
Explanation of the Formation of Complexes between Representatives of Oxazolidinones and HDAS-β-CD Using Molecular Modeling as a Complementary Technique to cEKC and NMR


Abstract
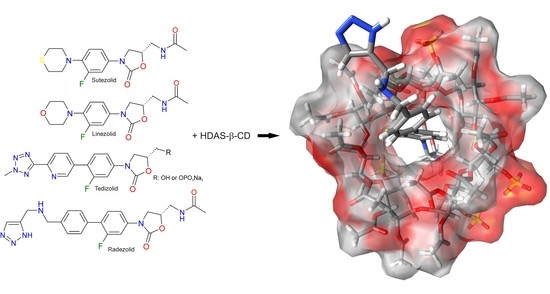
:
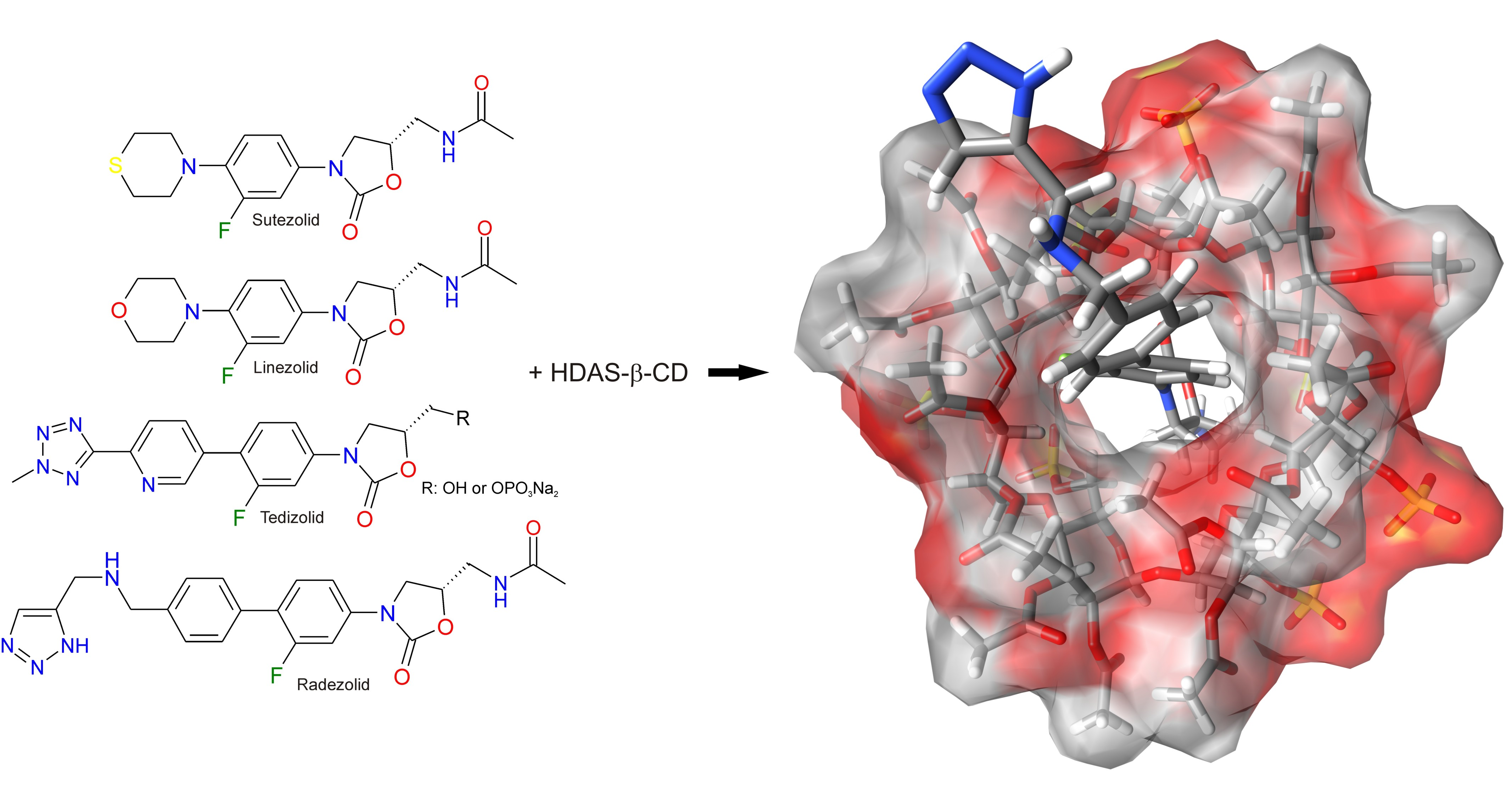
1. Introduction
1.1. General Concept
1.2. Evolution of Views on the Mechanisms of Chiral Recognition
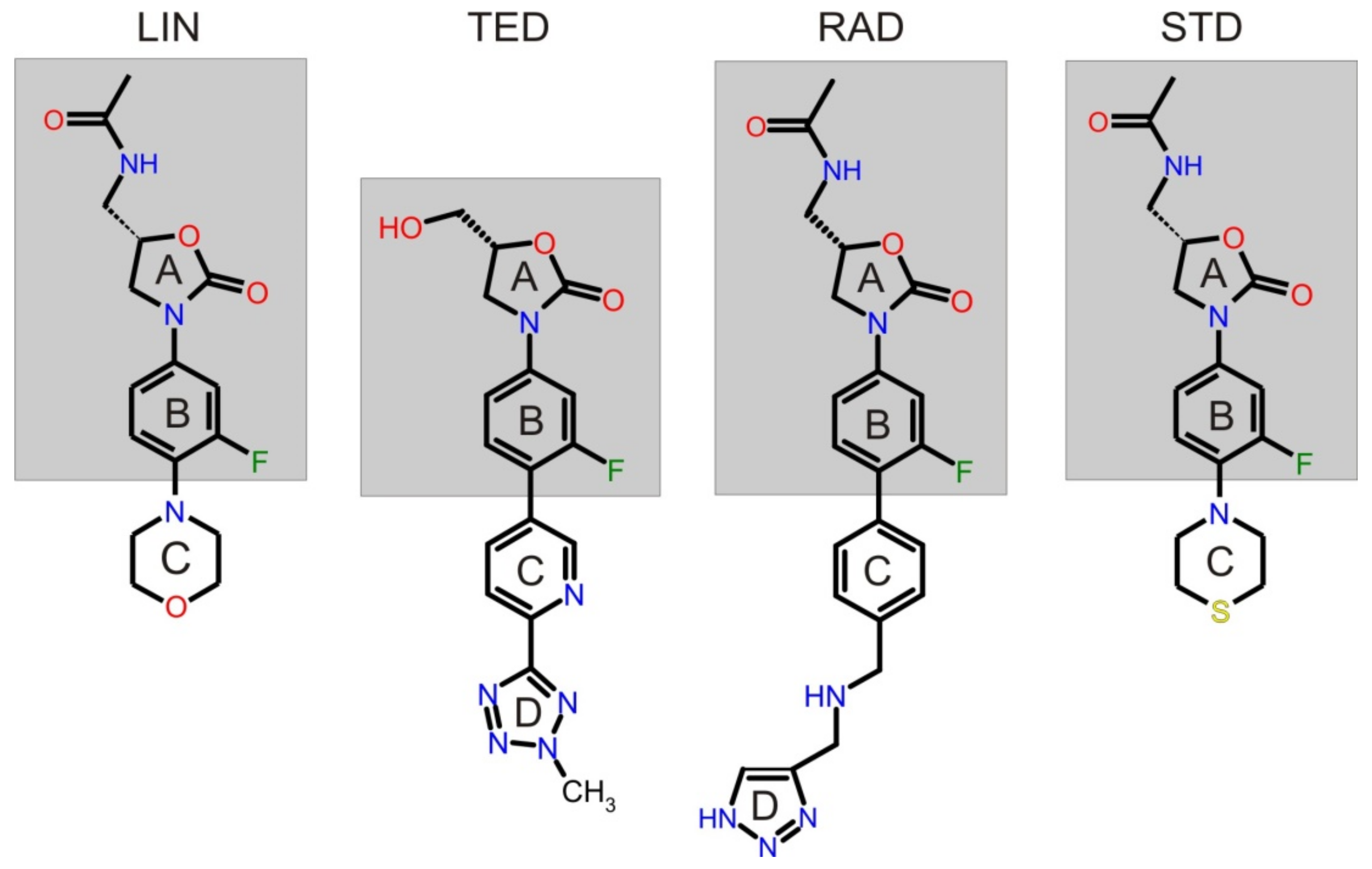
1.3. Published Results of cEKC Experiments and NMR Measurement of New Oxazolidinone Analogues
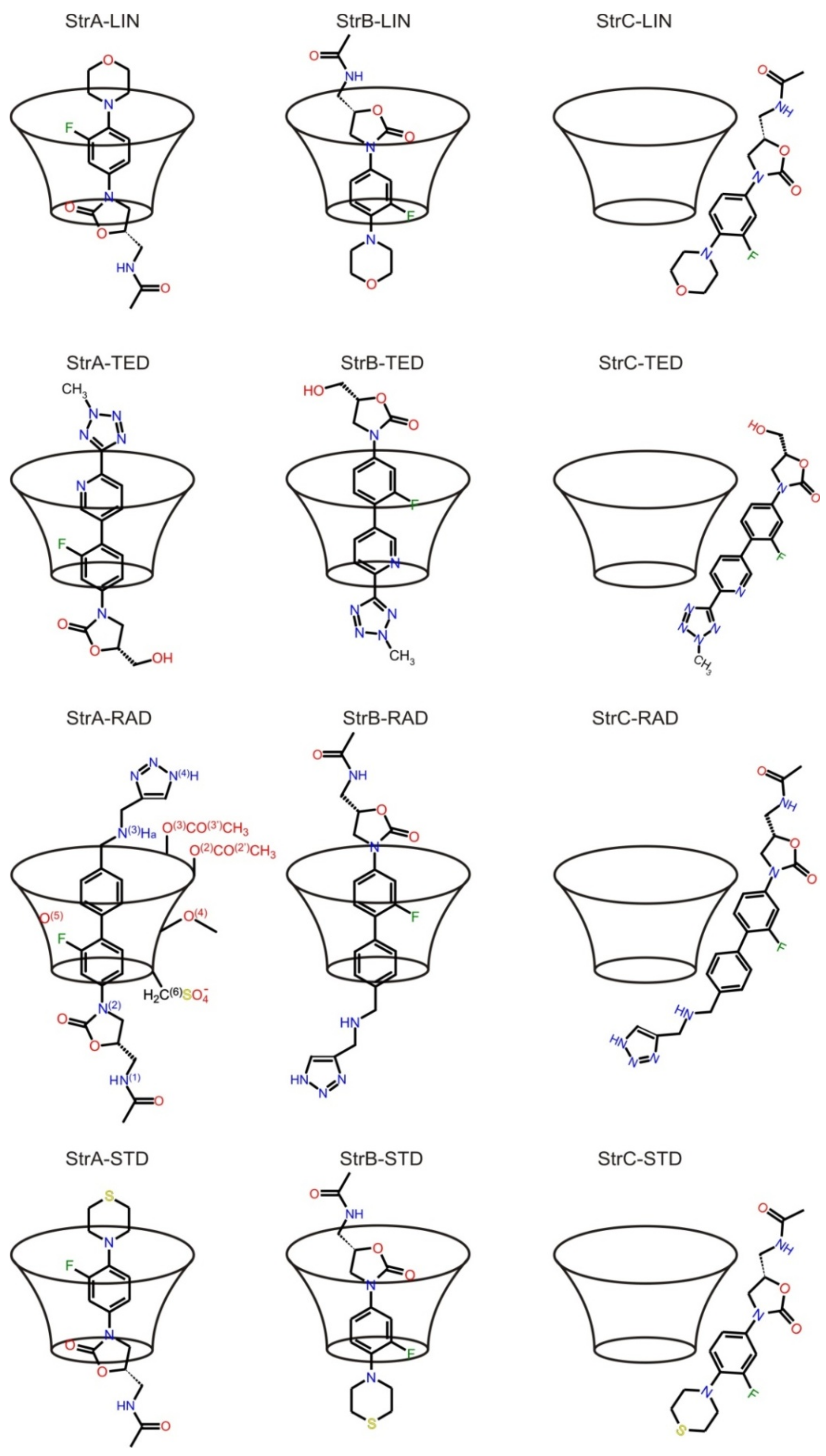
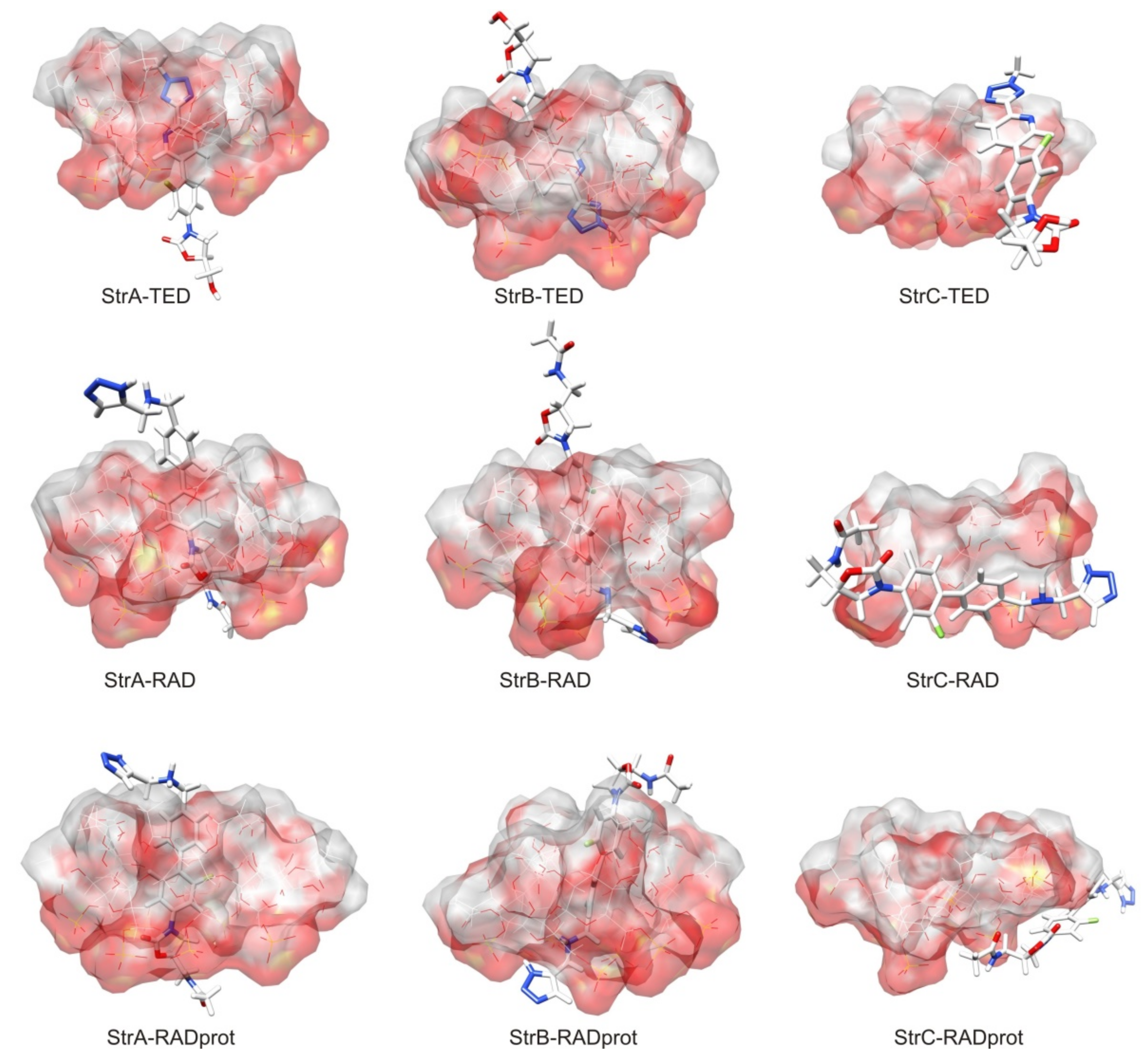
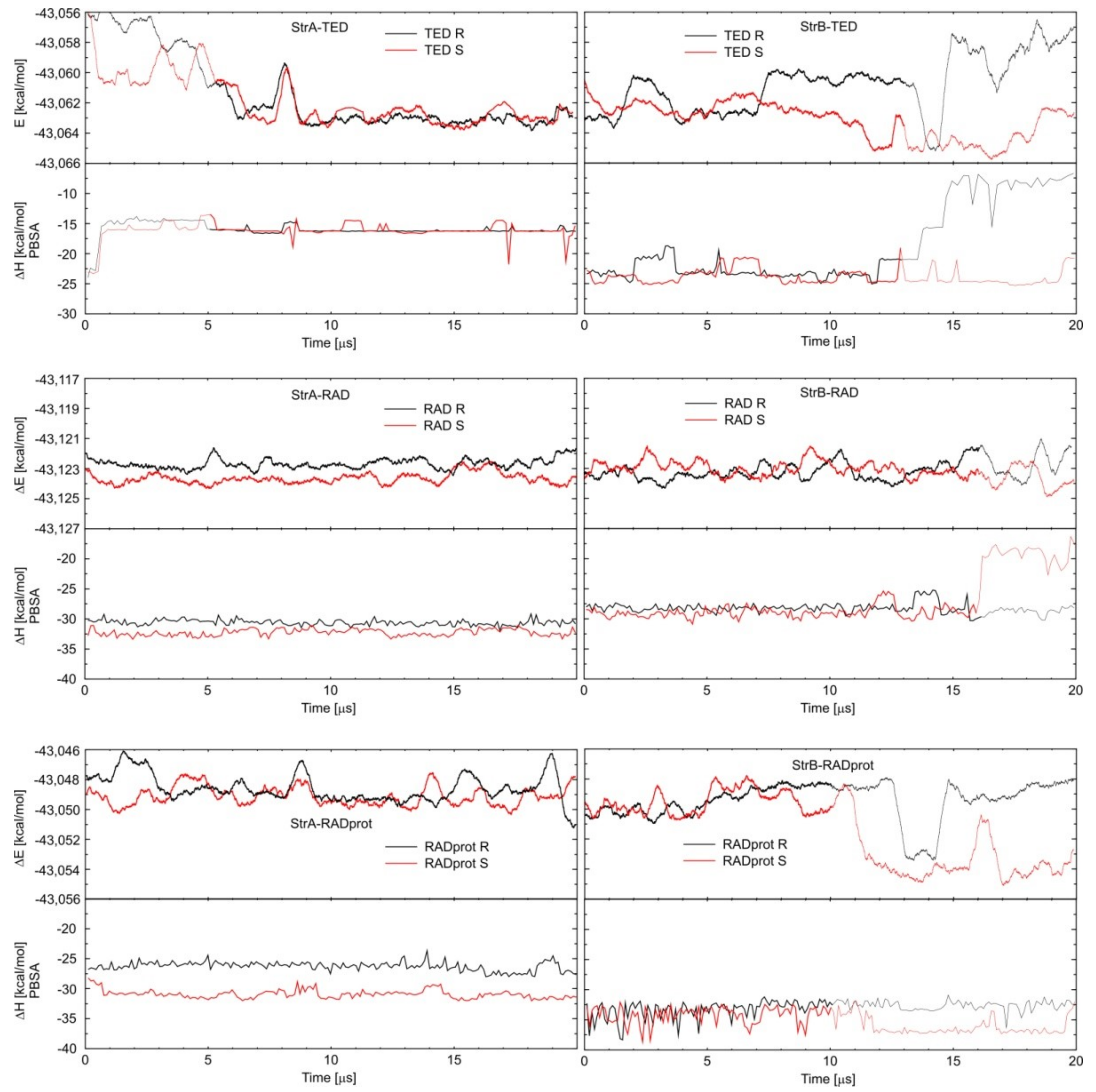
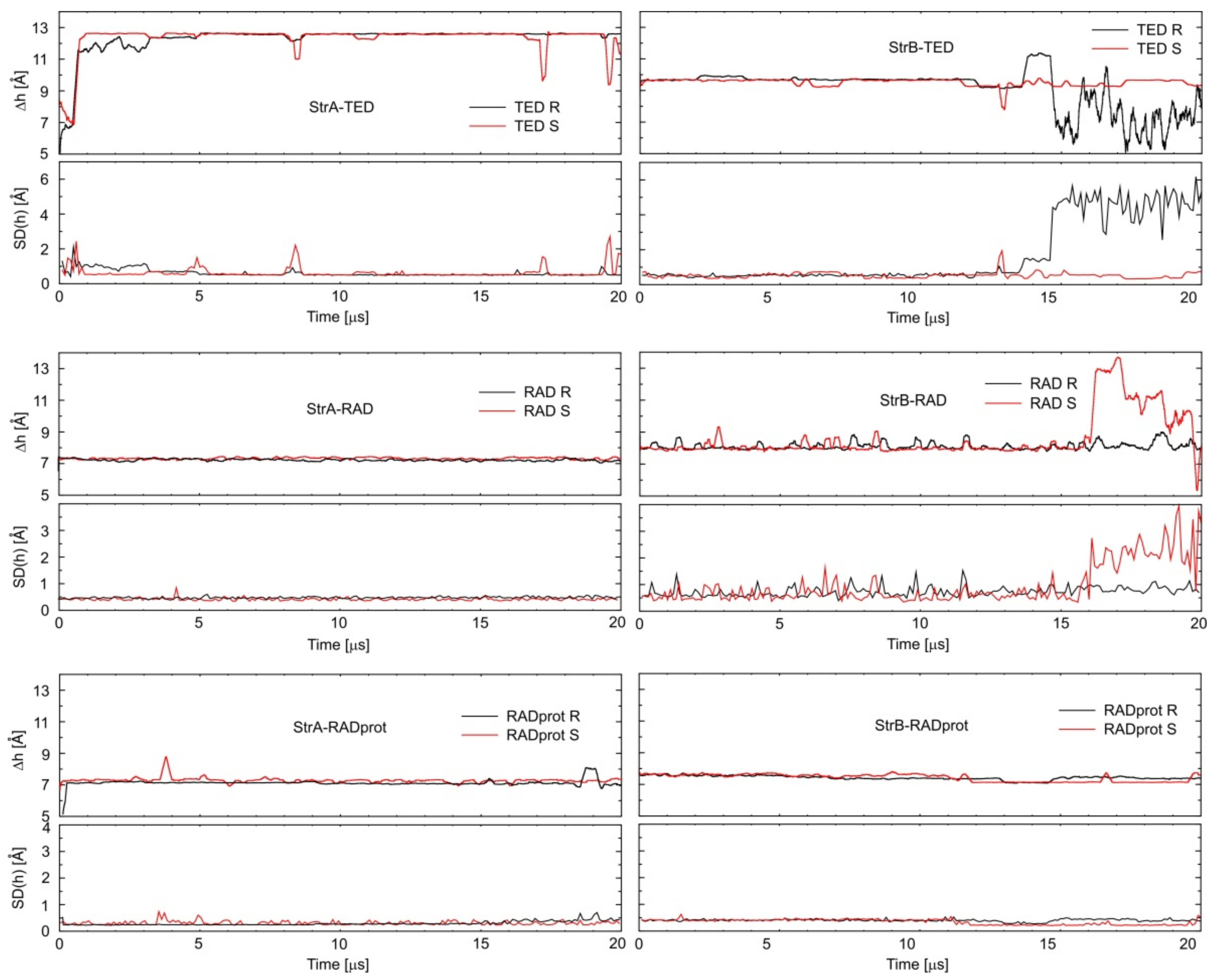
2. Results and Discussion
3. Computational Methods
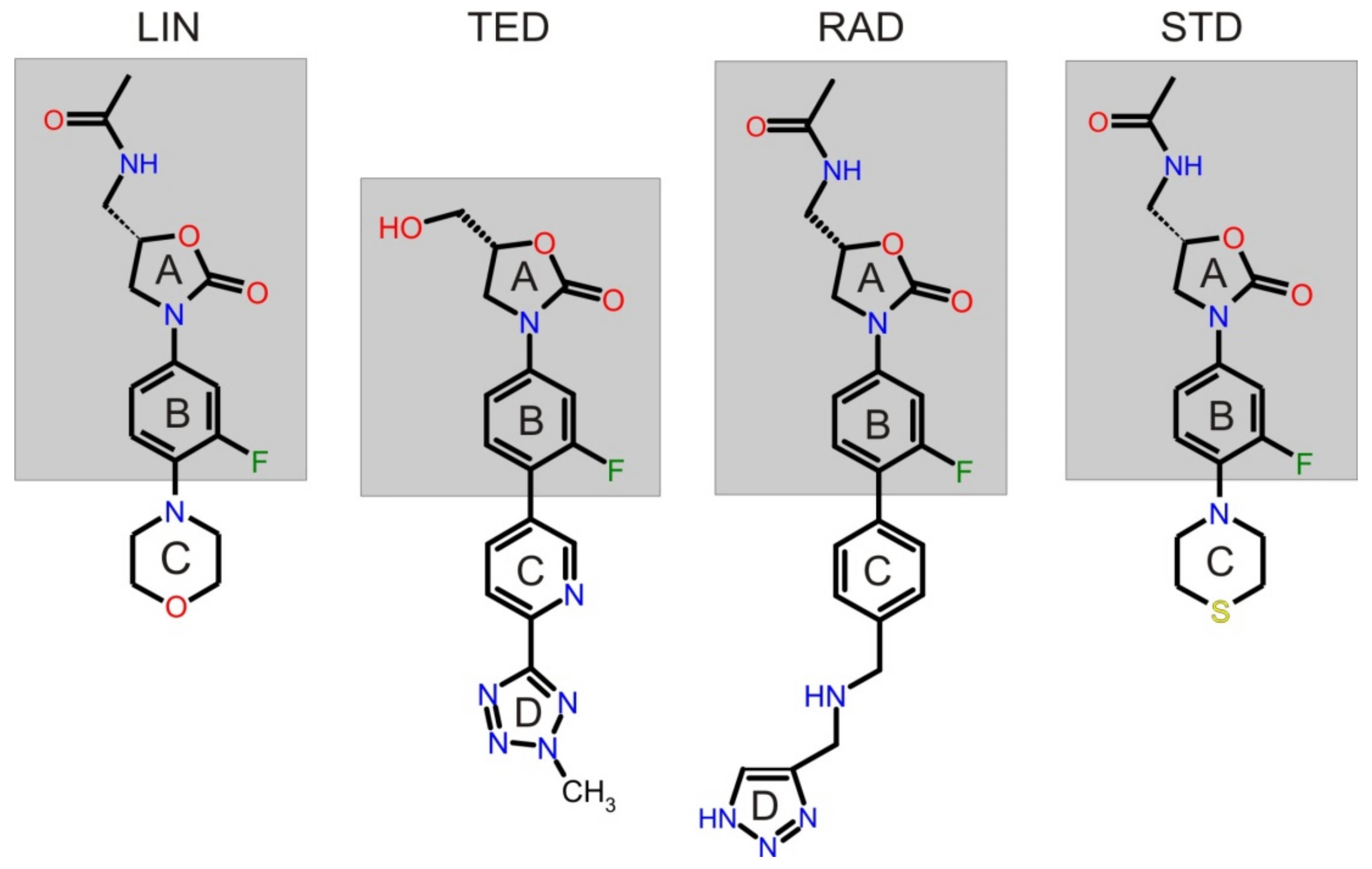
3.1. Preparing the Molecular Dynamic Calculations for TED and RAD
3.2. Calculating Binding Enthalpies Using Explicit Water Molecular Dynamics Simulations (Absolute Binding Free Energy)
3.3. Calculating Binding Free Energies (Enthalpies) Using MM-PBSA and MM-GBSA Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michalska, K.; Karpiuk, I.; Król, M.; Tyski, S. Recent development of potent analogues of oxazolidinone antibacterial agents. Bioorg. Med. Chem. 2013, 21, 577–591. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=sutezolid&cntry=&state=&city=&dist= (accessed on 12 May 2021).
- The PEW Charitable Trusts. Antibiotics Currently in Global Clinical Development. Available online: https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development (accessed on 12 May 2021).
- Michalska, K.; Pajchel, G.; Tyski, S. Determination of enantiomer impurity of linezolid by capillary electrophoresis using heptakis-(2,3-diacetyl-6-sulfato)-β-cyclodextrin. J. Chromatogr. A 2008, 1180, 179–186. [Google Scholar] [CrossRef]
- Bednarek, E.; Bocian, W.; Michalska, K. NMR and molecular modeling study, as complementary techniques to capillary electrophoresis method to elucidate the separation mechanism of linezolid enantiomers. J. Chromatogr. A 2008, 1193, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Michalska, K.; Gruba, E.; Cielecka-Piontek, J.; Bednarek, E. Chiral separation of tedizolid using charge single isomer derivatives of cyclodextrins by capillary electrokinetic chromatography. J. Pharm. Biomed. Anal. 2016, 120, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Michalska, K.; Mizera, M.; Lewandowska, K.; Cielecka-Piontek, J. Infrared, Raman and ultraviolet with circular dichroism analysis and theoretical calculations of tedizolid. J. Mol. Struc. 