
Small-Molecule Therapeutic Perspectives for the Treatment of Progeria



Abstract
:1. Introduction
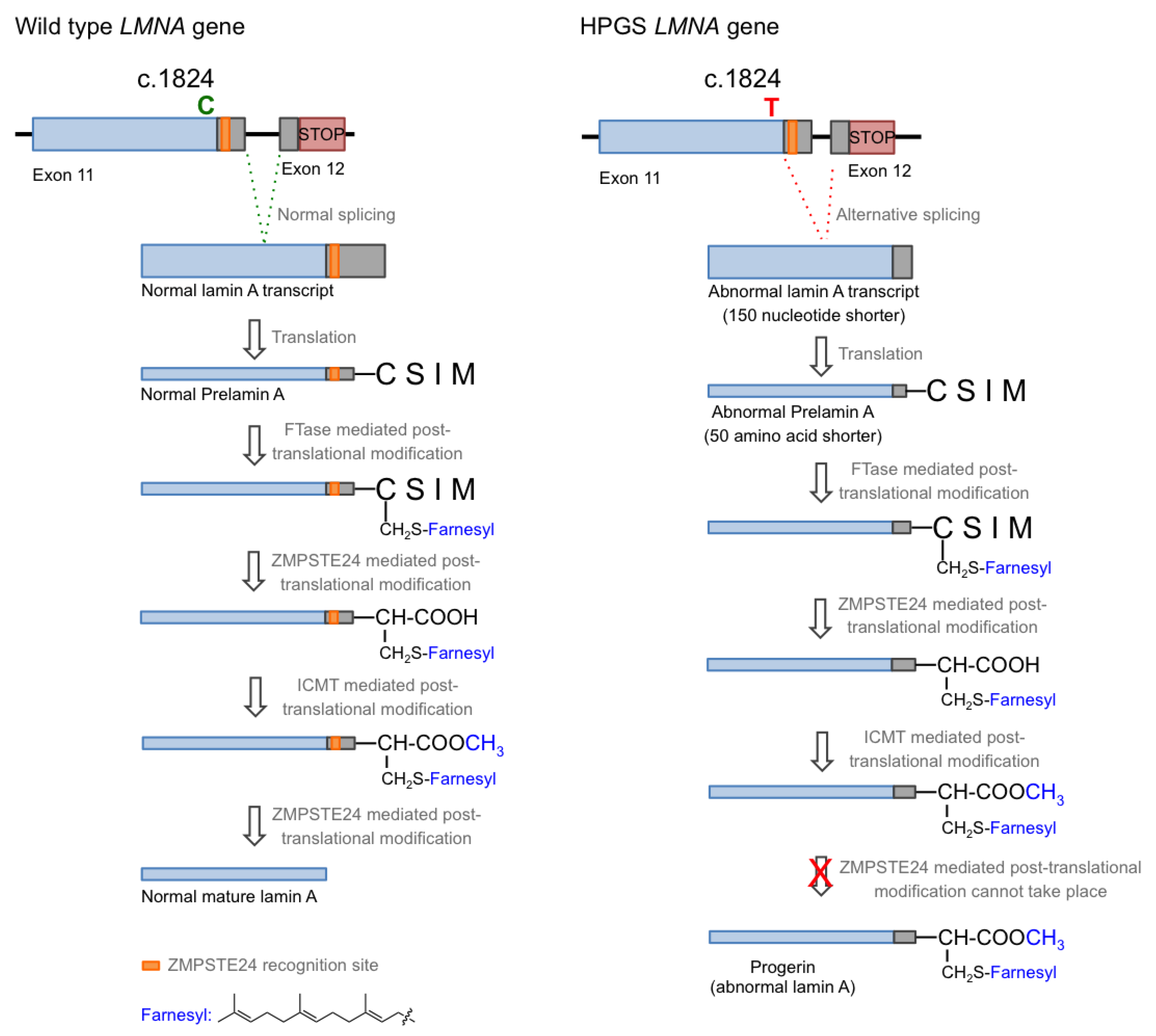
1.1. Hutchison–Gilford Progeria Syndrome and Its Molecular Causes
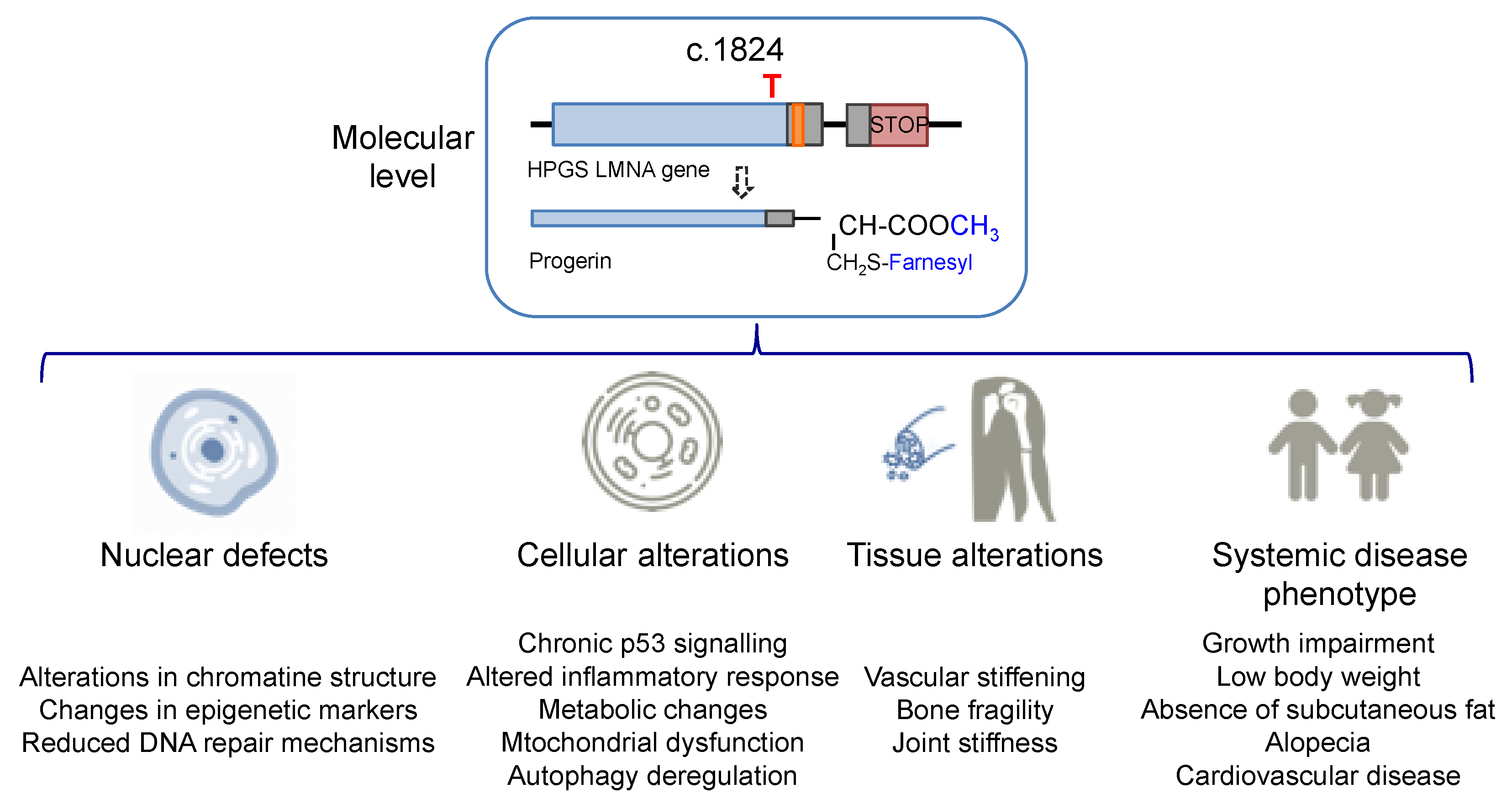
1.2. Progeria Phenotype: Nuclear, Cell, and Tissue Defects
1.3. Animal Models
2. Therapeutic Strategies for Treating Progeria
2.1. Gene Therapy Approaches
2.2. Biologicals
2.3. Small Molecules
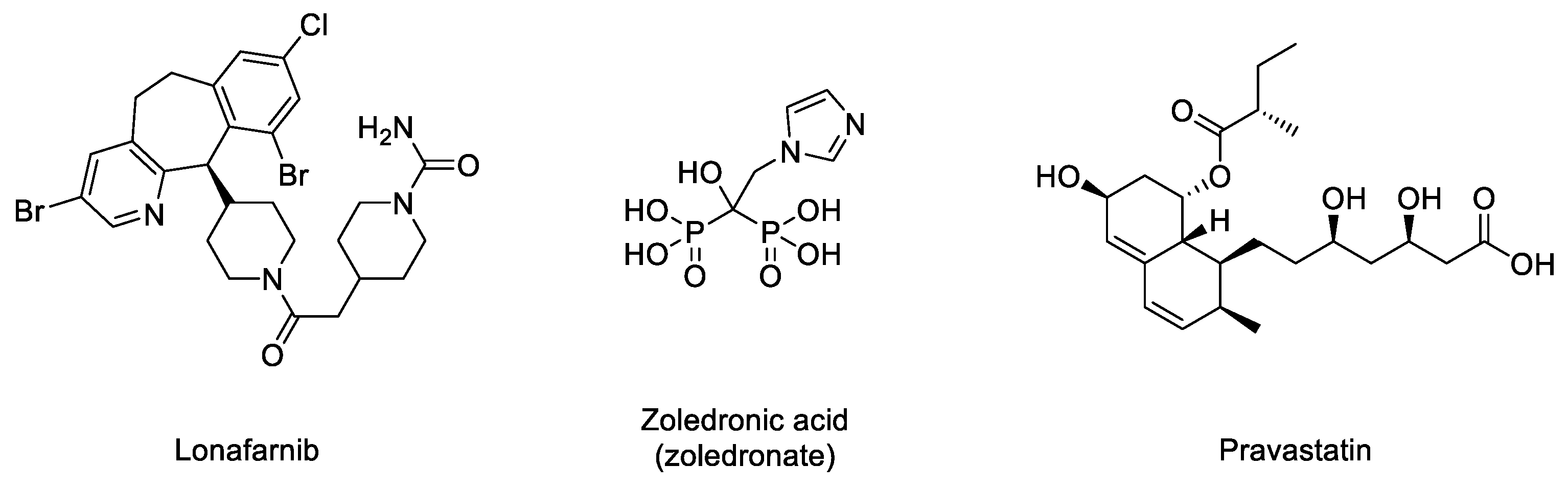
2.3.1. Inhibitors of the Prenylation Pathway
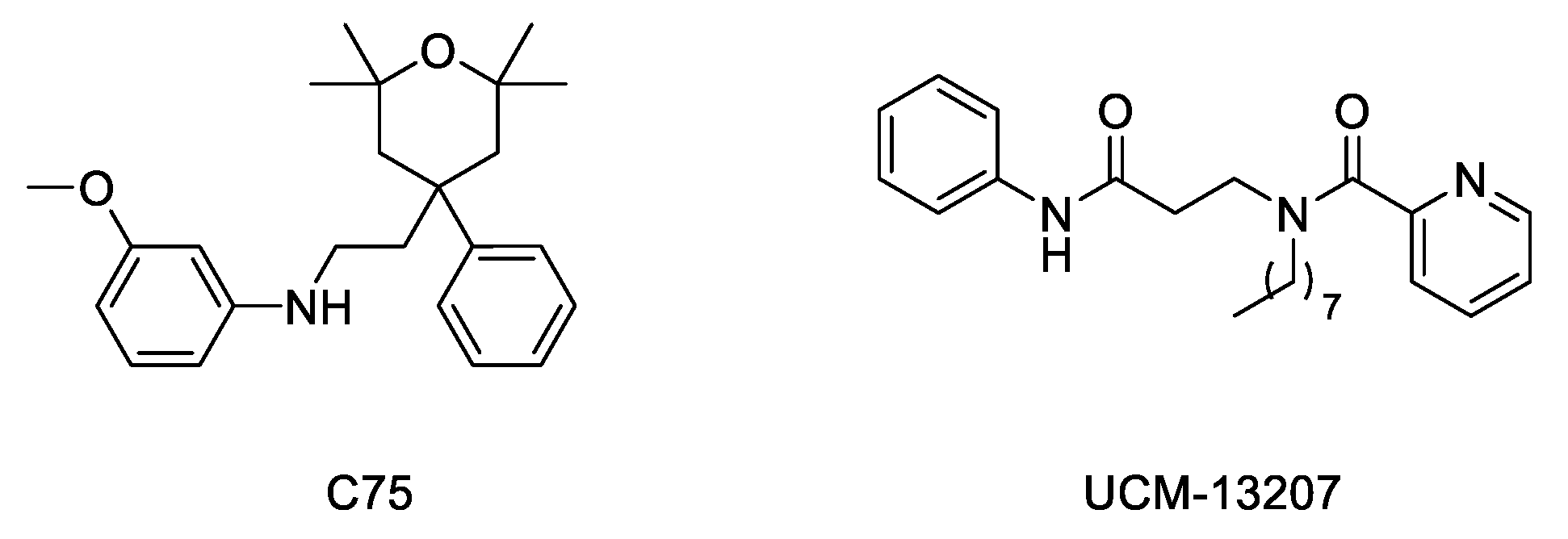
2.3.2. Methylation Inhibitors
2.3.3. Inhibitors of Progerin–Lamin A Interaction
2.3.4. Modulators of the Downstream Deleterious Effects Linked to Progerin Accumulation
3. Ongoing Clinical Trials
4. Future Perspectives
Funding
Conflicts of Interest
References
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin A truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Gordon, L.B.; Rothman, F.G.; Lopez-Otin, C.; Misteli, T. Progeria: A paradigm for translational medicine. Cell 2014, 156, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.G.; Fong, L.G.; Michaelis, S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria-new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 2005, 46, 2531–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, X.; Stewart, C.L. The laminopathies and the insights they provide into the structural and functional organization of the nucleus. Annu. Rev. Genomics Hum. Genet. 2020, 21, 263–288. [Google Scholar] [CrossRef]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Lee, L.; Kudlow, B.A.; Dos Santos, H.G.; Sletvold, O.; Shafeghati, Y.; Botha, E.G.; Garg, A.; Hanson, N.B.; Martin, G.M.; et al. LMNA mutations in atypical Werner’s syndrome. Lancet 2003, 362, 440–445. [Google Scholar] [CrossRef]
- Garg, A.; Subramanyam, L.; Agarwal, A.K.; Simha, V.; Levine, B.; D’Apice, M.R.; Novelli, G.; Crow, Y. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J. Clin. Endocrinol. Metab. 2009, 94, 4971–4983. [Google Scholar] [CrossRef] [PubMed]
- Mory, P.B.; Crispim, F.; Freire, M.B.; Salles, J.E.; Valério, C.M.; Godoy-Matos, A.F.; Dib, S.A.; Moisés, R.S. Phenotypic diversity in patients with lipodystrophy associated with LMNA mutations. Eur. J. Endocrinol. 2012, 167, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [Green Version]
