
Endophytic Microbiome Responses to Sulfur Availability in Beta vulgaris (L.)



 , ,
, , 
 and
and 
Abstract
:1. Introduction
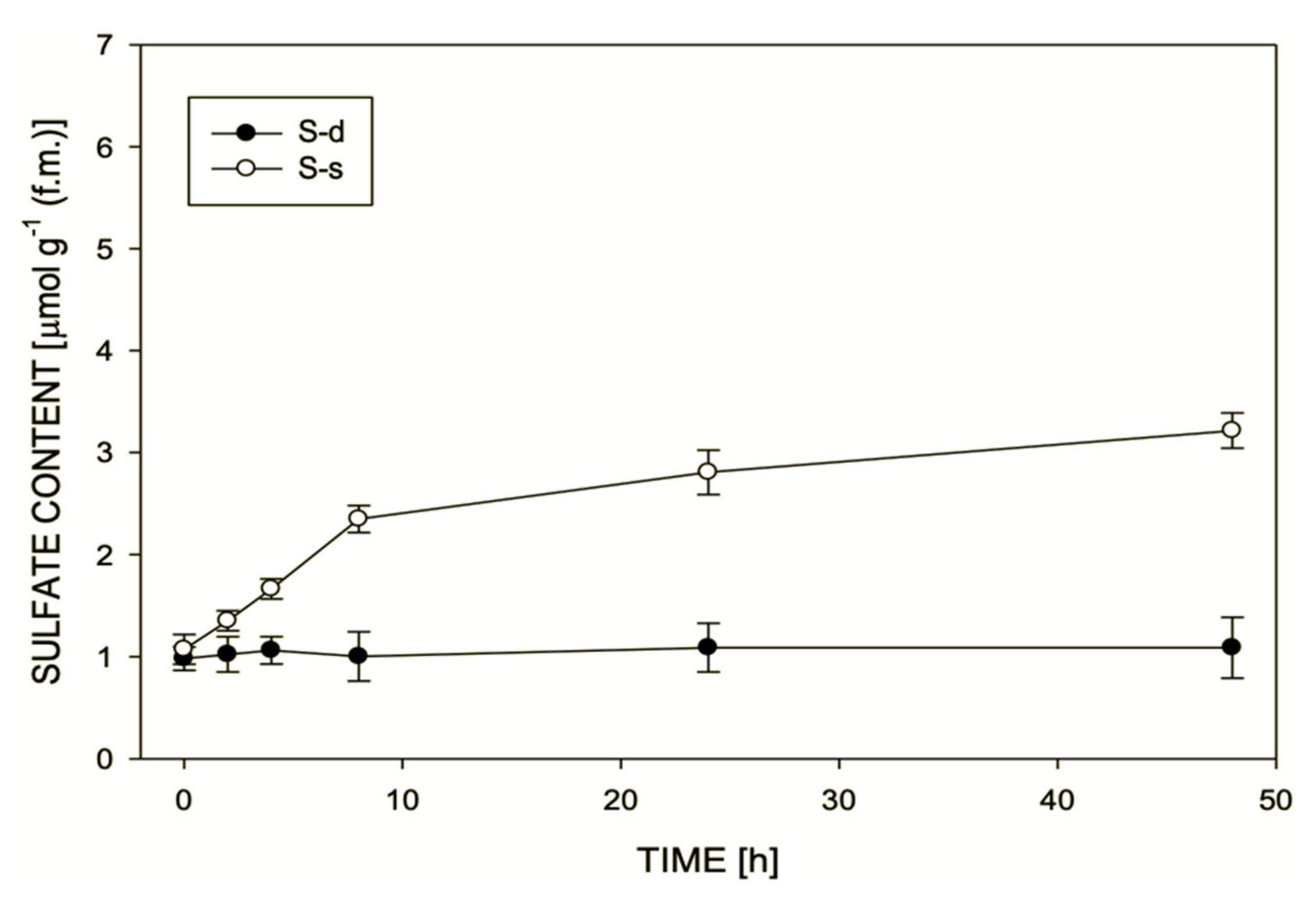
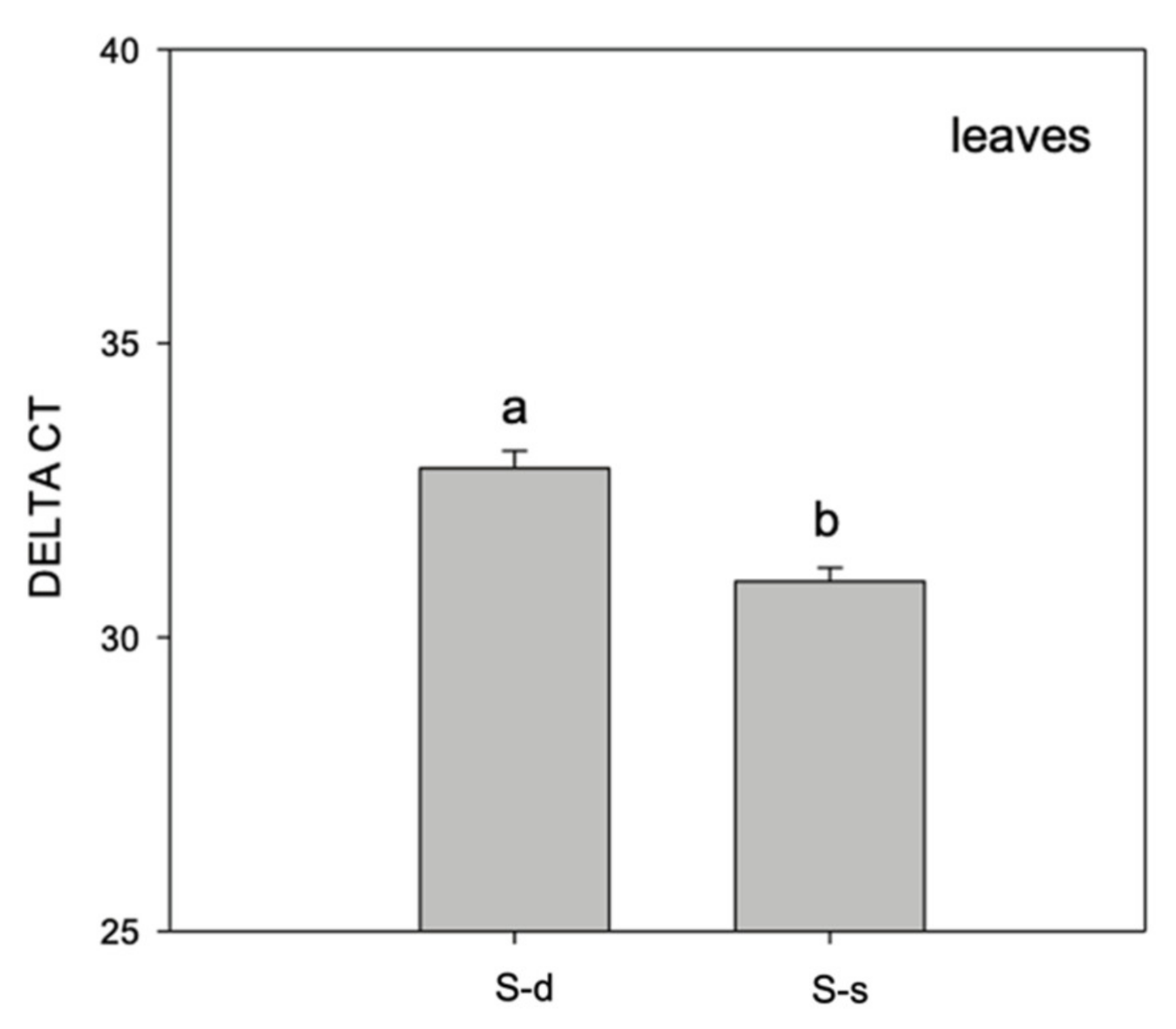
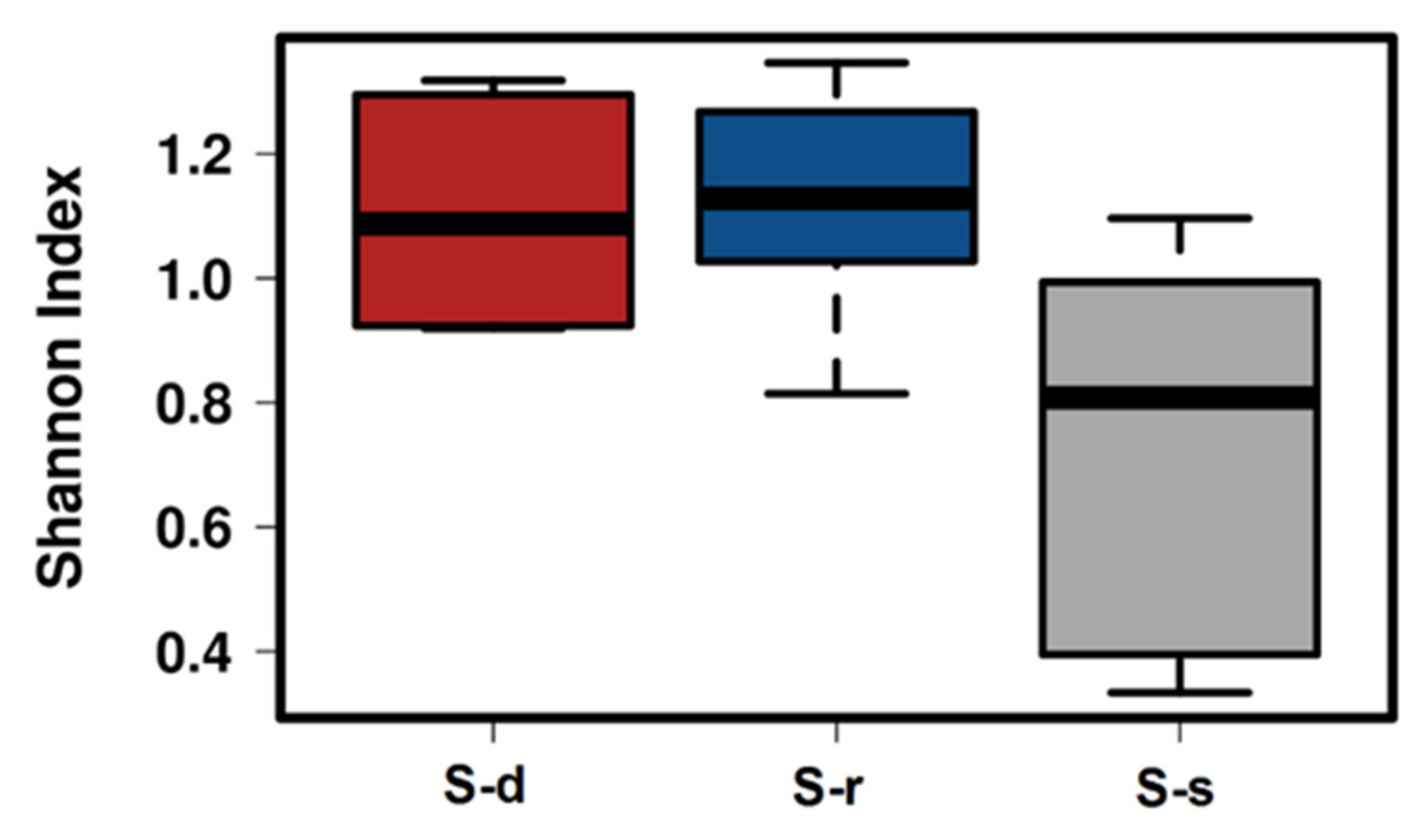
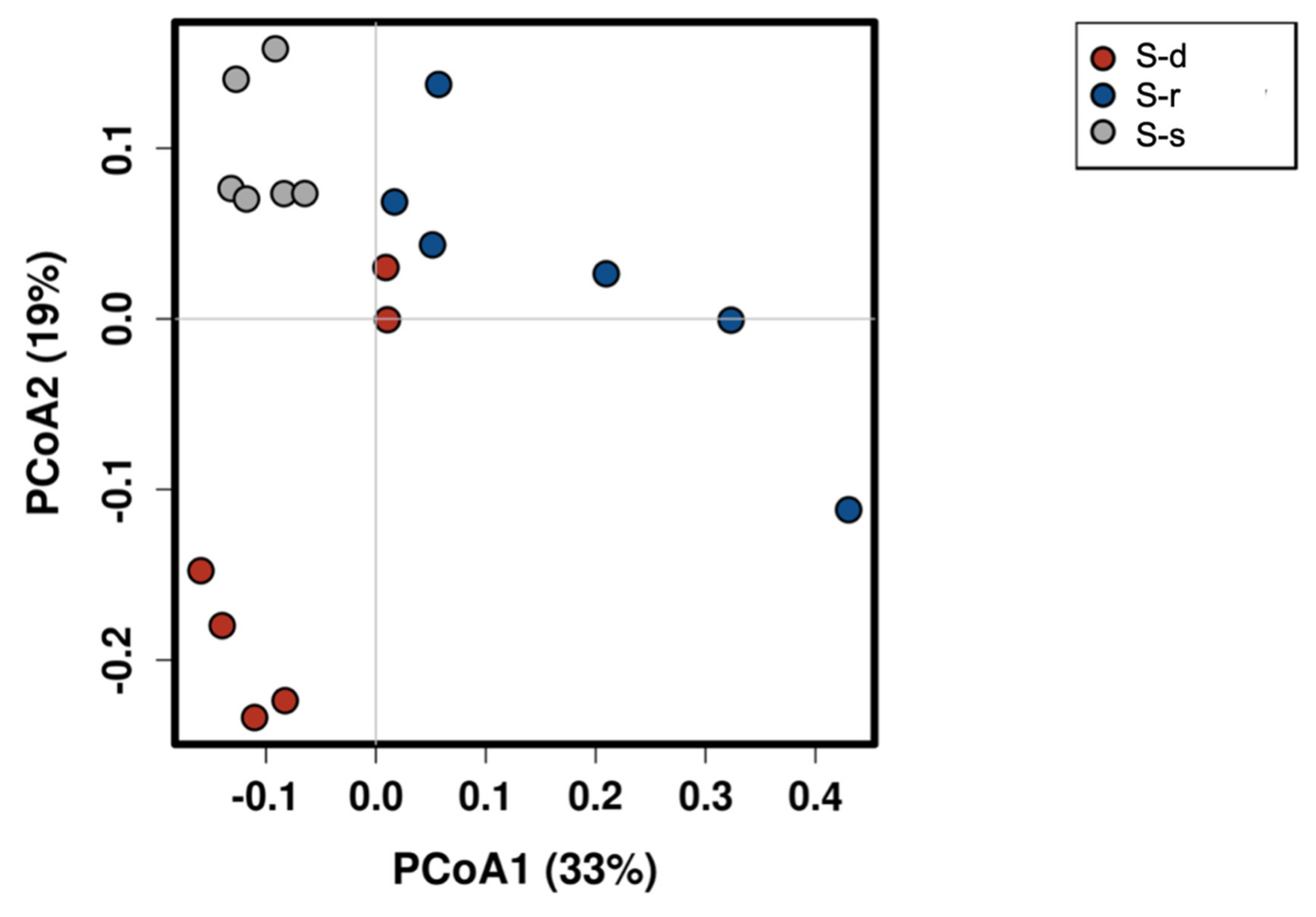
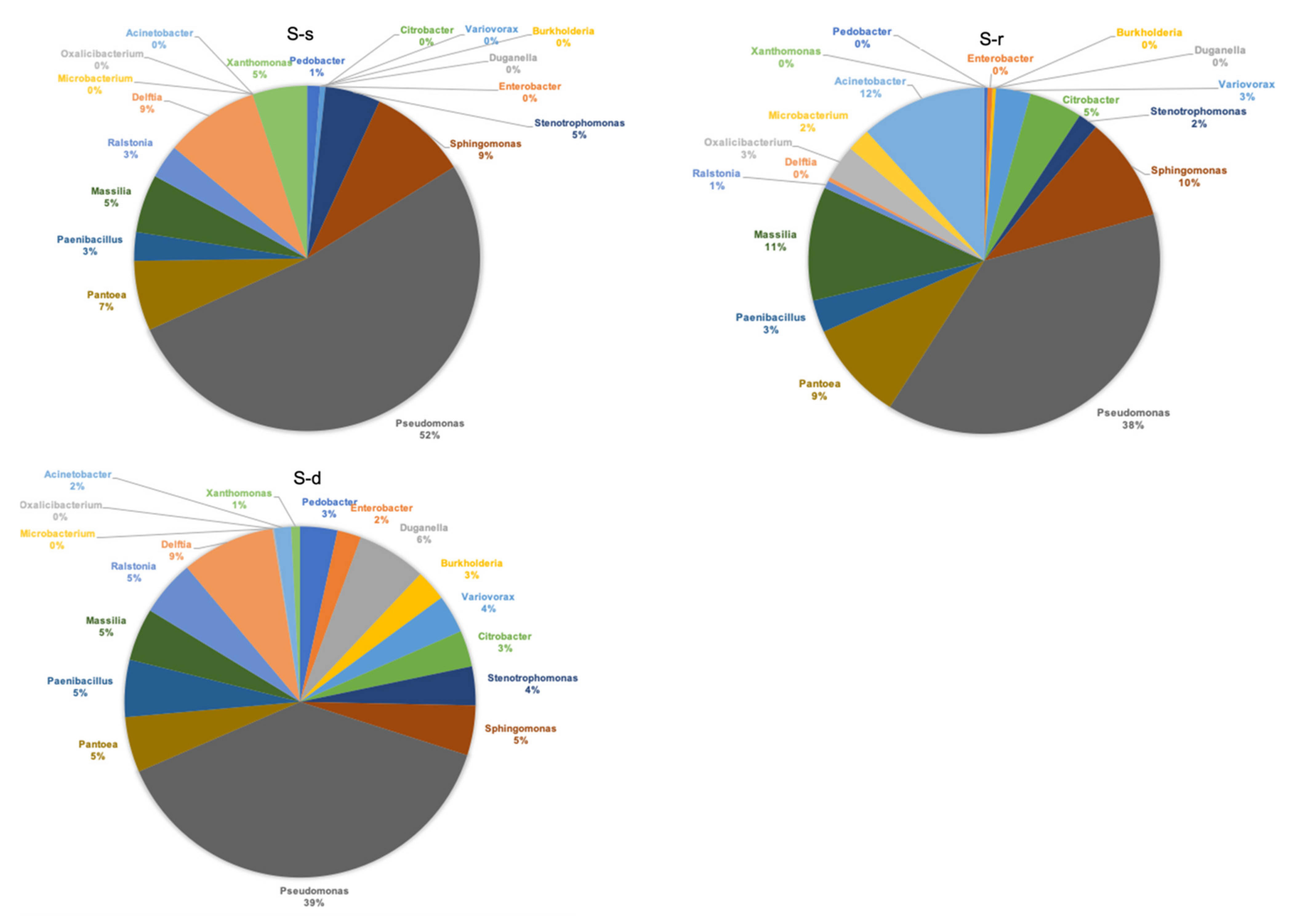
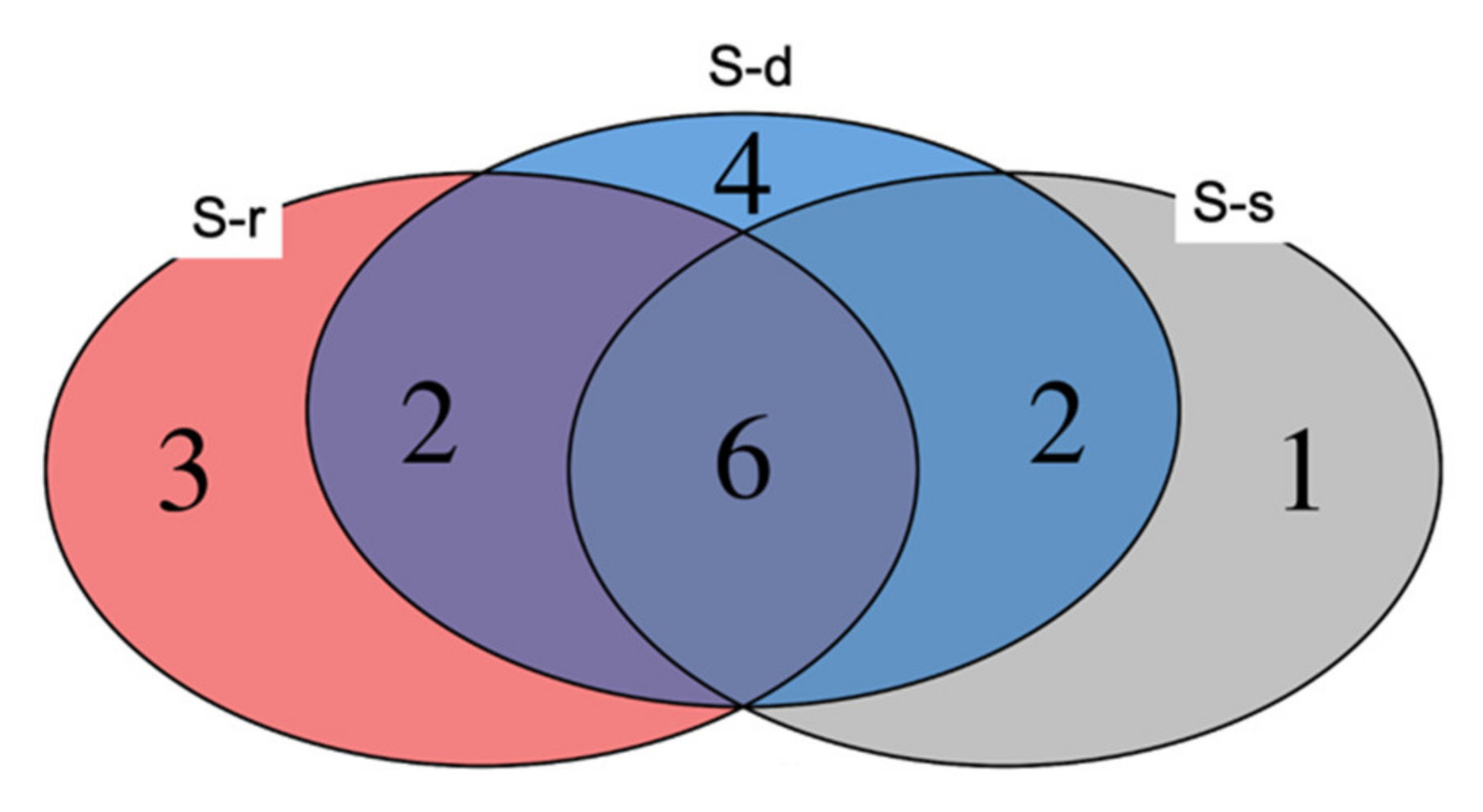
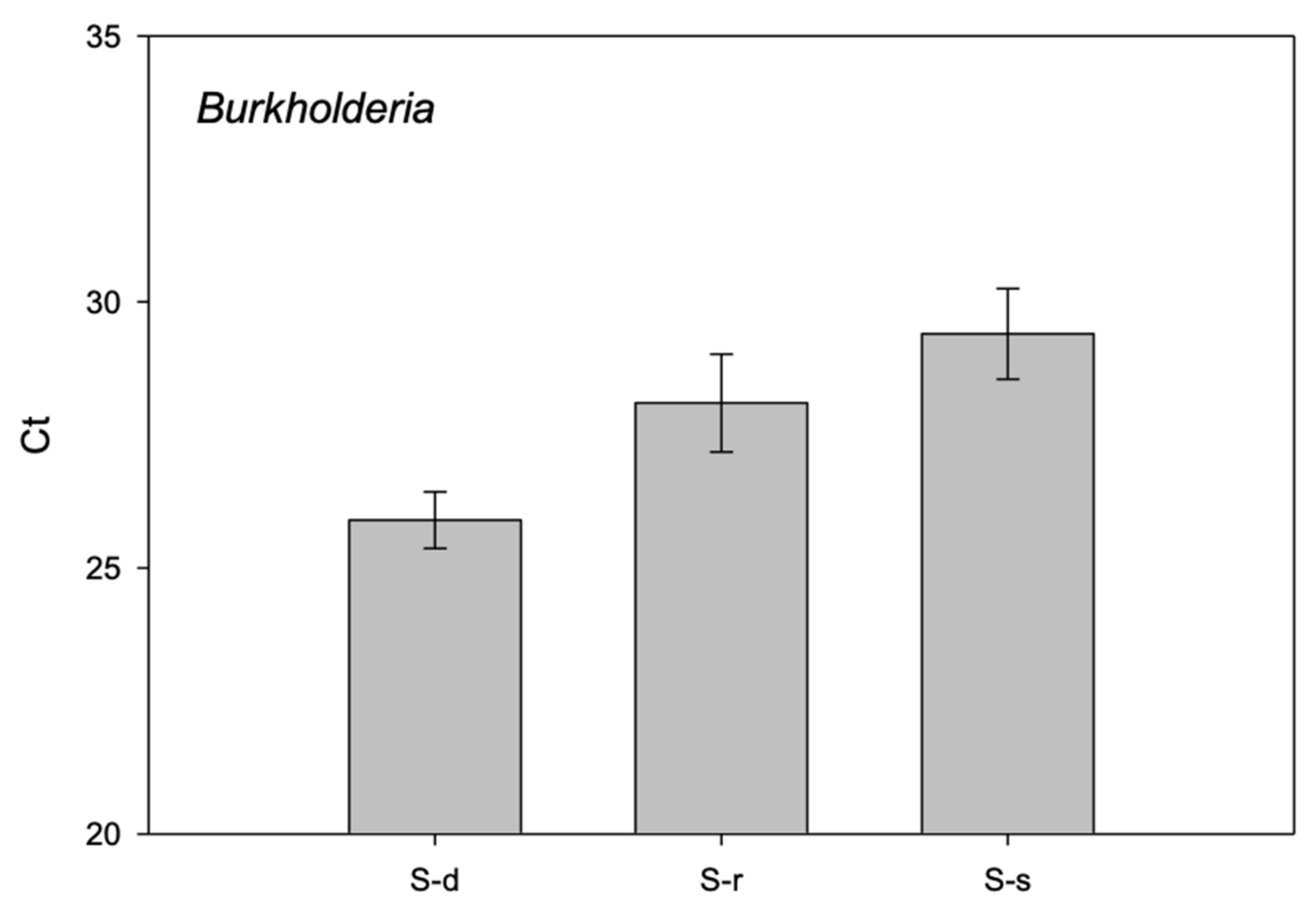
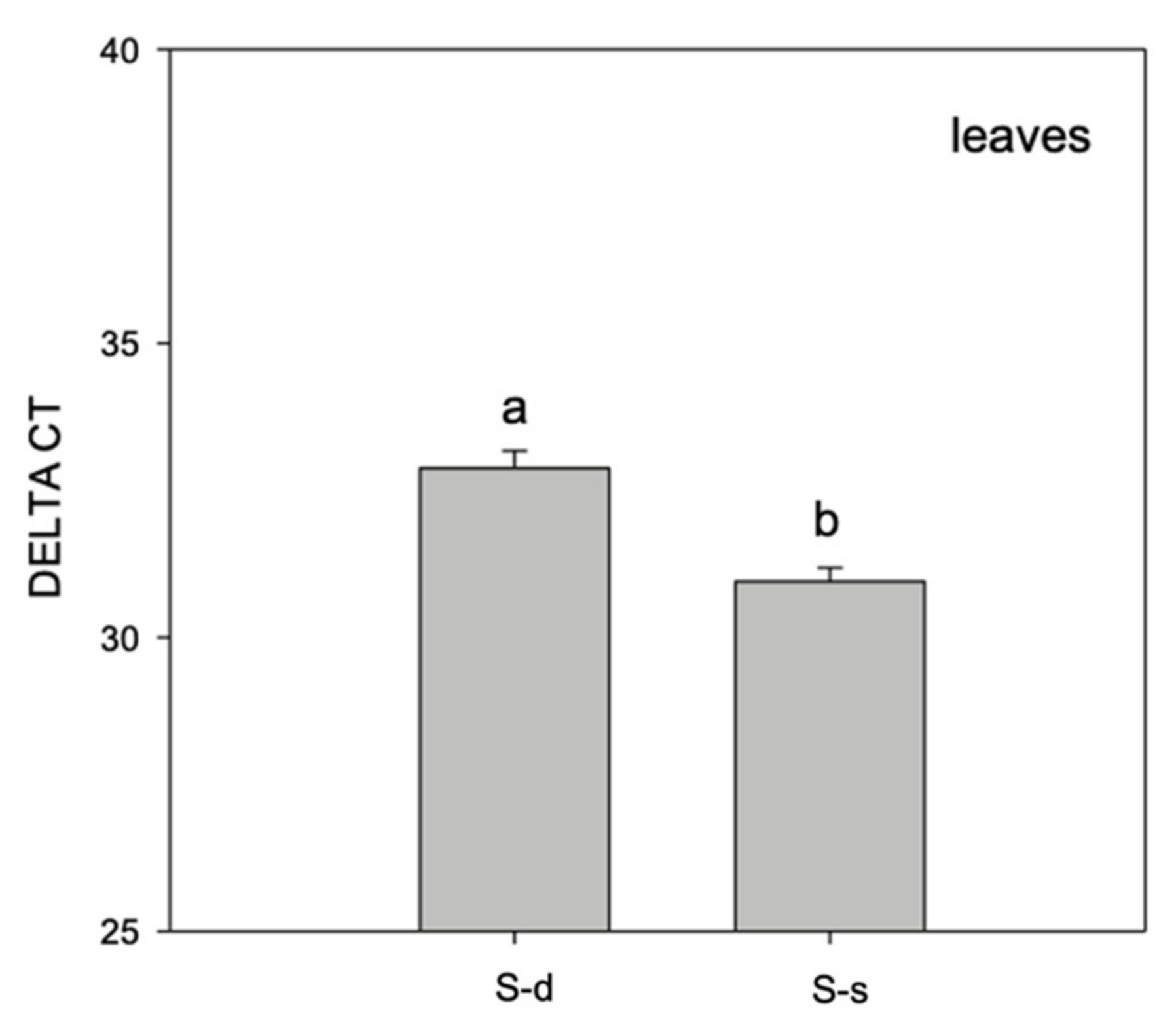
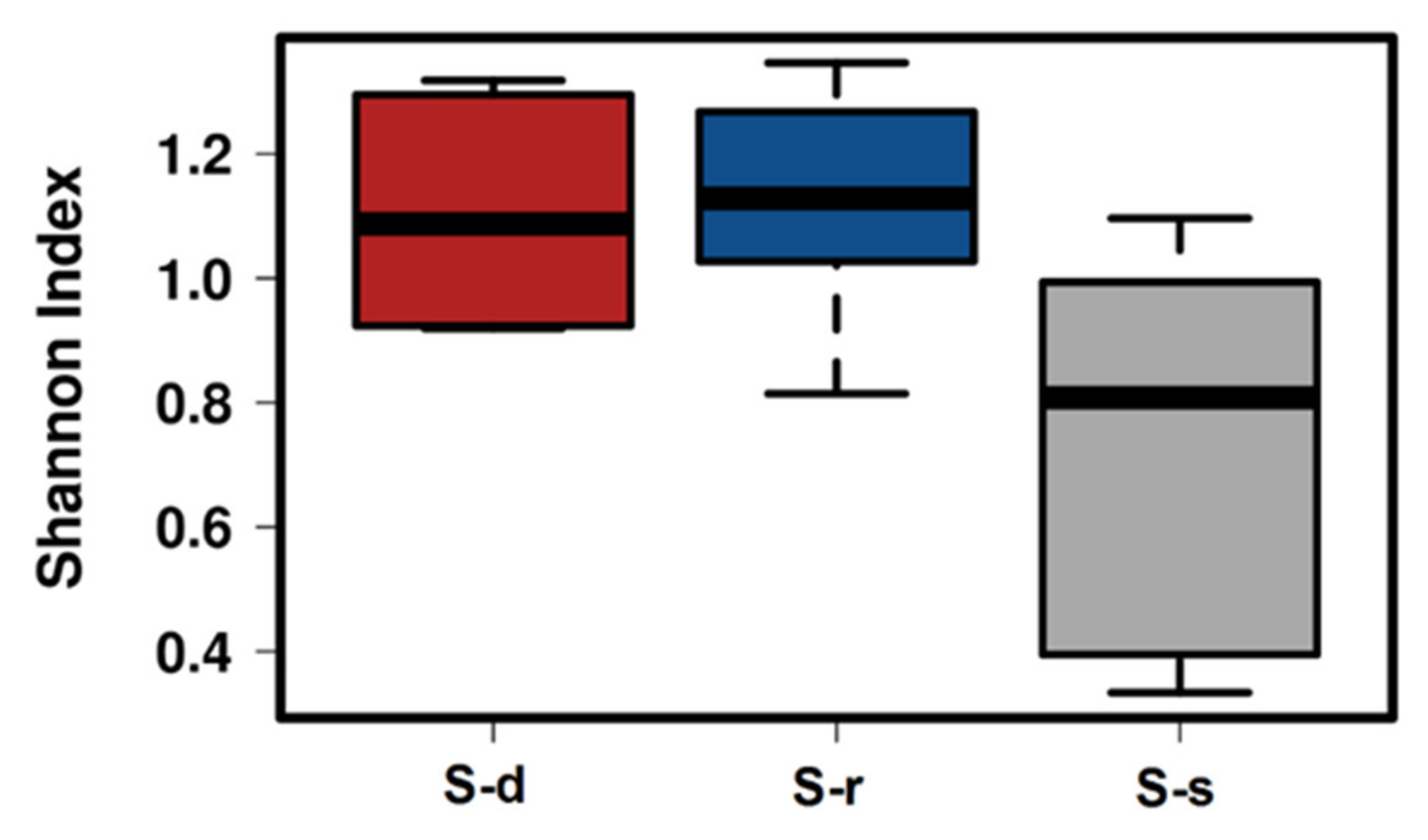
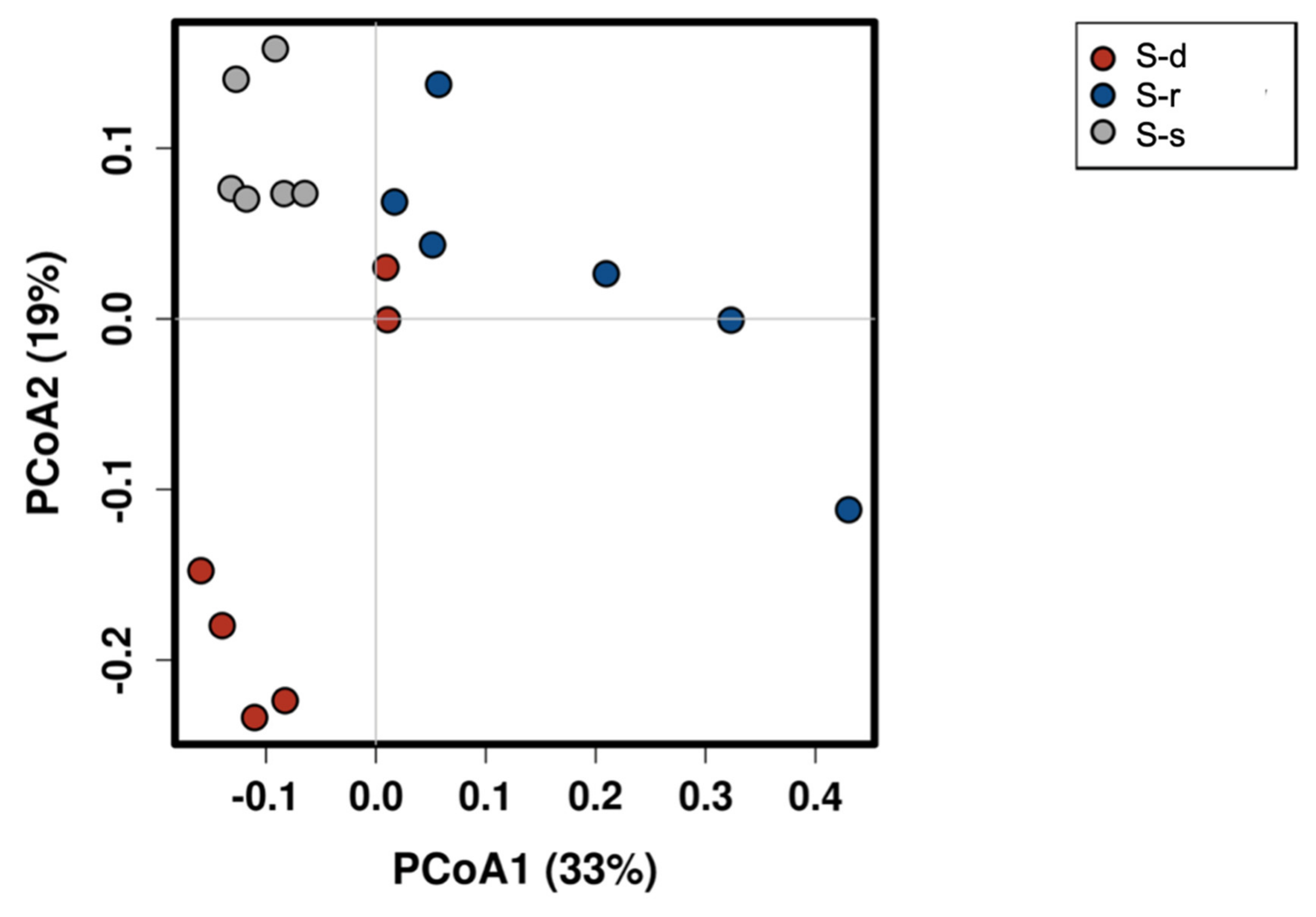
2. Results
3. Discussion
4. Material and Methods
4.1. Plant Material
4.2. Growing Conditions
4.3. Ionomic Analysis
4.4. RNA Extraction
4.5. Real-Time Quantitative RT–PCR
4.6. DNA Extraction
4.7. Metabarcoding of Bacterial 16S rRNA Gene Sequencing and Data Analysis
4.8. Primer Design and Real-Time PCR
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burba, M. Der schädliche stickstoff als kriterium der rübenqualität. Zuckerind 1996, 121, 165–173. [Google Scholar]
- Hoffmann, C.; Stockfisch, N.; Koch, H.-J. Influence of sulfur supply on yield and quality of sugar beet (Beta vulgaris L.)—determination of a threshold value. Eur. J. Agron. 2004, 21, 69–80. [Google Scholar] [CrossRef]