2016, 1115, 136–143. [Google Scholar] [CrossRef]
- Bednarek, E.; Bocian, W.; Michalska, K. Nuclear Magnetic Resonance Spectroscopic Study of the Inclusion Complex of (R)-Tedizolid with HDAS-β-CD, β-CD, and γ-Cyclodextrin in Aqueous Solution. J. Pharm. Biomed. Anal. 2019, 169, 170–180. [Google Scholar] [CrossRef]
- Michalska, K.; Gruba, E.; Bocian, W.; Cielecka-Piontek, J. Enantioselective recognition of radezolid by cyclodextrin modified capillary electrokinetic chromatography and electronic circular dichroism. J. Pharm. Biomed. Anal. 2017, 139, 98–108. [Google Scholar] [CrossRef]
- Michalska, K.; Bednarek, E.; Gruba, E.; Lewandowska, K.; Mizera, M.; Cielecka-Piontek, J. Comprehensive spectral identification of key intermediates to the final product of the chiral pool synthesis of radezolid. Chem. Cent. J. 2017, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Michalska, K.; Gruba, E.; Mizera, M.; Lewandowska, K.; Bednarek, E.; Bocian, W. Cielecka-Piontek, Application of spectroscopic methods (FT-IR, Raman, ECD and NMR) in studies of identification and optical purity of radezolid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2017, 183, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Michalska, K.; Bocian, W.; Bednarek, E.; Pałys, B.; Cielecka-Piontek, J. Enantioselective recognition of sutezolid by cyclodextrin modified non-aqueous capillary electrophoresis and explanation of complex formation by means of infrared spectroscopy, NMR and molecular modelling. J. Pharm. Biomed. Anal. 2019, 169, 49–59. [Google Scholar] [CrossRef]
- Chankvetadze, B. Enantioseparations by using capillary electrophoretic techniques. The story of 20 and a few more years. J. Chromatogr. A 2007, 1169, 45–70. [Google Scholar] [CrossRef]
- Lämmerhofer, M. Chiral recognition by enantioselective liquid chromatography: Mechanisms and modern chiral stationary phases. J. Chromatogr. A 2010, 1217, 814–856. [Google Scholar] [CrossRef] [PubMed]