- Mallampalli, M.P.; Huyer, G.; Bendale, P.; Gelb, M.H.; Michaelis, S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 14416–14421. [Google Scholar] [CrossRef] [Green Version]
- Dahl, K.N.; Scaffidi, P.; Islam, M.F.; Yodh, A.G.; Wilson, K.L.; Misteli, T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 10271–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.H.; Luu, J.; Heizer, P.; Tu, Y.; Weston, T.A.; Chen, N.; Lim, C.; Li, R.L.; Lin, P.Y.; Dunn, J.C.Y.; et al. Disrupting the LINC complex in smooth muscle cells reduces aortic disease in a mouse model of Hutchinson-Gilford progeria syndrome. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Dorado, B.; Andres, V. A-type lamins and cardiovascular disease in premature aging syndromes. Curr. Opin. Cell Biol. 2017, 46, 17–25. [Google Scholar] [CrossRef]
- Cao, K.; Blair, C.D.; Faddah, D.A.; Kieckhaefer, J.E.; Olive, M.; Erdos, M.R.; Nabel, E.G.; Collins, F.S. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J. Clin. Investig. 2011, 121, 2833–2844. [Google Scholar] [CrossRef] [Green Version]
- Benson, E.K.; Lee, S.W.; Aaronson, S.A. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J. Cell Sci. 2010, 123, 2605–2612. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Torres, J.; Acin-Perez, R.; Cabezas-Sanchez, P.; Osorio, F.G.; Gonzalez-Gomez, C.; Megias, D.; Camara, C.; Lopez-Otin, C.; Enriquez, J.A.; Luque-Garcia, J.L.; et al. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteomics 2013, 91, 466–477. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Del Campo, L.; Andres, V. Aging in the cardiovascular system: Lessons from Hutchinson-Gilford progeria syndrome. Annu. Rev. Physiol. 2018, 80, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef] [Green Version]
- Pendás, A.M.; Zhou, Z.; Cadiñanos, J.; Freije, J.M.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodríguez, F.; Tryggvason, K.; et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Torres, J.; Calvo, C.J.; Llach, A.; Guzman-Martinez, G.; Caballero, R.; Gonzalez-Gomez, C.; Jimenez-Borreguero, L.J.; Guadix, J.A.; Osorio, F.G.; Lopez-Otin, C.; et al. Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations. Proc. Natl. Acad. Sci. USA 2016, 113, E7250–E7259. [Google Scholar] [CrossRef] [Green Version]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef] [PubMed]