- Thomas, S.G.; Bilsborrow, P.E.; Hocking, T.J.; Bennett, J. Effect of sulphur deficiency on the growth and metabolism of sugar beet (Beta vulgaris cv druid). J. Sci. Food Agric. 2000, 80, 2057–2062. [Google Scholar] [CrossRef]
- Bloem, E.; Haneklaus, S.; Schnug, E. Significance of sulfur compounds in the protection of plants against pests and diseases. J. Plant Nutr. 2005, 28, 763–784. [Google Scholar] [CrossRef]
- Hernández, J.A.; Barba-Espín, G.; Diaz-Vivancos, P. Glutathione-mediated biotic stress tolerance in plants. In Glutathione in Plant Growth, Development, and Stress Tolerance; Springer: Cham, Switzerland, 2017; pp. 309–329. [Google Scholar]
- Brader, G.; Compant, S.; Vescio, K.; Mitter, B.; Trognitz, F.; Ma, L.-J.; Sessitsch, A. Ecology and genomic insights into plant-pathogenic and plant-nonpathogenic endophytes. Annu. Rev. Phytopathol. 2017, 55, 61–83. [Google Scholar] [CrossRef]
- Singh, D.; Raina, T.K.; Kumar, A.; Singh, J.; Prasad, R. Plant microbiome: A reservoir of novel genes and metabolites. Plant Gene 2019, 18, 100177. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef]
- Jones, P.; Garcia, B.; Furches, A.; Tuskan, G.; Jacobson, D. Plant host-associated mechanisms for microbial selection. Front. Plant Sci. 2019, 10, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, R.L.; Vismans, G.; Yu, K.; Song, Y.; de Jonge, R.; Burgman, W.P.; Burmølle, M.; Herschend, J.; Bakker, P.A.H.M.; Pieterse, C.M.J. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 2018, 12, 1496–1507. [Google Scholar] [CrossRef] [Green Version]
- Thapa, S.; Prasanna, R. Prospecting the characteristics and significance of the phyllosphere microbiome. Ann. Microbiol. 2018, 68, 229–245. [Google Scholar] [CrossRef]
- Lindow, S.E.; Brandl, M.T. Microbiology of the phyllosphere. Appl. Environ. Microb. 2003, 69, 1875–1883. [Google Scholar] [CrossRef] [Green Version]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Van Themaat, E.V.L.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Carvalhais, L.C.; Crawford, M.; Singh, E.; Dennis, P.G.; Pieterse, C.M.; Schenk, P.M. Inner plant values: Diversity, colonization and benefits from endophytic bacteria. Front. Microbiol. 2017, 8, 2552. [Google Scholar] [CrossRef]
- Guttman, D.; McHardy, A.C.; Schulze-Lefert, P. Microbial genome enabled insights into plant–microorganism interactions. Nat. Rev. Genet. 2014, 15, 797–813. [Google Scholar] [CrossRef]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [Green Version]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.H.; de Bang, T.C.; Laursen, K.H.; Pedas, P.; Husted, S.; Schjørring, J.K. Multielement plant tissue analysis using ICP spectrometry. In Plant Mineral Nutrients; Humana Press: Totowa, NJ, USA, 2013; pp. 121–141. [Google Scholar]
