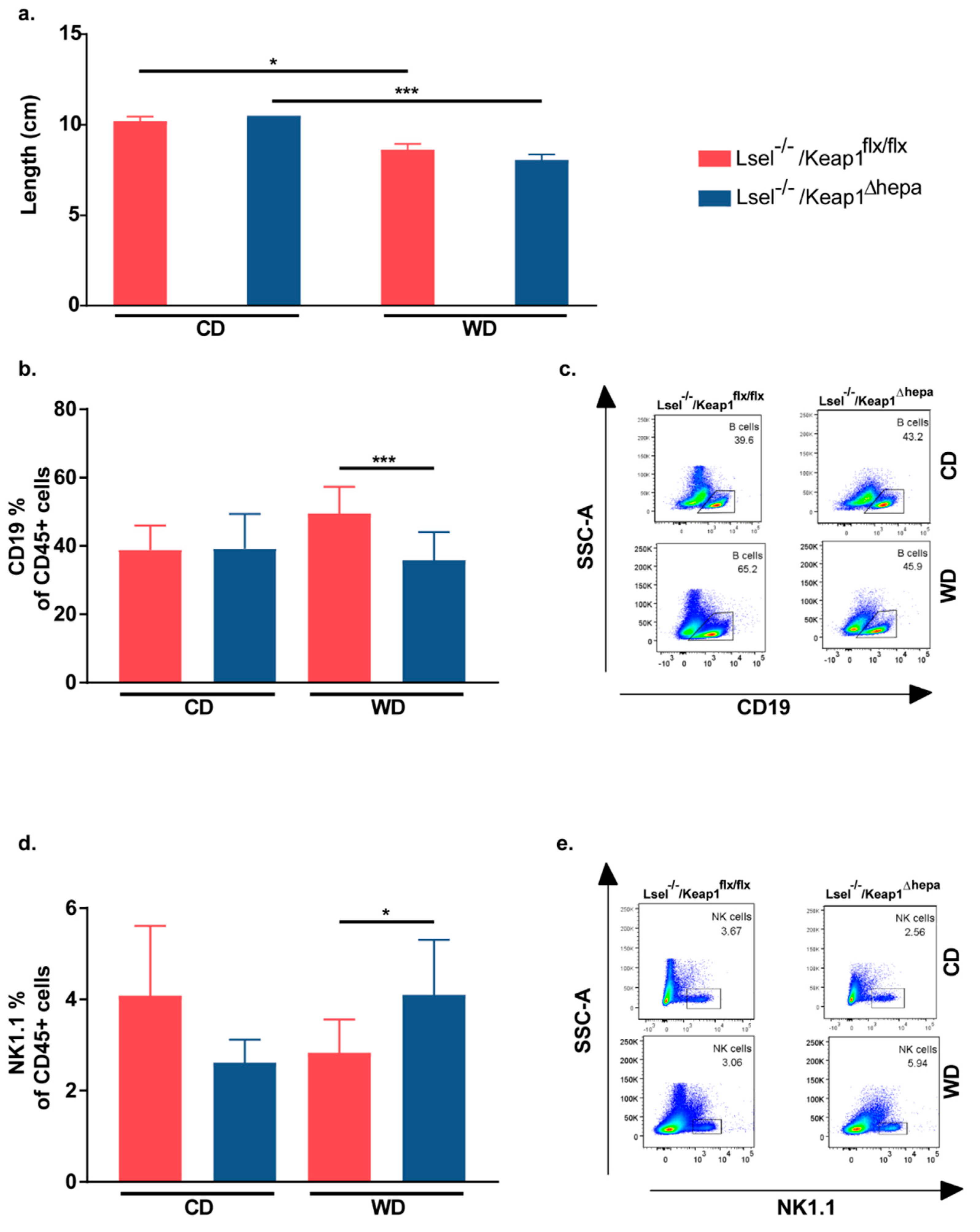
2.5. Nrf2 Activation in Hepatocytes of L-Selectin-Deficient Mice Has Only Minor Effects on WD-Induced Gut Inflammation
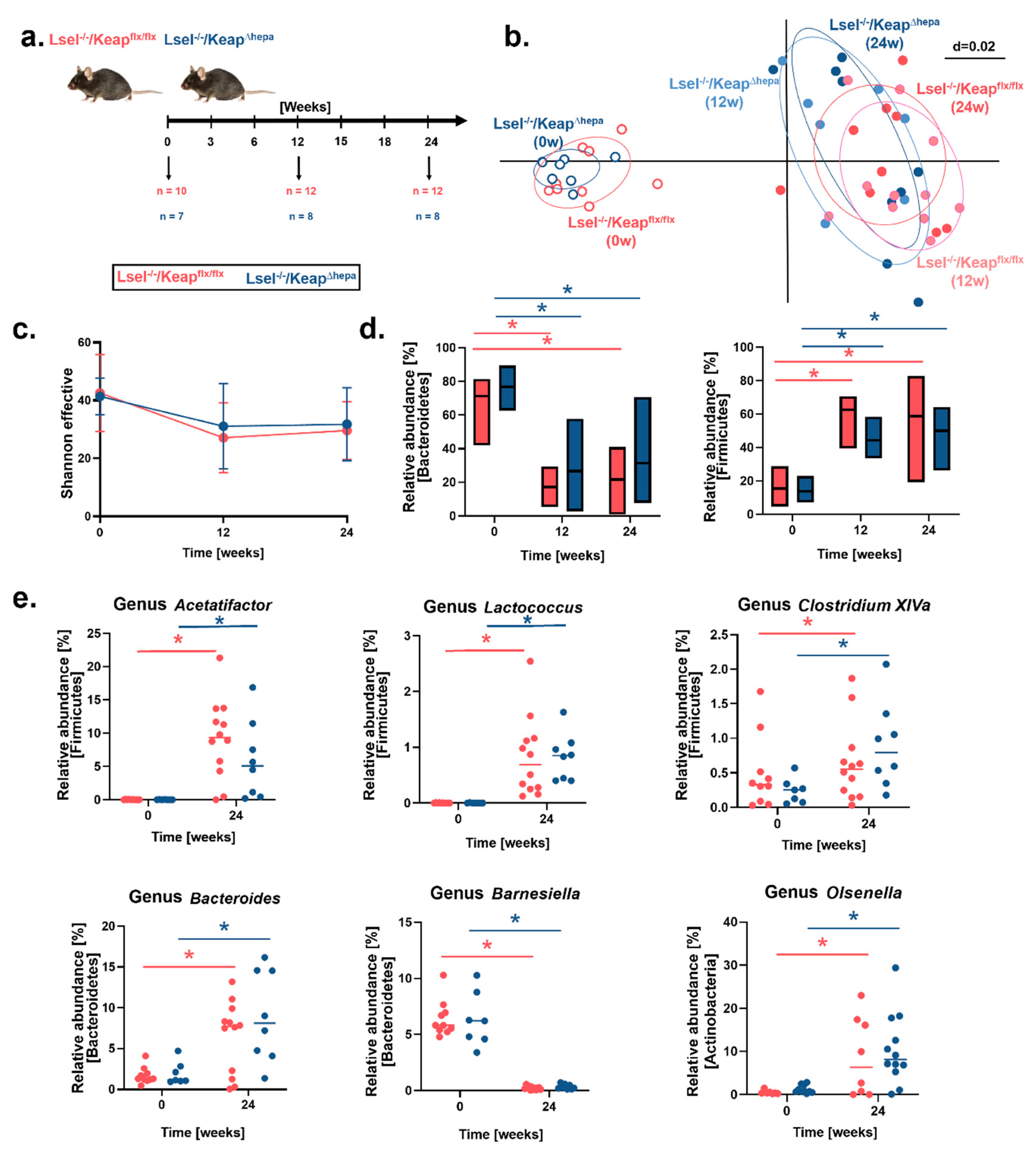
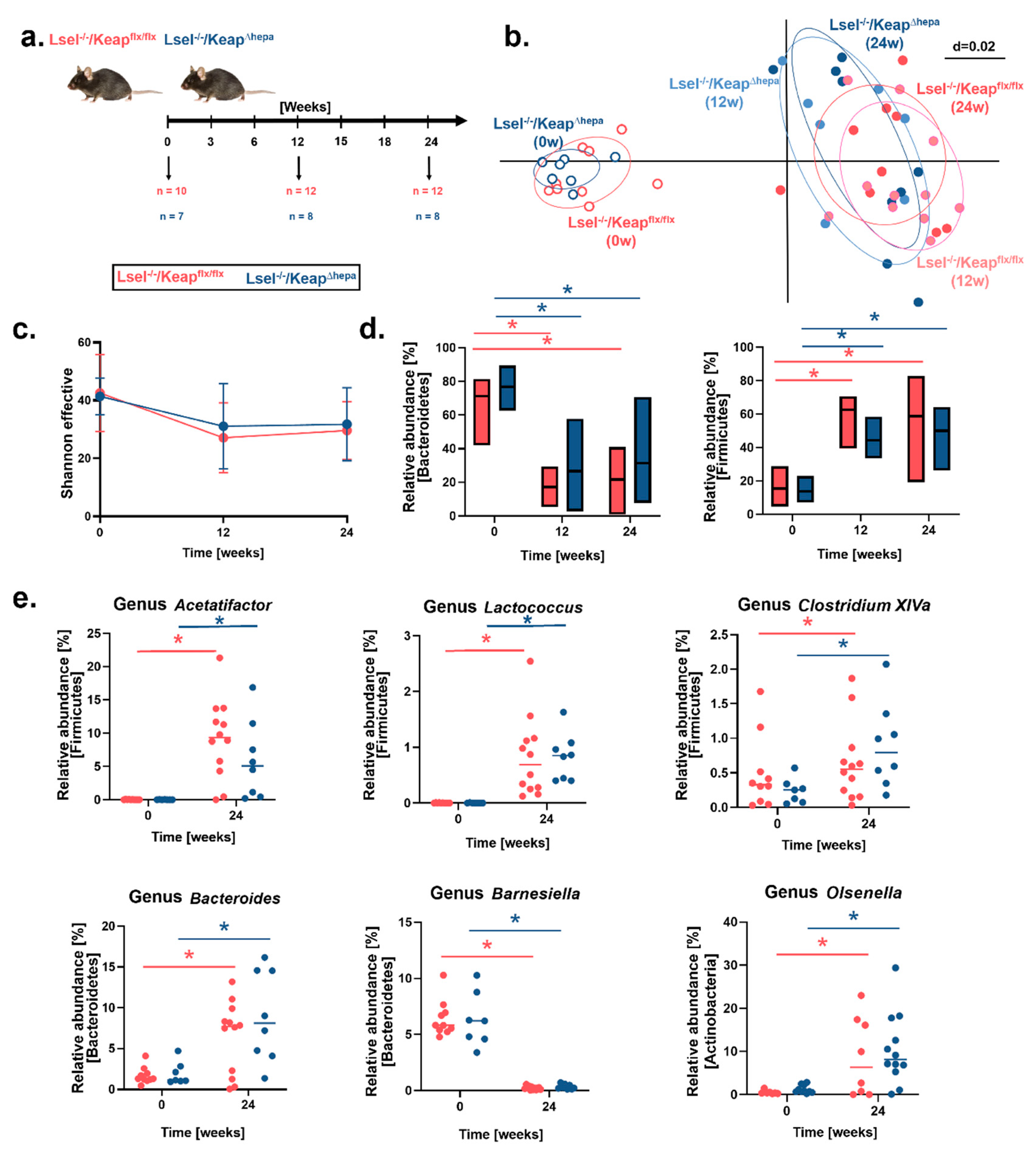
2.6. Fecal Microbiota Profiles Are Altered by WD-Feeding but Not by Hepatic Nrf2 Activation
3. Discussion
4. Materials and Methods
4.1. Housing, Generation of Mice, and Dietary Treatments
4.2. Glucose Tolerance Test (GTT)
4.3. Serum and Liver Biochemical Measurements
4.4. Histological Stainings
4.5. Isolation of Cells and Flow Cytometry
4.6. Gene Expression Analysis by Real-Time PCR
4.7. DNA Isolation from Feces, 16S rRNA Gene Amplicon Sequencing, and Data Analysis
4.8. Statistical Analysis of Microbiota Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF488 | Alexa Fluor 488 |
| APC-Cy7 | Allophycocyanin-cyanine 7 |
| CD | Chow diet |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GCLM | Glutamate-cysteine ligase modifier |
| GLRX | Glutaredoxin |
| GSTM1 | Glutathion S-transferase1 |
| HO-1 | Heme oxygenase-1 |
| IFN-γ | Interferon γ |
| IL-6 | Interleukin-6 |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NK | Natural killer |
| NQO-1 | NAD(P)H quinone dehydrogenase 1 |
| PE | Phycoerythrin |
| PerCP-Cy5.5 | Peridinin chlorophyll protein-cyanine5.5 |
| ROS | Reactive oxygen species |
| TNF-α | Tumor necrosis factor- α |
| TRX | Thioredoixin |
| WD | Western diet |
References
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, F.; George, J. The role of diet and nutritional intervention for the management of patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [Green Version]
- Gallatin, W.M.; Weissman, I.L.; Butcher, E.C. A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature 1983, 304, 30–34. [Google Scholar] [CrossRef]
- Martinez-Mier, G.; Toledo-Pereyra, L.H.; McDuffie, E.; Warner, R.L.; Ward, P.A. L-Selectin and chemokine response after liver ischemia and reperfusion. J. Surg. Res. 2000, 93, 156–162. [Google Scholar] [CrossRef]
- Lawson, J.A.; Burns, A.R.; Farhood, A.; Lynn Bajt, M.; Collins, R.G.; Smith, C.W.; Jaeschke, H. Pathophysiologic importance of E- and L-selectin for neutrophil-induced liver injury during endotoxemia in mice. Hepatology 2000, 32, 990–998. [Google Scholar] [CrossRef]