- Cubria, M.B.; Suarez, S.; Masoudi, A.; Oftadeh, R.; Kamalapathy, P.; DuBose, A.; Erdos, M.R.; Cabral, W.A.; Karim, L.; Collins, F.S.; et al. Evaluation of musculoskeletal phenotype of the G608G progeria mouse model with lonafarnib, pravastatin, and zoledronic acid as treatment groups. Proc. Natl. Acad. Sci. USA 2020, 117, 12029–12040. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andres-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; Lopez-Otin, C.; Andres, V. Vascular smooth muscle-specific progerin expression accelerates atherosclerosis and death in a mouse model of Hutchinson-Gilford progeria syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorado, B.; Pløen, G.G.; Barettino, A.; Macías, A.; Gonzalo, P.; Andrés-Manzano, M.J.; González-Gómez, C.; Galán-Arriola, C.; Alfonso, J.M.; Lobo, M.; et al. Generation and characterization of a novel knockin minipig model of Hutchinson-Gilford progeria syndrome. Cell Discov. 2019, 5, 16. [Google Scholar] [CrossRef]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Levy, N. An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.F.; Wong, W.T. Progress and trends in the development of therapies for Hutchinson-Gilford progeria syndrome. Aging Cell 2020, 19, e13175. [Google Scholar] [CrossRef]
- Piekarowicz, K.; Machowska, M.; Dzianisava, V.; Rzepecki, R. Hutchinson-Gilford progeria syndrome-current status and prospects for gene therapy treatment. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Fernandez, O.; Osorio, F.G.; Quesada, V.; Rodriguez, F.; Basso, S.; Maeso, D.; Rolas, L.; Barkaway, A.; Nourshargh, S.; Folgueras, A.R.; et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. [Google Scholar] [CrossRef]
- Beyret, E.; Liao, H.K.; Yamamoto, M.; Hernandez-Benitez, R.; Fu, Y.; Erikson, G.; Reddy, P.; Izpisua-Belmonte, J.C. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 419–422. [Google Scholar] [CrossRef]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense oligonucleotides: An emerging area in drug discovery and development. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.H. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Puttaraju, M.; Jackson, M.; Klein, S.; Shilo, A.; Bennett, C.F.; Gordon, L.; Rigo, F.; Misteli, T. Systematic screening identifies therapeutic antisense oligonucleotides for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021, 27, 526–535. [Google Scholar] [CrossRef]
- Erdos, M.R.; Cabral, W.A.; Tavarez, U.L.; Cao, K.; Gvozdenovic-Jeremic, J.; Narisu, N.; Zerfas, P.M.; Crumley, S.; Boku, Y.; Hanson, G.; et al. A targeted antisense therapeutic approach for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021, 27, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Bergo, M.O.; Toth, J.I.; Qiao, X.; Hu, Y.; Sandoval, S.; Meta, M.; Bendale, P.; Gelb, M.H.; Young, S.G.; et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 10291–10296. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.G.; Frost, D.; Meta, M.; Qiao, X.; Yang, S.H.; Coffinier, C.; Young, S.G. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006, 311, 1621–1623. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Yang, S.H.; Davies, B.S.; Jung, H.J.; Fong, L.G. Targeting protein prenylation in progeria. Sci. Transl. Med. 2013, 5, 171ps173. [Google Scholar] [CrossRef] [Green Version]
- Marín-Ramos, N.I.; Piñar, C.; Vázquez-Villa, H.; Martín-Fontecha, M.; González, Á.; Canales, Á.; Algar, S.; Mayo, P.P.; Jiménez-Barbero, J.; Gajate, C.; et al. Development of a nucleotide exchange inhibitor that impairs Ras oncogenic signaling. Chem. Eur. J. 2017, 23, 1676–1685. [Google Scholar] [CrossRef]
- Marin-Ramos, N.I.; Ortega-Gutierrez, S.; Lopez-Rodriguez, M.L. Blocking Ras inhibition as an antitumor strategy. Semin. Cancer Biol. 2019, 54, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B., Sr.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S. Lonafarnib: First Approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef]