- Stevanato, P.; Fedito, P.; Trebbi, D.; Cagnin, M.; Saccomani, M.; Cacco, G. Effect of sulfate availability on root traits and microRNA395 expression in sugar beet. Biol. Plant 2015, 59, 491–496. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, Y.; Jagadeeswaran, G.; Li, Y.; Gowdu, K.; Sunkar, R. Identification and temporal expression analysis of conserved and novel microRNAs in Sorghum. Genomics 2011, 98, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeeswaran, G.; Li, Y.F.; Sunkar, R. Redox signaling mediates the expression of a sulfate-deprivation-inducible microRNA395 in Arabidopsis. Plant J. 2014, 77, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Stone, B.W.; Weingarten, E.A.; Jackson, C.R. The role of the phyllosphere microbiome in plant health and function. Ann. Plant Rev. Online 2018, 1, 533–556. [Google Scholar]
- Goswami, D.; Vaghela, H.; Parmar, S.; Dhandhukia, P.; Thakker, J.N. Plant growth promoting potentials of Pseudomonas spp. strain OG isolated from marine water. J. Plant Interact. 2013, 8, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef]
- Khan, A.L.; Waqas, M.; Asaf, S.; Kamran, M.; Shahzad, R.; Bilal, S.; Aaqil Khan, M.; Kang, S.-M.; Kim, Y.-H.; Yun, B.-W.; et al. Plant growth-promoting endophyte Sphingomonas sp. LK11 alleviates salinity stress in Solanum pimpinellifolium. Environ. Exp. Bot. 2017, 133, 58–69. [Google Scholar] [CrossRef]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Coenye, T.; Vandamme, P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiol. 2003, 5, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Compant, S.; Nowak, J.; Coenye, T.; Clément, C.; Ait Barka, E. Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol. Rev. 2008, 32, 607–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, M.; Ali, S.Z.; Sandhya, V.; Rasul, A.; Venkateswarlu, B. Role of microorganisms in adaptation of agriculture crops to abiotic stresses. World J. Microb. Biot. 2011, 27, 1231–1240. [Google Scholar] [CrossRef]
- Suárez-Moreno, Z.; Caballero-Mellado, J.; Coutinho, B.G.; Mendonça-Previato, L.; James, E.K.; Venturi, V. Common features of environmental and potentially beneficial plant-associated Burkholderia. Microb. Ecol. 2012, 63, 249–266. [Google Scholar] [CrossRef]
- Castro-Gonzalez, R.; Martinez-Aguilar, L.; Ramirez-Trujillo, A.; Estrada-de los Santos, P.; Caballero-Mellado, J. High diversity of culturable Burkholderia species associated with sugarcane. Plant Soil 2011, 345, 155–169. [Google Scholar] [CrossRef]