- Mitsui, A.; Tada, Y.; Shibata, S.; Kamata, M.; Hau, C.; Asahina, A.; Sato, S. Deficiency of both L-selectin and ICAM-1 exacerbates imiquimod-induced psoriasis-like skin inflammation through increased infiltration of antigen presenting cells. Clin. Immunol. 2015, 157, 43–55. [Google Scholar] [CrossRef]
- Oishi, K.; Hamaguchi, Y.; Matsushita, T.; Hasegawa, M.; Okiyama, N.; Dernedde, J.; Weinhart, M.; Haag, R.; Tedder, T.F.; Takehara, K.; et al. A crucial role of L-selectin in C protein-induced experimental polymyositis in mice. Arthritis Rheumatol. 2014, 66, 1864–1871. [Google Scholar] [CrossRef] [Green Version]
- Kolachala, V.L.; Palle, S.; Shen, M.; Feng, A.; Shayakhmetov, D.; Gupta, N.A. Loss of L-selectin-guided CD8(+), but not CD4(+), cells protects against ischemia reperfusion injury in a steatotic liver. Hepatology 2017, 66, 1258–1274. [Google Scholar] [CrossRef] [Green Version]
- Drescher, H.K.; Schippers, A.; Rosenhain, S.; Gremse, F.; Bongiovanni, L.; Bruin, A.; Eswaran, S.; Gallage, S.U.; Pfister, D.; Szydlowska, M.; et al. L-Selectin/CD62L is a Key Driver of Non-Alcoholic Steatohepatitis in Mice and Men. Cells 2020, 9, 1106. [Google Scholar] [CrossRef]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Da Costa, R.M.; Fais, R.S.; Dechandt, C.R.P.; Louzada-Junior, P.; Alberici, L.C.; Lobato, N.S.; Tostes, R.C. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice. Br. J. Pharmacol. 2017, 174, 3527–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, A.; Svegliati-Baroni, G.; Ceni, E.; Milani, S.; Ridolfi, F.; Salzano, R.; Tarocchi, M.; Grappone, C.; Pellegrini, G.; Benedetti, A.; et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology 2005, 41, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Van Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver 2001, 21, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Lee, J.M.; Li, J.; Johnson, D.A.; Stein, T.D.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Johnson, J.A. Nrf2, a multi-organ protector? FASEB J. 2005, 19, 1061–1066. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova-Kostova, A.T.; Georgiev, M.I. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef] [Green Version]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 2010, 245, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Ramadori, P.; Drescher, H.; Erschfeld, S.; Schumacher, F.; Berger, C.; Fragoulis, A.; Schenkel, J.; Kensler, T.W.; Wruck, C.J.; Trautwein, C.; et al. Hepatocyte-specific Keap1 deletion reduces liver steatosis but not inflammation during non-alcoholic steatohepatitis development. Free Radic. Biol. Med. 2016, 91, 114–126. [Google Scholar] [CrossRef]
- Ramadori, P.; Drescher, H.; Erschfeld, S.; Fragoulis, A.; Kensler, T.W.; Wruck, C.J.; Cubero, F.J.; Trautwein, C.; Streetz, K.L.; Kroy, D.C. Genetic Nrf2 Overactivation Inhibits the Deleterious Effects Induced by Hepatocyte-Specific c-met Deletion during the Progression of NASH. Oxid. Med. Cell. Longev. 2017, 2017, 3420286. [Google Scholar] [CrossRef]
- Vasconcelos, A.R.; Dos Santos, N.B.; Scavone, C.; Munhoz, C.D. Nrf2/ARE Pathway Modulation by Dietary Energy Regulation in Neurological Disorders. Front. Pharmacol. 2019, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Sah, S.K.; Lee, C.; Jang, J.H.; Park, G.H. Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 493, 731–736. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef] [Green Version]
- Mulder, P.; Morrison, M.C.; Wielinga, P.Y.; Van Duyvenvoorde, W.; Kooistra, T.; Kleemann, R. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non-alcoholic steatohepatitis in obesity. Int. J. Obes. 2016, 40, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayangsari, Y.; Suzuki, T. Resveratrol Ameliorates Intestinal Barrier Defects and Inflammation in Colitic Mice and Intestinal Cells. J. Agric. Food Chem. 2018, 66, 12666–12674. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, P.A. Hepatic bile acid uptake in humans and mice: Multiple pathways and expanding potential role for gut-liver signaling. Hepatology 2017, 66, 1384–1386. [Google Scholar] [CrossRef]