- Scriba, G.K.E. Fundamental aspects of chiral electromigration techniques and application in pharmaceutical and biomedical analysis. J. Pharm. Biomed. Anal. 2011, 55, 688–701. [Google Scholar] [CrossRef]
- Chankvetadze, B.; Fanali, S. Special issue, Enantioseparations. J. Chromatogr. A 2014, 1363, 1–372. [Google Scholar] [CrossRef]
- Chankvetadze, B.; Fanali, S. Special issue, Enantioseparations. J. Chromatogr. A 2016, 1467, 1–496. [Google Scholar] [CrossRef]
- Lapizco-Encinas, B.H.; Wätzig, H. Special issue, Enantioseparations 2019. Electrophoresis 2019, 40, 1863–1996. [Google Scholar]
- Peluso, P.; Dessì, A.; Dallocchio, R.; Mamane, V.; Cossu, S. Recent studies of docking and molecular dynamics simulation for liquid-phase enantioseparations. Electrophoresis 2019, 40, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Chankvetadze, B. Contemporary theory of enantioseparations in capillary electrophoresis. J. Chromatogr. A 2018, 1567, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.B.; Quirino, J.P. Chiral Selectors in Capillary Electrophoresis: Trends during 2017–2018. Molecules 2019, 24, 1135. [Google Scholar] [CrossRef] [Green Version]
- Scriba, G.K.E. Chiral recognition in separation sciences. Part I: Polysaccharide and cyclodextrin selectors. Trends Anal. Chem. 2019, 120, 115639. [Google Scholar] [CrossRef]
- Bernardo-Bermejo, S.; Sánchez-López, E.; Castro-Puyana, M.; Marina, M.L. Chiral capillary electrophoresis. TrAC Trends Anal. Chem. 2020, 124, 115807. [Google Scholar] [CrossRef]
- József, S. Past, present, and future of cyclodextrin research. Pure Appl. Chem. 2004, 76, 1825–1845. [Google Scholar]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Majdecki, M.; Krzak, A.; Żelechowska, S.O. Monosubstituted hydrazone β-cyclodextrin derivatives for pH-sensitive complex formation with aromatic drugs. J. Incl. Phenom. Macrocycl. Chem. 2019, 93, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Fanali, S. Identification of chiral drug isomers by capillary electrophoresis. J. Chromatogr. A 1996, 735, 77–121. [Google Scholar] [CrossRef]