- Varela, I.; Pereira, S.; Ugalde, A.P.; Navarro, C.L.; Suarez, M.F.; Cau, P.; Cadinanos, J.; Osorio, F.G.; Foray, N.; Cobo, J.; et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008, 14, 767–772. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B., Sr.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, V.L.; Peckham, L.A.; Olive, M.; Capell, B.C.; Collins, F.S.; Nabel, E.G.; Young, S.G.; Fong, L.G.; Lammerding, J. Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc. Natl. Acad. Sci. USA 2011, 108, 4997–5002. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.M.; Yoon, M.H.; Ahn, J.; Kim, J.E.; Kim, S.Y.; Kang, S.Y.; Joo, J.; Park, S.; Cho, J.H.; Woo, T.G.; et al. Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Commun. Biol. 2021, 4. [Google Scholar] [CrossRef]
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational modifications of Ras proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a031484. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Kamen, B.A.; Bergo, M.O.; Young, S.G.; Melnyk, S.; James, S.J.; Casey, P.J. Targeting Ras signaling through inhibition of carboxyl methylation: An unexpected property of methotrexate. Proc. Natl. Acad. Sci. USA 2003, 100, 6529–6534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter-Vann, A.M.; Baron, R.A.; Wong, W.; dela Cruz, J.; York, J.D.; Gooden, D.M.; Bergo, M.O.; Young, S.G.; Toone, E.J.; Casey, P.J. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4336–4341. [Google Scholar] [CrossRef] [Green Version]
- Judd, W.R.; Slattum, P.M.; Hoang, K.C.; Bhoite, L.; Valppu, L.; Alberts, G.; Brown, B.; Roth, B.; Ostanin, K.; Huang, L.; et al. Discovery and SAR of methylated tetrahydropyranyl derivatives as inhibitors of isoprenylcysteine carboxyl methyltransferase (ICMT). J. Med. Chem. 2011, 54, 5031–5047. [Google Scholar] [CrossRef]
- Marin-Ramos, N.I.; Balabasquer, M.; Ortega-Nogales, F.J.; Torrecillas, I.R.; Gil-Ordonez, A.; Marcos-Ramiro, B.; Aguilar-Garrido, P.; Cushman, I.; Romero, A.; Medrano, F.J.; et al. A potent isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor improves survival in Ras-driven acute myeloid leukemia. J. Med. Chem. 2019, 62, 6035–6046. [Google Scholar] [CrossRef]
- López-Rodríguez, M.L.; Ortega-Gutiérrez, S.; Martín-Fontecha, M.; Balabasquer, M.; Ortega-Nogales, F.J.; Marín-Ramos, N.I. Novel Inhibitors of the Enzyme Isoprenylcysteine Carboxyl Methyltransferase (ICMT). European Patent PCT No. 2014118418A1, 7 August 2014. [Google Scholar]
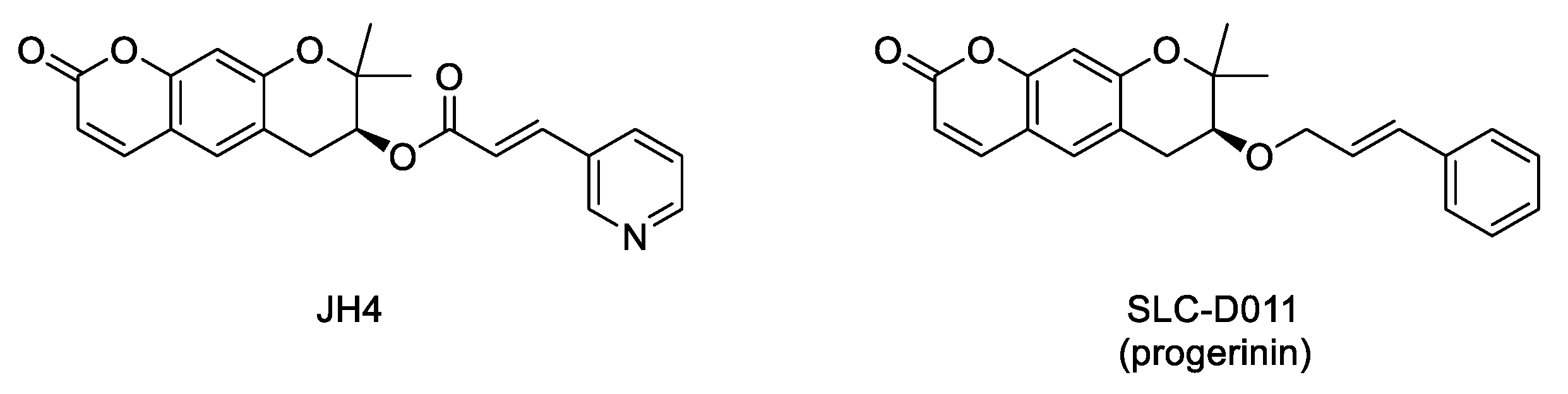