- Slocum, S.L.; Skoko, J.J.; Wakabayashi, N.; Aja, S.; Yamamoto, M.; Kensler, T.W.; Chartoumpekis, D.V. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch. Biochem. Biophys. 2016, 591, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Berger, F.; Ramirez-Hernandez, M.H.; Ziegler, M. The new life of a centenarian: Signalling functions of NAD(P). Trends Biochem. Sci. 2004, 29, 111–118. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Hwang, J.H.; Kim, D.W.; Jo, E.J.; Kim, Y.K.; Jo, Y.S.; Park, J.H.; Yoo, S.K.; Park, M.K.; Kwak, T.H.; Kho, Y.L.; et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes 2009, 58, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.I.; Zou, X.; Ijaz, M.U.; Hussain, M.; Liu, C.; Xu, X.; Zhou, G.; Li, C. Processed Meat Protein Promoted Inflammation and Hepatic Lipogenesis by Upregulating Nrf2/Keap1 Signaling Pathway in Glrx-Deficient Mice. J. Agric. Food Chem. 2019, 67, 8794–8809. [Google Scholar] [CrossRef]
- Beyer, T.A.; Xu, W.; Teupser, D.; Auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: Role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef]
- Barroso, E.; Rodriguez-Rodriguez, R.; Chacon, M.R.; Maymo-Masip, E.; Ferrer, L.; Salvado, L.; Salmeron, E.; Wabistch, M.; Palomer, X.; Vendrell, J.; et al. PPARbeta/delta ameliorates fructose-induced insulin resistance in adipocytes by preventing Nrf2 activation. Biochim. Biophys. Acta 2015, 1852, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar]
- Chartoumpekis, D.V.; Yagishita, Y.; Fazzari, M.; Palliyaguru, D.L.; Rao, U.N.; Zaravinos, A.; Khoo, N.K.; Schopfer, F.J.; Weiss, K.R.; Michalopoulos, G.K.; et al. Nrf2 prevents Notch-induced insulin resistance and tumorigenesis in mice. JCI Insight 2018, 3, e97735. [Google Scholar] [CrossRef]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Kubeck, R.; Bonet-Ripoll, C.; Hoffmann, C.; Walker, A.; Muller, V.M.; Schuppel, V.L.; Lagkouvardos, I.; Scholz, B.; Engel, K.H.; Daniel, H.; et al. Dietary fat and gut microbiota interactions determine diet-induced obesity in mice. Mol. Metab. 2016, 5, 1162–1174. [Google Scholar] [CrossRef]
- Xiao, L.; Sonne, S.B.; Feng, Q.; Chen, N.; Xia, Z.; Li, X.; Fang, Z.; Zhang, D.; Fjaere, E.; Midtbo, L.K.; et al. High-fat feeding rather than obesity drives taxonomical and functional changes in the gut microbiota in mice. Microbiome 2017, 5, 43. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, L.; Goig, G.A.; Cardona, P.; Torres-Puente, M.; Diaz, J.; Rosales, Y.; Garcia, E.; Tapia, G.; Comas, I.; Vilaplana, C.; et al. Influence of Gut Microbiota on Progression to Tuberculosis Generated by High Fat Diet-Induced Obesity in C3HeB/FeJ Mice. Front. Immunol. 2019, 10, 2464. [Google Scholar] [CrossRef] [PubMed]