- Ait Bakra, E.; Nowak, J.; Clement, C. Enhancement of chilling resistance of inoculated grapevine plantlets with a plant growth promoting rhizobacterium, Burkholderia phytofirmans strain PsJN. Appl. Environ. Microbiol. 2006, 72, 7246–7252. [Google Scholar]
- Naveed, M.; Mitter, B.; Reichenauer, T.G.; Wieczorek, K.; Sessitsch, A. Increased drought stress resilience of maize through endophytic colonization by Burkholderia phytofirmans PsJN and Enterobacter sp. FD17. Environ. Exp. Bot. 2014, 97, 30–39. [Google Scholar] [CrossRef]
- Pinedo, I.; Ledger, T.; Greve, M.; Poupin, M.J. Burkholderia phytofirmans PsJN induces long-term metabolic and transcriptional changes involved in Arabidopsis thaliana salt tolerance. Front. Plant Sci. 2015, 6, 466. [Google Scholar] [CrossRef] [Green Version]
- Nafees, M.; Ali, S.; Naveed, M.; Rizwan, M. Efficiency of biogas slurry and Burkholderia phytofirmans PsJN to improve growth, physiology, and antioxidant activity of Brassica napus L. in chromium-contaminated soil. Environ. Sci. Pollut. Res. 2018, 25, 6387–6397. [Google Scholar] [CrossRef]