- Marcos-Ramiro, B.; Gil-Ordóñez, A.; Marin-Ramos, N.I.; Ortega-Nogales, F.J.; Balabasquer, M.; Gonzalo, P.; Khiar-Fernández, N.; Rolas, L.; Barkaway, A.; Nourshargh, S.; et al. Isoprenylcysteine carboxylmethyltransferase-based therapy for Hutchinson–Gilford progeria syndrome. ACS Cent. Sci. 2021. [Google Scholar] [CrossRef]
- Chen, X.; Yao, H.; Kashif, M.; Revêchon, G.; Eriksson, M.; Hu, J.; Wang, T.; Liu, Y.; Tüksammel, E.; Strömblad, S.; et al. A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. Elife 2021, 10. [Google Scholar] [CrossRef]
- Lee, S.J.; Jung, Y.S.; Yoon, M.H.; Kang, S.M.; Oh, A.Y.; Lee, J.H.; Jun, S.Y.; Woo, T.G.; Chun, H.Y.; Kim, S.K.; et al. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J. Clin. Investig. 2016, 126, 3879–3893. [Google Scholar] [CrossRef] [Green Version]
- Park, B.J.; Song, G.Y.; Yu, S.O.; Lee, J.H.; Yun, E.J. Pharmaceutical Composition for Preventing or Treating Aging-Related Diseases Containing Decursin Derivative as Active Ingredient. U.S. Patent US11008332B2, 18 May 2021. [Google Scholar]
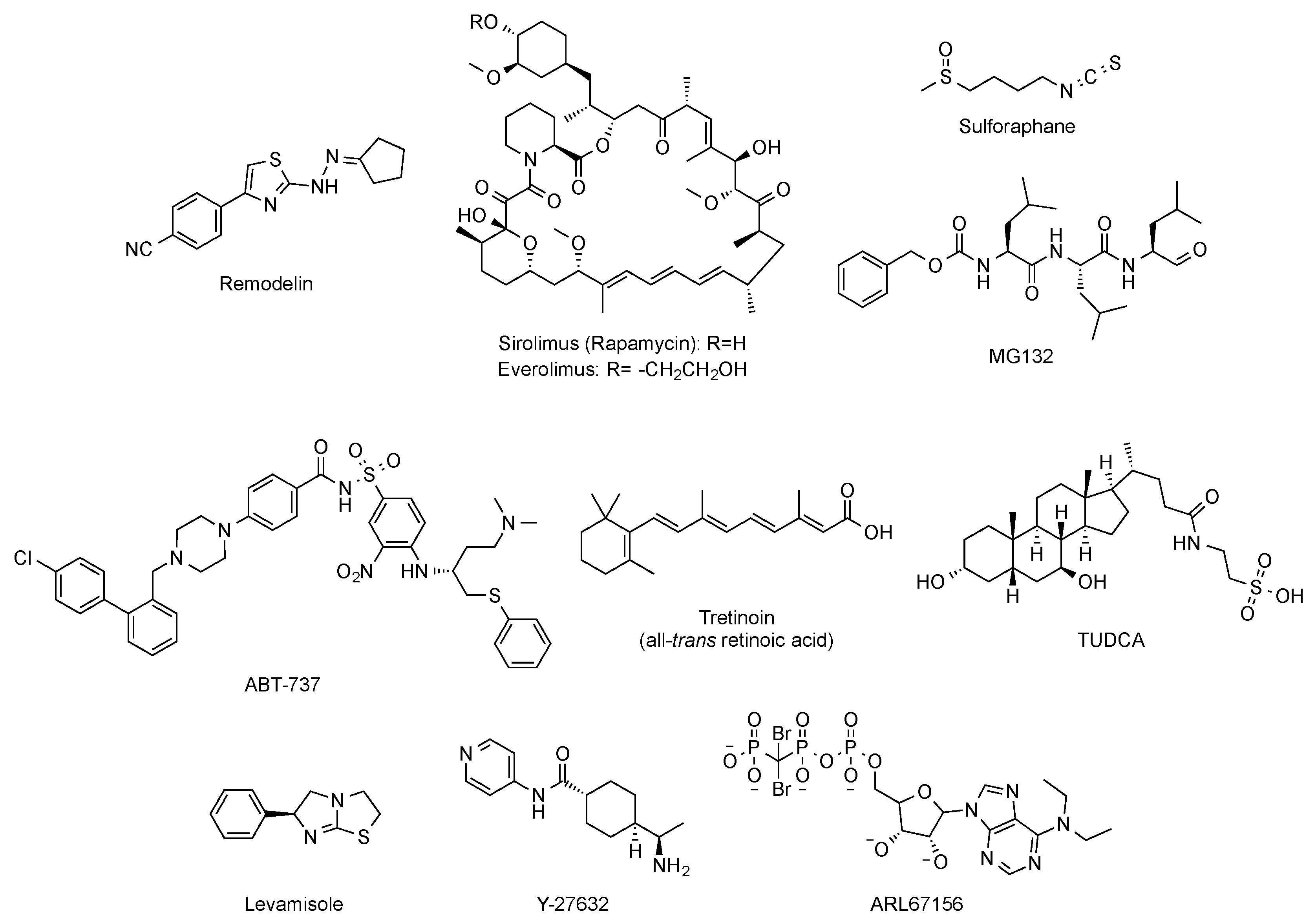
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 2014, 344, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Balmus, G.; Larrieu, D.; Barros, A.C.; Collins, C.; Abrudan, M.; Demir, M.; Geisler, N.J.; Lelliott, C.J.; White, J.K.; Karp, N.A.; et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 2018, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Evangelisti, C.; Cenni, V.; Fazio, A.; Paganelli, F.; Martelli, A.M.; Lattanzi, G. The cutting edge: The role of mTOR signaling in laminopathies. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef]
- Gabriel, D.; Shafry, D.D.; Gordon, L.B.; Djabali, K. Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in Hutchinson-Gilford progeria fibroblasts. Oncotarget 2017, 8, 64809–64826. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.G.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Lévy, N. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol. Med. 2017, 9, 1294–1313. [Google Scholar] [CrossRef]