- Fleissner, C.K.; Huebel, N.; Abd El-Bary, M.M.; Loh, G.; Klaus, S.; Blaut, M. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br. J. Nutr. 2010, 104, 919–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, B.W.; Nam, E.; Org, E.; Kostem, E.; Norheim, F.; Hui, S.T.; Pan, C.; Civelek, M.; Rau, C.D.; Bennett, B.J.; et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013, 17, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Abdullah, Z.; Deng, L.; Zhao, M.; Tang, J.; Liu, T.; Zhang, H.; Feng, F. Probiotic-fermented blueberry juice prevents obesity and hyperglycemia in high fat diet-fed mice in association with modulating the gut microbiota. Food Funct. 2020, 11, 9192–9207. [Google Scholar] [CrossRef]
- Anhe, F.F.; Nachbar, R.T.; Varin, T.V.; Trottier, J.; Dudonne, S.; Le Barz, M.; Feutry, P.; Pilon, G.; Barbier, O.; Desjardins, Y.; et al. Treatment with camu camu (Myrciaria dubia) prevents obesity by altering the gut microbiota and increasing energy expenditure in diet-induced obese mice. Gut 2019, 68, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Li, H.; Wang, R.; Tang, J.; Huang, T.; et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: A 6-month randomised controlled-feeding trial. Gut 2019, 68, 1417–1429. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.Y.; Rho, M.; Song, Y.M.; Lee, K.; Sung, J.; Ko, G. Stability of gut enterotypes in Korean monozygotic twins and their association with biomarkers and diet. Sci. Rep. 2014, 4, 7348. [Google Scholar] [CrossRef] [Green Version]
- Bloom, S.M.; Bijanki, V.N.; Nava, G.M.; Sun, L.; Malvin, N.P.; Donermeyer, D.L.; Dunne, W.M., Jr.; Allen, P.M.; Stappenbeck, T.S. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 2011, 9, 390–403. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 1716–1724. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Salazar, E.O.; Ortiz-Lopez, M.G.; Granados-Silvestre, M.L.A.; Palacios-Gonzalez, B.; Menjivar, M. Altered Gut Microbiota and Compositional Changes in Firmicutes and Proteobacteria in Mexican Undernourished and Obese Children. Front. Microbiol. 