- Servais, A.-C.; Rousseau, A.; Fillet, M.; Lomasadze, K.; Salgado, A.; Crommen, J.; Chankvetadze, B. Separation of propranolol enantiomers by CE using sulphated-β-CD derivatives in aqueous and non-aqueous electrolytes: Comparative CE and NMR study. Electrophoresis 2010, 31, 1467–1474. [Google Scholar]
- Chankvetadze, L.; Servais, A.-C.; Fillet, M.; Salgado, A.; Crommen, J.; Chankvetadze, B. Comparative enantioseparation of talinolol in aqueous and non-aqueous capillary electrophoresis and study of related selector-selectand interactions by nuclear magnetic resonance spectroscopy. J. Chromatogr. A 2012, 1267, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Fejős, I.; Kazsoki, A.; Sohajda, T.; Márványos, E.; Volk, B.; Szente, L.; Béni, S.Z. Interactions of non-charged tadalafil stereoisomers with cyclodextrin: Capillary electrophoresis and nuclear magnetic resonance studies. J. Chromatogr. A 2014, 1363, 348–355. [Google Scholar] [CrossRef]
- Servais, A.-C.; Rousseau, A.; Dive, G.; Frederich, M.; Crommen, J.; Fillet, M. Combination of capillary electrophoresis, molecular modelling and nuclear magnetic resonance to study the interaction mechanisms between single-isomer anionic cyclodextrin derivatives and basic drug enantiomers in a methanolic background electrolyte. J. Chromatogr. A 2012, 1232, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Beneš, M.; Riesová, M.; Svobodová, J.; Tesařová, E.; Dubský, P.; Gaš, B. Complexation of Buffer Constituents with Neutral Complexation Agents: Part II. Practical Impact in Capillary Zone Electrophoresis. Anal. Chem. 2013, 85, 8526–8534. [Google Scholar] [CrossRef]
- Salgado, A.; Chankvetadze, B. Application of nuclear magnetic resonance spectroscopy for the understanding of enantiomer separation mechanisms in capillary electrophoresis. J. Chromatogr. A 2016, 1467, 95–144. [Google Scholar] [CrossRef] [PubMed]
- Scriba, G.K.E. Chiral Recognition in Separation Sciences: An Overview. In Chiral Separations: Methods and Protocols; Methods in Molecular Biology Series; Humana Press: Totowa, NJ, USA, 2013; Volume 970, pp. 1–27. [Google Scholar]
- Biedermann, F.; Nau, W.M.; Schneider, H.-J. The hydrophobic effect revisited-studies with supramolecular complexes imply high-energy water as noncovalent driving force. Angew. Chem. Int. 2014, 53, 11158–11171. [Google Scholar] [CrossRef] [PubMed]