- Arnon, D.I.; Hoagland, D.R. Crop production in artificial culture solution and in soils with special reference to factors influencing yields and absorption of inorganic nutrients. Soil Sci. 1940, 50, 463–483. [Google Scholar]
- Axtell, M.J.; Bartel, D.P. Antiquity of microRNAs and their targets in land plants. Plant Cell 2005, 17, 1658–1673. [Google Scholar] [CrossRef] [Green Version]
- Hajizadeh, H.; Razavi, K.; Mostofi, Y.; Musavi, A.; Cacco, G.; Zamani, Z.; Stevanato, P. Identification and characterization of genes differentially displayed in Rosa hybrida petals during flower senescence. Sci. Hort. 2011, 128, 320–324. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer Forward 5′,3′ | Primer Reverse 5′,3′ |
|---|---|---|
| Pseudomonas | GCGCGTAGGTGGCTTGATAA | GGATGCAGTTCCCAGGTTGA |
| Pedobacter | CTGGTCCGGTGTCTCAGTAC | GGGGATCTGAGAGGATGACC |
| Enterobacter | CATGGGAGTGGGTTGCAAAA | TCACAAAAGTGGTAAGCGCC |
| Duganella | GCTACTGATCGATGCCTTGG | GCGGCCGATATCTGATTAGC |
| Burkholderia | CCTCTGCCATACTCTAGCCC | ATGTGAAATCCCCGGGCTTA |
| Microbacterium | GGTACCGTCACTTTCGCTTC | GTGAGGGATGACGGCCTT |
| Oxalicibacterium | GCGCAACCCTTGTCATTAGT | TGTCACCGGCAGTCTCATTA |
| Acinetobacter | GCGAGGAGGAGGCTACTTTA | CGGTGCTTATTCTGCGAGTA |