2018, 9, 2494. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Andoh, A.; Nishida, A.; Takahashi, K.; Inatomi, O.; Imaeda, H.; Bamba, S.; Kito, K.; Sugimoto, M.; Kobayashi, T. Comparison of the gut microbial community between obese and lean peoples using 16S gene sequencing in a Japanese population. J Clin. Biochem. Nutr. 2016, 59, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Kong, C.; Gao, R.; Yan, X.; Huang, L.; Qin, H. Probiotics improve gut microbiota dysbiosis in obese mice fed a high-fat or high-sucrose diet. Nutrition 2019, 60, 175–184. [Google Scholar] [CrossRef]
- Wang, J.; Tang, H.; Zhang, C.; Zhao, Y.; Derrien, M.; Rocher, E.; van-Hylckama Vlieg, J.E.; Strissel, K.; Zhao, L.; Obin, M.; et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. 2015, 9, 1–15. [Google Scholar] [CrossRef]
- Arbones, M.L.; Ord, D.C.; Ley, K.; Ratech, H.; Maynard-Curry, C.; Otten, G.; Capon, D.J.; Tedder, T.F. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity 1994, 1, 247–260. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Pils, M.C.; Bleich, A.; Prinz, I.; Fasnacht, N.; Bollati-Fogolin, M.; Schippers, A.; Rozell, B.; Muller, W. Commensal gut flora reduces susceptibility to experimentally induced colitis via T-cell-derived interleukin-10. Inflamm. Bowel Dis. 2011, 17, 2038–2046. [Google Scholar] [CrossRef]
- Erben, U.; Loddenkemper, C.; Doerfel, K.; Spieckermann, S.; Haller, D.; Heimesaat, M.M.; Zeitz, M.; Siegmund, B.; Kuhl, A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014, 7, 4557–4576. [Google Scholar]
- Tran, H.Q.; Bretin, A.; Adeshirlarijaney, A.; Yeoh, B.S.; Vijay-Kumar, M.; Zou, J.; Denning, T.L.; Chassaing, B.; Gewirtz, A.T. “Western Diet”-Induced Adipose Inflammation Requires a Complex Gut Microbiota. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 313–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clahsen, T.; Pabst, O.; Tenbrock, K.; Schippers, A.; Wagner, N. Localization of dendritic cells in the gut epithelium requires MAdCAM-1. Clin. Immunol. 