- Richards, S.A.; Muter, J.; Ritchie, P.; Lattanzi, G.; Hutchison, C.J. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum. Mol. Genet. 2011, 20, 3997–4004. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.T.; Park, J.T.; Choi, K.; Choi, H.J.C.; Jung, C.W.; Kim, G.R.; Lee, Y.S.; Park, S.C. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell 2017, 16, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.M.; Choi, J.Y.; Wang, K.; Zhang, H.; Tariq, Z.; Wu, D.; Ko, E.; LaDana, C.; Sesaki, H.; Cao, K. Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell 2016, 15, 279–290. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [Green Version]
- Kubben, N.; Brimacombe, K.R.; Donegan, M.; Li, Z.; Misteli, T. A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods 2016, 96, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Columbaro, M.; Capanni, C.; D’Apice, M.R.; Cavallo, C.; Murdocca, M.; Lattanzi, G.; Squarzoni, S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget 2015, 6, 29914–29928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finley, J. Cellular stress and AMPK activation as a common mechanism of action linking the effects of metformin and diverse compounds that alleviate accelerated aging defects in Hutchinson-Gilford progeria syndrome. Med. Hypotheses 2018, 118, 151–162. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Andrés-Manzano, M.J.; Misteli, T.; López-Otín, C.; Andrés, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11, e9736. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R. ATP-based therapy prevents vascular calcification and extends longevity in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 23698–23704. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R. Dietary magnesium supplementation improves lifespan in a mouse model of progeria. EMBO Mol. Med. 2020, 12, e12423. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Main Phenotypic Features | Main Limitations | Ref. |
|---|---|---|---|
| Zmpste24−/− mice | Bone fragility, reduced weight and growth, defective prelamin A processing, early death, muscular weakness, age-dependent cardiac electrical defects | No severe vascular alterations | [19,20,21] |
| Heterozygous LMNAp.G608G/+ mice | Ubiquitous progerin accumulation, vascular abnormalities | Lack rest of features of progeria phenotype | [22] |
| LmnaG609G/G609G mice | Ubiquitous progerin accumulation, shortened lifespan, reduced weight, main metabolic, bone, and cardiovascular alterations | Mice do not develop atherosclerosis | [23] |