- Scriba, G.K.E. Chiral recognition in separation science—An update. J. Chromatogr. A 2016, 1467, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Waibel, B.; Schreiber, J.; Meier, C.; Hammitzsch, M.; Baumann, K.; Scriba, G.K.E.; Holzgrabe, U. Comparison of cyclodextrin-dipeptide inclusion complexes in the absence and presence of urea by means of capillary electrophoresis, nuclear magnetic resonance and molecular modelling. Eur. J. Org. Chem. 2007, 18, 2921–2930. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, S.; Chao, J.; Wang, S.; Cong, C.; Shuang, S.; Paau, M.C.; Choi, M.M.F. An evidence for the chiral discrimination of naproxen enantiomers: A combined experimental and theoretical study. J. Phys. Chem. C. 2011, 115, 4033–4040. [Google Scholar] [CrossRef]
- Ali, I.; Sanagi, M.M.; Aboul-Enein, H.Y. Advances in chiral separtions by nonaqueous capillary electrophoresis in pharmaceutical and biomedical analysis. Electrophoresis 2014, 35, 926–936. [Google Scholar] [CrossRef]
- Lindsey, R.K.; Rafferty, J.L.; Eggimann, B.L.; Siepmann, J.I.; Schure, M.R. Molecular simulation studies of reversed-phase liquid chromatography. J. Chromatogr. A 2013, 1287, 60–82. [Google Scholar] [CrossRef]
- Wyczalkowski, M.A.; Vitalis, A.; Pappu, R.V. New Estimators for Calculating Solvation Entropy and Enthalpy and Comparative Assessments of Their Accuracy and Precision. J. Phys. Chem. B 2010, 114, 8166–8180. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.L.; Gilson, M.K. Predicting Binding Free Energies. Annu. Rev. Biophys. 2017, 46, 531–558. [Google Scholar] [CrossRef] [Green Version]
- Alvira, E. Molecular Simulation of the Separation of Some Amino Acid Enantiomers by beta-Cyclodextrin in Gas-Phase. Front. Chem. 2020, 8, 823. [Google Scholar] [CrossRef]
- Alvira, E. Molecular Simulation of the Separation of Isoleucine Enantiomers by β-Cyclodextrin. Molecules 2019, 24, 1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Andrew, T.F.; Michael, K.G. Host-Guest in Explicit Water Example, Calculating the Binding Enthalpy of Guest B2 with Host Cucurbit[7]Uril (CB7); AMBER 14 Advanced Tutorial 21; Available online: http://ambermd.org/tutorials/advanced/tutorial21 (accessed on 12 May 2021).