2015, 156, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Muller, W.; Sparwasser, T.; Forster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Drescher, H.K.; Schippers, A.; Clahsen, T.; Sahin, H.; Noels, H.; Hornef, M.; Wagner, N.; Trautwein, C.; Streetz, K.L.; Kroy, D.C. β7-Integrin and MAdCAM-1 play opposing roles during the development of non-alcoholic steatohepatitis. J. Hepatol. 2017, 66, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.; Ben Mahfoudh, K.; Wagner, M.; Loy, A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 2011, 77, 7846–7849. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Bartram, J.; Mountjoy, E.; Brooks, T.; Hancock, J.; Williamson, H.; Wright, G.; Moppett, J.; Goulden, N.; Hubank, M. Accurate Sample Assignment in a Multiplexed, Ultrasensitive, High-Throughput Sequencing Assay for Minimal Residual Disease. J. Mol. Diagn. 2016, 18, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Lagkouvardos, I.; Pukall, R.; Abt, B.; Foesel, B.U.; Meier-Kolthoff, J.P.; Kumar, N.; Bresciani, A.; Martinez, I.; Just, S.; Ziegler, C.; et al. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016, 1, 16131. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Fischer, S.; Kumar, N.; Clavel, T. Rhea: A transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ 2017, 5, e2836. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Channel | Manufacturer | Clone | Dilution |
|---|---|---|---|---|
| NK 1.1 | AF 488 | Biolegend | PK136 | 1:400 |
| CD4 | PE | BD biosciences | GK1.5 | 1:400 |
| CD45 | APC-Cy7 | BD biosciences | 30-F11 | 1:400 |
| CD19 | eFluor 450 | eBioscience | 1D3 | 1:200 |
| CD8a | Amcyan | Biolegend | 53-6.7 | 1:400 |
| CD3e | PerCp-Cy5.5 | eBioscience | 145-2C11 | 1:100 |
| F4/80 | eFluor 450 | Serotec | CI:A3-1 | 1:200 |
| CD11b | Amcyan | BD Biosciences | M1/70 | 1:400 |
| Ly6G | FITC | BD Biosciences | 1A8 | 1:400 |
| Calibrite counting beads | APC | BD Biosciences | 1 million beads/mL PBS | |
| CD146 | APC | Miltenyi | ME-9F1 | 1:400 |
| Nrf2 | GeneTex | N2C2 | 1:400 | |
| Goat anti-rabbit IgG | Cy3 | Invitrogen | 1:500 | |
| Goat anti-rabbit IgG | AF 633 | Invitrogen | 1:500 |
| Primer | Forward | Reverse |
|---|---|---|
| GAPDH | ACCTGCCAAGTATGATGACATCA | GGTCCTCAGTGTAGCCCAAGAT |
| NQO-1 | AGAGAGTGCTCGTAGCAGGAT | CTACCCCCAGTGGTGATAGAAA |