| Xanthomonas | AAGGTGGGGATGACGTCAAG | TGTGTAGCCCTGGTCGTAAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertoldo, G.; Della Lucia, M.C.; Squartini, A.; Concheri, G.; Broccanello, C.; Romano, A.; Ravi, S.; Cagnin, M.; Baglieri, A.; Stevanato, P. Endophytic Microbiome Responses to Sulfur Availability in Beta vulgaris (L.). Int. J. Mol. Sci. 2021, 22, 7184. https://doi.org/10.3390/ijms22137184
Bertoldo G, Della Lucia MC, Squartini A, Concheri G, Broccanello C, Romano A, Ravi S, Cagnin M, Baglieri A, Stevanato P. Endophytic Microbiome Responses to Sulfur Availability in Beta vulgaris (L.). International Journal of Molecular Sciences. 2021; 22(13):7184. https://doi.org/10.3390/ijms22137184
Chicago/Turabian StyleBertoldo, Giovanni, Maria Cristina Della Lucia, Andrea Squartini, Giuseppe Concheri, Chiara Broccanello, Alessandro Romano, Samathmika Ravi, Massimo Cagnin, Andrea Baglieri, and Piergiorgio Stevanato. 2021. "Endophytic Microbiome Responses to Sulfur Availability in Beta vulgaris (L.)" International Journal of Molecular Sciences 22, no. 13: 7184. https://doi.org/10.3390/ijms22137184
APA StyleBertoldo, G., Della Lucia, M. C., Squartini, A., Concheri, G., Broccanello, C., Romano, A., Ravi, S., Cagnin, M., Baglieri, A., & Stevanato, P. (2021). Endophytic Microbiome Responses to Sulfur Availability in Beta vulgaris (L.). International Journal of Molecular Sciences, 22(13), 7184. https://doi.org/10.3390/ijms22137184