- Fenley, A.T.; Henriksen, N.M.; Muddana, H.S.; Gilson, M.K. Bridging Calorimetry and Simulation Through Precise Calculations of Cucurbituril-Guest Binding Enthalpies. J. Chem. Theory Comput. 2014, 10, 4069–4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
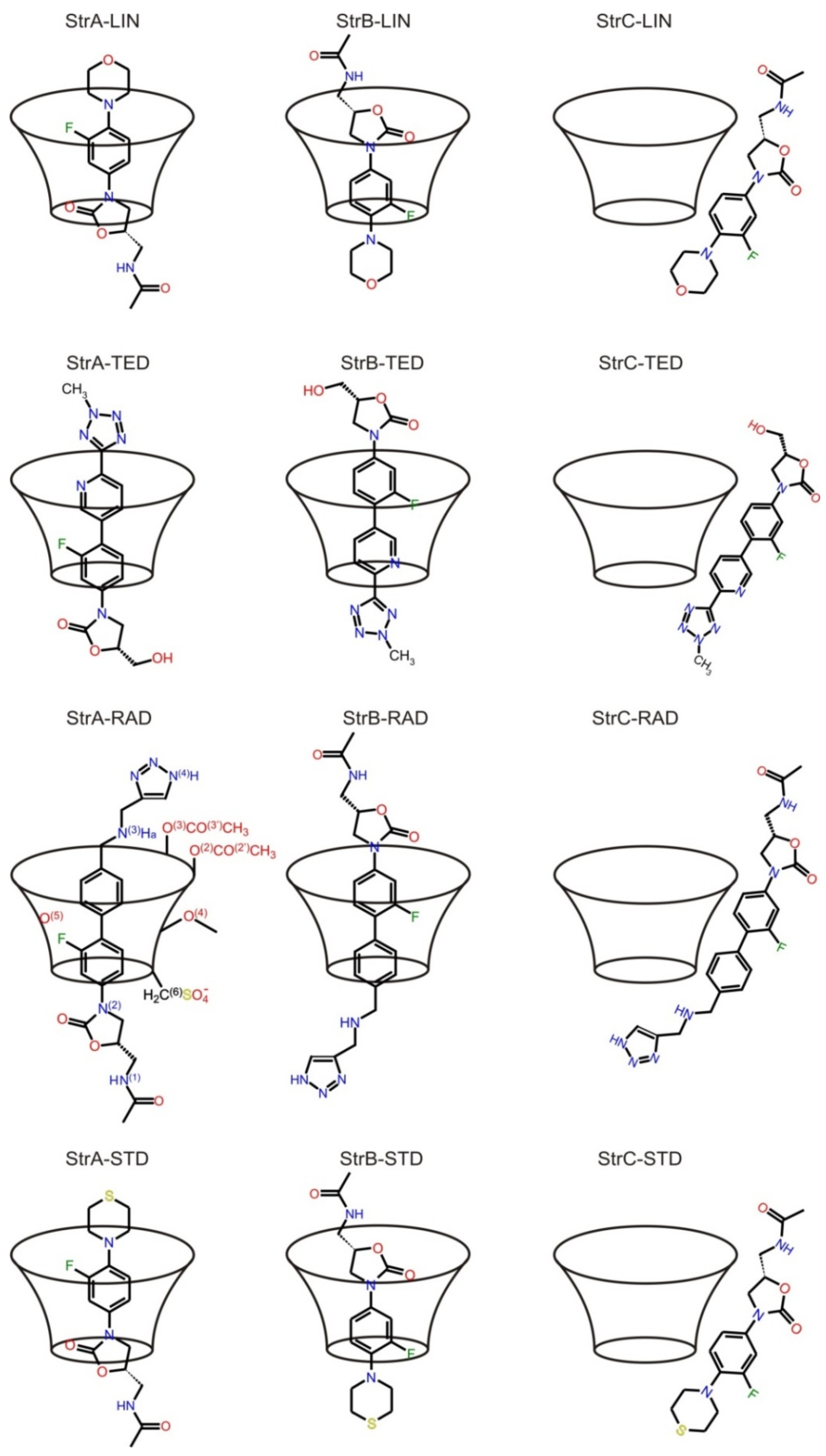{kind=link}
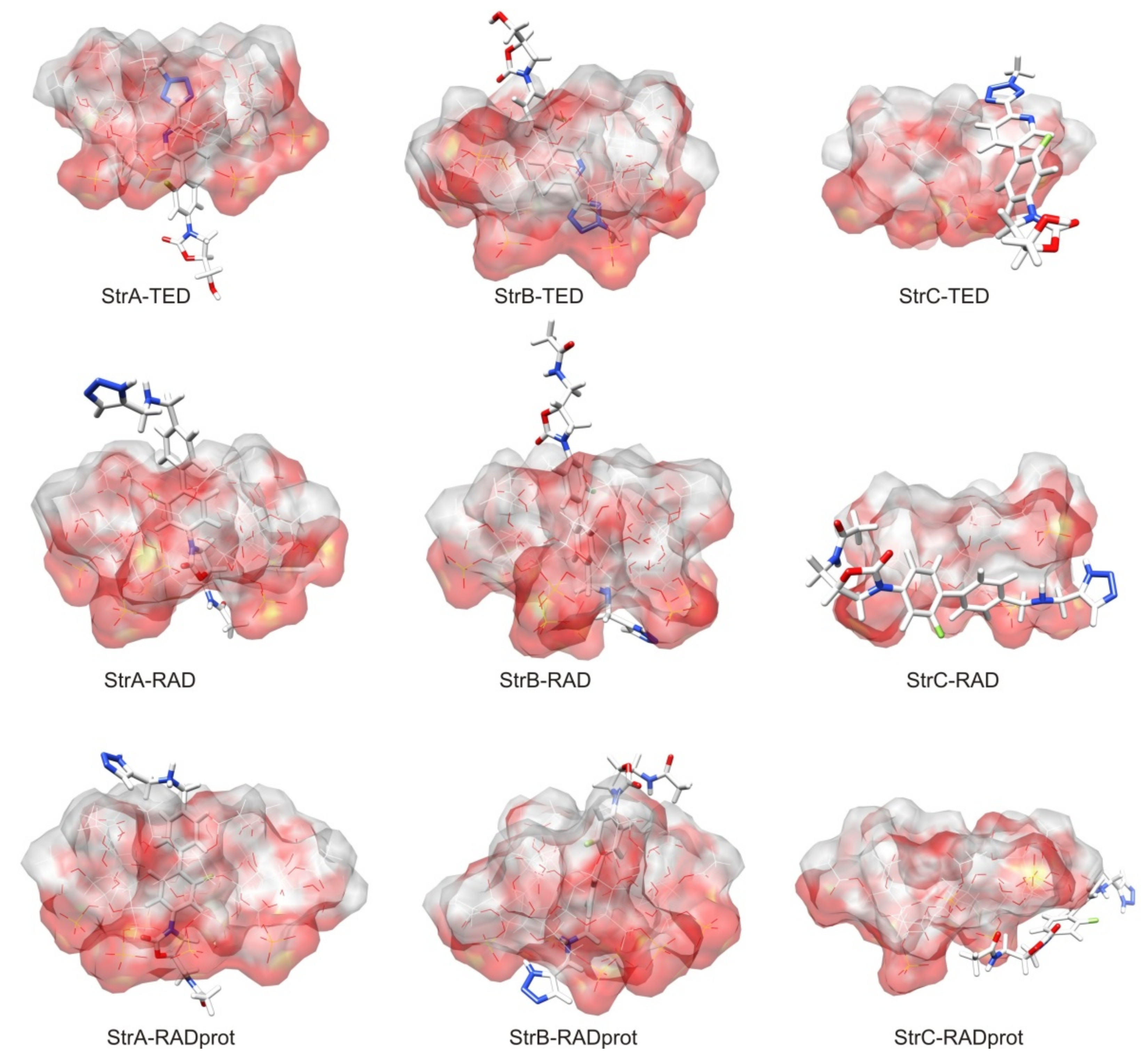{kind=link}
{kind=link}
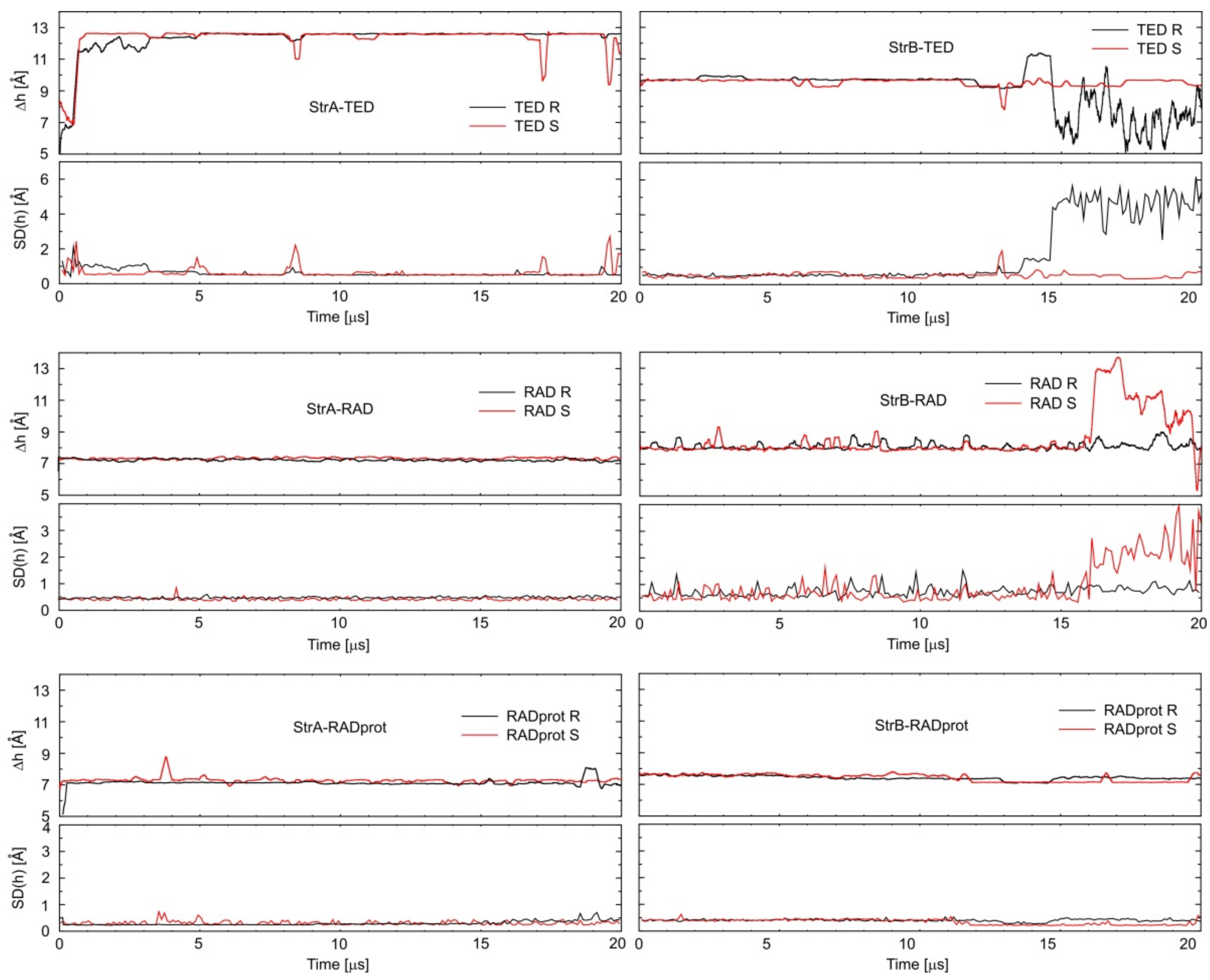{kind=link}
| Analyte | Type of BGE | Separation Conditions | EMO Reversal for a Leading Compound | Ref. | Method of Determination of Binding Constant | pH | Binding Constant Ka * [M−1] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| -(R) | -(S) | ||||||||
| LIN leading | ACE | 27.5 mM HDAS-β-CD in 50 mM borate buffer, pH 9.0, 15 °C, capillary (1), NP, 15 kV | 18.75 mM HDAS-β-CD in 50 mM borate buffer, pH 8.0, 15 °C, capillary (2), RP, 15 kV | [4] | NMR diffusion, D2O buffer | 2.4 | 76 ± 5 | 66 ± 5 | [5] |
| 7.0 | 52 ± 2 | 35 ± 2 | |||||||
| LIN | ACE | 37.5 mM HDAS-β-CD in 50 mM formic buffer, pH 4.0 with ACN (81.4:18.6, v/v), 27 °C, capillary (1), 12 kV, NP/ or 35 mM HDAS-β-CD in 50 mM borate buffer, pH 9.0 with ACN (81.4:18.6, v/v), 27 °C, capillary (1), 12 kV, NP | not observed | [6] | cEKC | 4.0 | 1311 ± 256 | 1507 ± 307 | [6] |
| 9.0 | 571 ± 80 | 625 ± 88 | |||||||
| TED leading | 4.0 | 747 ± 155 | 728 ± 145 | ||||||
| 9.0 | 319 ± 51 | 326 ± 50 | |||||||
| TED | - | - | - | NMR diffusion, D2O | - | 140 ± 30 | - | [8] | |
| TED-PO4 | - | 98 ± 20 | - | ||||||
| LIN | ACE | 6 mM HDAS-β-CD in 50 mM phosphoric buffer, pH 2.5, capillary (1), 28 kV, 17–27 °C, NP | 18 mM HDAS-β-CD in 50 mM phosphate buffer, pH 6.6, 27 °C, capillary (1), NP | [9] | cEKC | 2.5 | 41.5 ± 10 | 42.9 ± 10 | [9] |
| 6.6 | 45.8 ± 10 | 53.4 ± 10 | |||||||
| RAD leading | 20 mM HDAS-β-CD in 50 mM phosphoric buffer, pH 2.5, capillary (1), 28 kV, 52 °C, RP (partial resolution) | 2.5 | 2940 ± 600 | 2817 ± 600 | |||||
| 6.6 | 999 ± 200 | 1082 ± 200 | |||||||
| LIN | NACE | not observed with HDAS-β-CD | 45 mM HDMS-β-CD in MeOH/ACN (85:15, v/v), 200 mM TFA/20 mM ammonium formate, capillary (1); 25 kV, 22 °C, NP | [12] nd | nd | nd | nd | - | |
| STD leading | 5 mM HDAS-β-CD in MeOH/ACN (85:15, v/v), 200 mM TFA/20 mM ammonium formate, capillary (1); 25 kV, 22 °C, NP | ||||||||
| Compound | Average Binding Enthalpies [kcal/mol] (b) | |||||||