| TNF-α | CATCTTCTCAAAATTCGAGTGACAA | TGGGAGTAGACAAGGTACAACCC |
| IFN-γ | GAGGTCAACAACCCACAGGTC | CGAATCAGCAGCGACTCCT |
| IL-6 | CTGGAGTCACAGAAGGAGTGG | GGTTTGCCGAGTAGATCTCAA |
| IL-1β | GATCCCAAGGCAATACCCAAA | GGGGAACTCTGCAGACTCAA |
| Foxp3 | GGCAAATGGAGTCTGCAAGTG | CAGGAGATGATCTGCTTGGCA |
| GLRX | TTATAAAAGGGGTGGCAGGCAG | GTTAAGCTTCTCGGCCCCAT |
| GSTM1 | CCTGGATGGAGAGACAGAGG | GACCTTGTCCCCTGCAAA |
| GCLC | GGCACAAGGACGTTCTCAAGT | CAAAGGGTAGGATGGTTTGGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eswaran, S.; Babbar, A.; Drescher, H.K.; Hitch, T.C.A.; Clavel, T.; Muschaweck, M.; Ritz, T.; Kroy, D.C.; Trautwein, C.; Wagner, N.; et al. Upregulation of Anti-Oxidative Stress Response Improves Metabolic Changes in L-Selectin-Deficient Mice but Does Not Prevent NAFLD Progression or Fecal Microbiota Shifts. Int. J. Mol. Sci. 2021, 22, 7314. https://doi.org/10.3390/ijms22147314
Eswaran S, Babbar A, Drescher HK, Hitch TCA, Clavel T, Muschaweck M, Ritz T, Kroy DC, Trautwein C, Wagner N, et al. Upregulation of Anti-Oxidative Stress Response Improves Metabolic Changes in L-Selectin-Deficient Mice but Does Not Prevent NAFLD Progression or Fecal Microbiota Shifts. International Journal of Molecular Sciences. 2021; 22(14):7314. https://doi.org/10.3390/ijms22147314
Chicago/Turabian StyleEswaran, Sreepradha, Anshu Babbar, Hannah K. Drescher, Thomas C. A. Hitch, Thomas Clavel, Moritz Muschaweck, Thomas Ritz, Daniela C. Kroy, Christian Trautwein, Norbert Wagner, and et al. 2021. "Upregulation of Anti-Oxidative Stress Response Improves Metabolic Changes in L-Selectin-Deficient Mice but Does Not Prevent NAFLD Progression or Fecal Microbiota Shifts" International Journal of Molecular Sciences 22, no. 14: 7314. https://doi.org/10.3390/ijms22147314
APA StyleEswaran, S., Babbar, A., Drescher, H. K., Hitch, T. C. A., Clavel, T., Muschaweck, M., Ritz, T., Kroy, D. C., Trautwein, C., Wagner, N., & Schippers, A. (2021). Upregulation of Anti-Oxidative Stress Response Improves Metabolic Changes in L-Selectin-Deficient Mice but Does Not Prevent NAFLD Progression or Fecal Microbiota Shifts. International Journal of Molecular Sciences, 22(14), 7314. https://doi.org/10.3390/ijms22147314