| Apoe−/− LmnaG609G/G609G mice | Same phenotype as LmnaG609G/G609G but including the development of atherosclerosis | - | [25] |
| Apoe−/−LmnaLCS/LCS SM22αCre mice | Progerin expression restricted to VSMCs. Mice recapitulate vascular features of progeria | Lack of overt growth defects and other disease symptoms compared to the phenotype observed in LmnaG609G/G609G mice | [25] |
| Apoe−/−LmnaLCS/LCS LysMCre mice | Progerin expression restricted to macrophages | Lack of overt growth defects and other disease symptoms compared to the phenotype observed in LmnaG609G/G609G mice and in Apoe−/−LmnaLCS/LCS SM22αCre | [25] |
| Prog-Tg mice | Progerin expression restricted to endothelium. Reduced growth, weight, and lifespan. Mice recapitulate many cardiovascular alterations such as profibrotic response and cardiac functional impairment | Lack of VSMC loss | [26] |
| Knockin heterozygous LMNA c.1824C > T Yucatan minipig | Expression of progerin and normal lamin A/C, growth retardation, lipodystrophy, skin and bone alterations, cardiovascular alterations, cardiovascular disease, and mortality around puberty | Difficulty to establish an HGPS minipig colony through conventional breeding | [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macicior, J.; Marcos-Ramiro, B.; Ortega-Gutiérrez, S. Small-Molecule Therapeutic Perspectives for the Treatment of Progeria. Int. J. Mol. Sci. 2021, 22, 7190. https://doi.org/10.3390/ijms22137190
Macicior J, Marcos-Ramiro B, Ortega-Gutiérrez S. Small-Molecule Therapeutic Perspectives for the Treatment of Progeria. International Journal of Molecular Sciences. 2021; 22(13):7190. https://doi.org/10.3390/ijms22137190
Chicago/Turabian StyleMacicior, Jon, Beatriz Marcos-Ramiro, and Silvia Ortega-Gutiérrez. 2021. "Small-Molecule Therapeutic Perspectives for the Treatment of Progeria" International Journal of Molecular Sciences 22, no. 13: 7190. https://doi.org/10.3390/ijms22137190
APA StyleMacicior, J., Marcos-Ramiro, B., & Ortega-Gutiérrez, S. (2021). Small-Molecule Therapeutic Perspectives for the Treatment of Progeria. International Journal of Molecular Sciences, 22(13), 7190. https://doi.org/10.3390/ijms22137190