|---|---|---|---|---|---|---|---|---|
| StrA | StrB | StrC | ||||||
| H2O | MeOH/ACN | H2O | MeOH/ACN | H2O | ||||
| MD | PBSA (GBSA) | PBSA (GBSA) | MD | PBSA (GBSA) | PBSA (GBSA) | MD | PBSA (GBSA) | |
| LIN-(R) (a) | − | −21.6 ± 4.8 (−21.1 ± 3.9) | − | − | −26.4 ± 4.5 (−31.7 ± 4.5) | - | - | - |
| LIN-S (a) | − | −21.7 ± 5.0 (−22.5 ± 3.3) | − | − | 24.7 ± 3.8 (−30.4 ± 2.6) | - | - | - |
| TED-(R) | −21.3 ± 1.7 | −16.2 ± 0.2 (−18.8 ± 0.2) | −16.4 ± 0.2 (−19.1 ± 0.2) | −21.4 ± 1.2 | −22.9 ± 1.0 (−27.9 ± 0.8) | −23.2 ± 1.1 (−28.2 ± 0.8) | −17.5 ± 2.2 | −10.6 ± 2.8 (−9.9 ± 2.7) |
| TED-(S) | −21.4 ± 1.5 | −16.1 ± 0.4 (−18.8 ± 0.4) | −16.4 ± 0.4 (−19.2 ± 0.4) | −21.9 ± 0.5 | −23.7 ± 1.1 (−28.3 ± 1.0) | −24.0 ± 1.1 (−28.6 ± 1.0) | - | - |
| RAD-(R) | −23.4 ± 0.3 | −28.6 ± 0.3 (−31.1 ± 0.3) | −29.3 ± 0.2 (−31.8 ± 0.3) | −23.6 ± 0.8 | −28.2 ± 0.7 (−31.1 ± 0.7) | −29.1 ± 0.7 (−31.9 ± 0.7) | −21.9 ± 1.9 | −17.2 ± 2.2 (−17.4 ± 2.9) |
| RAD-(S) | −24.3 ± 0.4 | −30.2 ± 0.4 (−32.3 ± 0.5) | −31.2 ± 0.4 (−33.2 ± 0.5) | −23.8 ± 0.6 | 27.0 ± 3.8 (−29.9 ± 4.1) | 27.8 ± 4.1 (−30.5 ± 4.1) | - | - |
| RADprot-(R) | −26.6 ± 0.9 | −26.6 ± 0.5 (−30.4 ± 0.4) | −30.8 ± 0.7 (−33.5± 0.3) | −27.7 ± 0.7 | −33.3 ± 0.8 (−36.1 ± 0.7) | −40.6 ± 0.8 (−40.8± 0.7) | −20.2 ± 3.2 | −16.4 ± 1.8 −15.8 ± 2.2 |
| RADprot-(S) | −27.1 ± 0.6 | −31.3 ± 0.5 (−34.4 ± 0.3) | −35.8 ± 0.7 (−37.6 ± 0.4) | −27.5 ± 0.8 | −34.2 ± 0.7 (−36.9 ± 0.5) | −41.3 ± 0.8 (−41.5 ± 0.6) | - | - |
| STD (a) | −18.9 ± 0.7 | −23.4 ± 0.7 (−25.9 ± 0.8) | −24.1 ± 0.7 (−26.6 ± 0.7) | −20.2 ± 0.8 | −25.8 ± 2.1 (−29.3 ± 1.0) | −26.6 ± 2.1 (−29.9 ± 1.0) | −16.5 ± 1.1 | −5.5 ± 4.0 −5.5 ± 3.5 |
| STDprot (a) | −18.8 ± 0.7 | −23.8 ± 0.9 (−26.1 ± 0.8) | −24.3 ± 1.2 (−26.7 ± 1.1) | −19.9 ± 0.6 | −26.2 ± 1.0 (−32.1 ± 0.9) | −32.8 ± 1.1 (−36.2 ± 1.2) | - | - |
| Complex Structure | Intermolecular H-Bonds | |||
|---|---|---|---|---|
| Name | Av. Distance [Å] | Population | Av. H-Bonds No. | |
| StrA–TED | - | - | No | 0 |
| StrB–TED | - | - | No | 0 |
| StrA–RAD | RAD-N(1)H … CD-SO4− | 2.29 +/− 0.47 | High | 1.30 (R) 2.06 (S) |
| RAD-N(3)Ha … CD-O3′ | 2.24 +/− 0.34 | High | ||
| RAD-N(4)H … CD-O3′ | 2.17 +/− 0.30 | High | ||
| RAD-N(4)H … CD-O2′ | 2.29 +/− 0.34 | Mid | ||
| RAD-N(3)Ha … CD-O3 | 2.60 +/− 0.28 | Mid | ||
| RAD-N(3)Ha … CD-O2 | 2.57 +/− 0.31 | Mid | ||
| RAD-N(3)Ha … CD-O2′ | 2.75 +/− 0.30 | Low | ||
| StrB–RAD | RAD-N(4)H … CD-SO4− | 2.24 +/− 0.35 | High | 2.08 (R) 2.01 (S) |
| RAD-N(3)Ha … CD-SO4− | 2.30 +/− 0.35 | High | ||
| RAD-N(3)Ha … CD-O5 | 2.44 +/− 0.31 | Mid | ||
| RAD-N(4)H … CD-O5 | 2.39 +/− 0.31 | Mid | ||
| RAD-N(4)H … CD-O6 | 2.44 +/− 0.29 | Low | ||
| StrA–RADprot | RADprot-N(1)H … CD-SO4− | 2.29 +/− 0.46 | High | 1.94 (R) 2.59 (S) |
| RADprot-N(3)Hb … CD-O2′ | 2.12 +/− 0.28 | High | ||
| RADprot-N(3)Hb … CD-O3′ | 2.18 +/− 0.35 | High | ||
| RADprot-N(3)Ha … CD-O2′ | 2.14 +/− 0.29 | High | ||
| RADprot-N(3)Ha … CD-O3′ | 2.08 +/− 0.23 | High | ||
| RADprot-N(4)H … CD-O2′ | 2.12 +/− 0.28 | Mid | ||
| RADprot-N(4)H … CD-O3′ | 2.22 +/− 0.32 | Mid | ||
| StrB–RADprot | RADprot-N(3)Ha … CD-SO4− | 2.27 +/− 0.37 | High | 3.25 (R) 3.21 (S) (*) 3.65 (S) |
| RADprot-N(3)Hb … CD-SO4− | 2.23 +/− 0.35 | High | ||
| RADprot-N(4)H … CD-SO4− | 2.23 +/− 0.37 | High | ||
| RADprot-N(1)H … CD-O2′ | 2.23 +/− 0.41 | Low | ||
| (*)RADprot-N(1)H … CD-O3′ | (*) 2.54 +/− 0.44 | (*)High | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocian, W.; Bednarek, E.; Michalska, K. Explanation of the Formation of Complexes between Representatives of Oxazolidinones and HDAS-β-CD Using Molecular Modeling as a Complementary Technique to cEKC and NMR. Int. J. Mol. Sci. 2021, 22, 7139. https://doi.org/10.3390/ijms22137139
Bocian W, Bednarek E, Michalska K. Explanation of the Formation of Complexes between Representatives of Oxazolidinones and HDAS-β-CD Using Molecular Modeling as a Complementary Technique to cEKC and NMR. International Journal of Molecular Sciences. 2021; 22(13):7139. https://doi.org/10.3390/ijms22137139
Chicago/Turabian StyleBocian, Wojciech, Elżbieta Bednarek, and Katarzyna Michalska. 2021. "Explanation of the Formation of Complexes between Representatives of Oxazolidinones and HDAS-β-CD Using Molecular Modeling as a Complementary Technique to cEKC and NMR" International Journal of Molecular Sciences 22, no. 13: 7139. https://doi.org/10.3390/ijms22137139
APA StyleBocian, W., Bednarek, E., & Michalska, K. (2021). Explanation of the Formation of Complexes between Representatives of Oxazolidinones and HDAS-β-CD Using Molecular Modeling as a Complementary Technique to cEKC and NMR. International Journal of Molecular Sciences, 22(13), 7139. https://doi.org/10.3390/ijms22137139