
Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4


Abstract
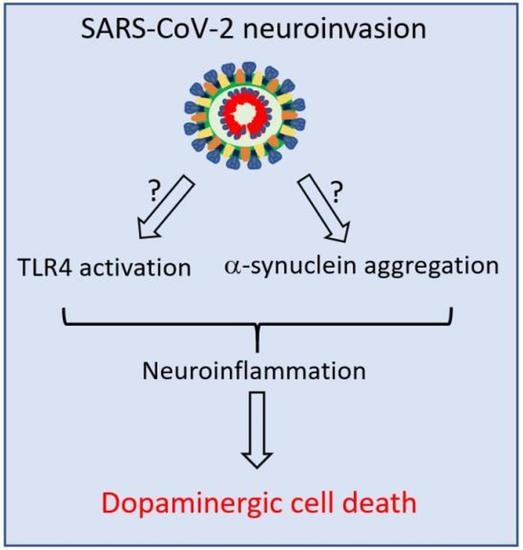
:
{kind=link}
{kind=link}
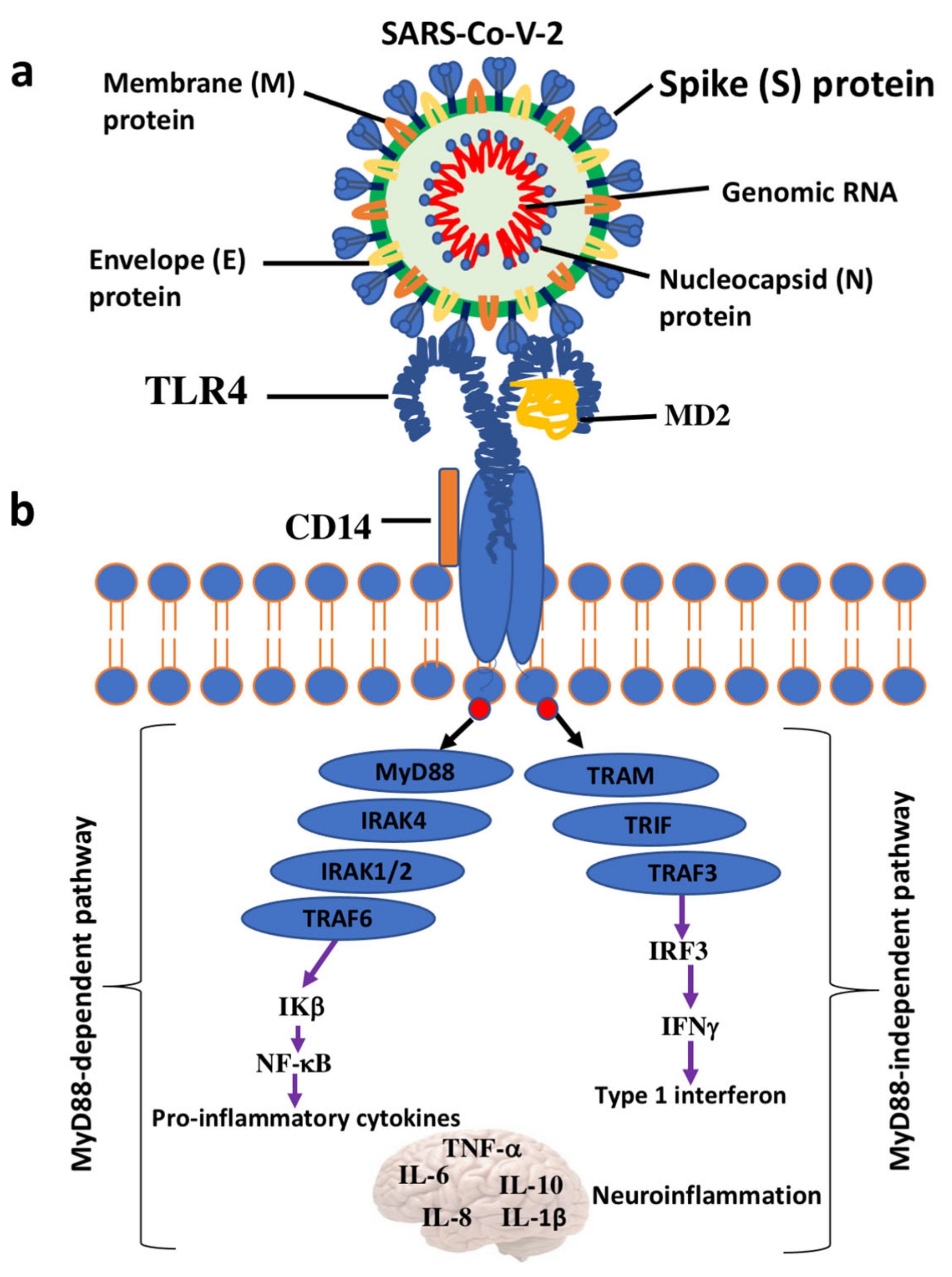{kind=link}
{kind=link}
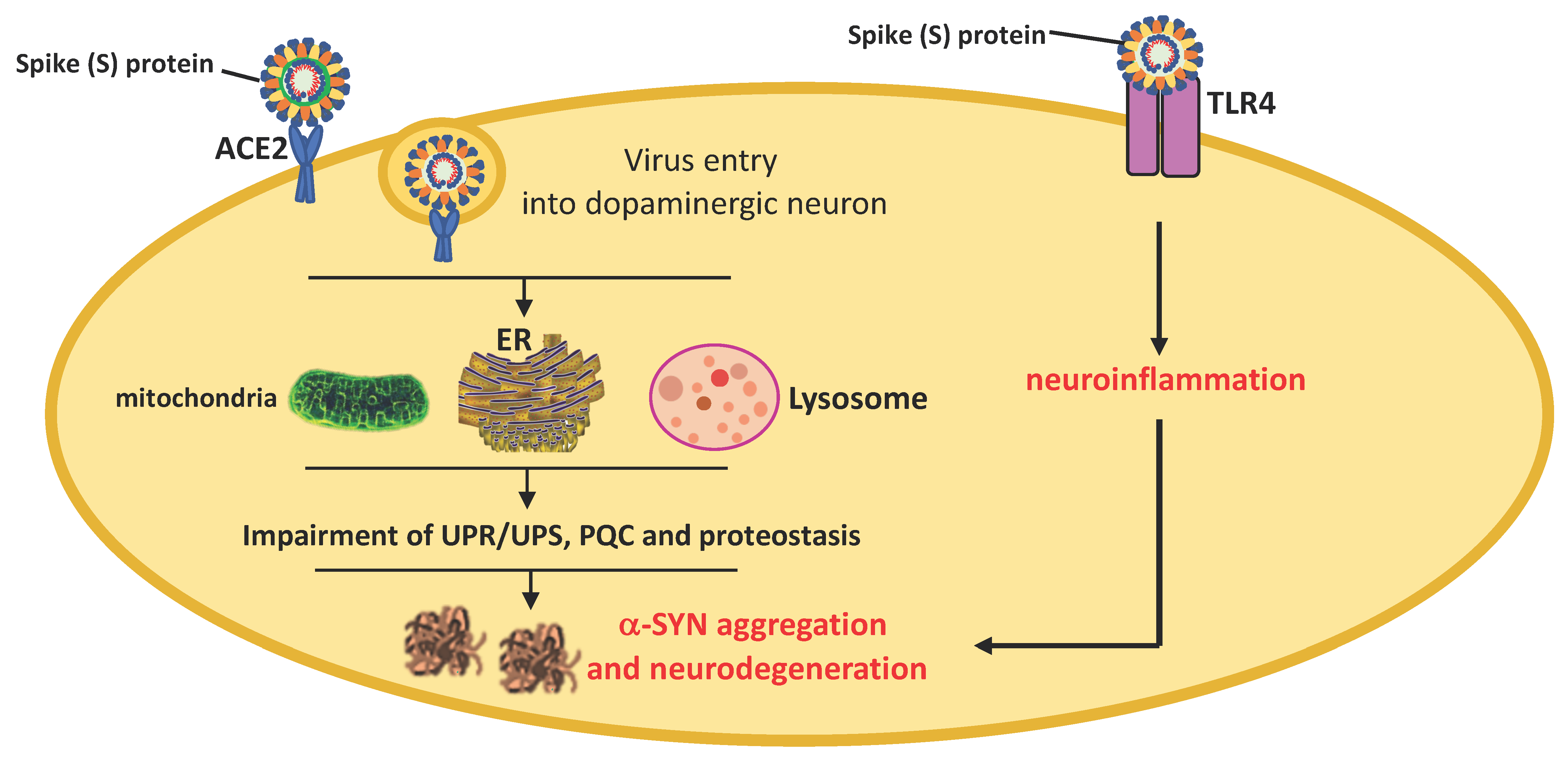
{kind=link}
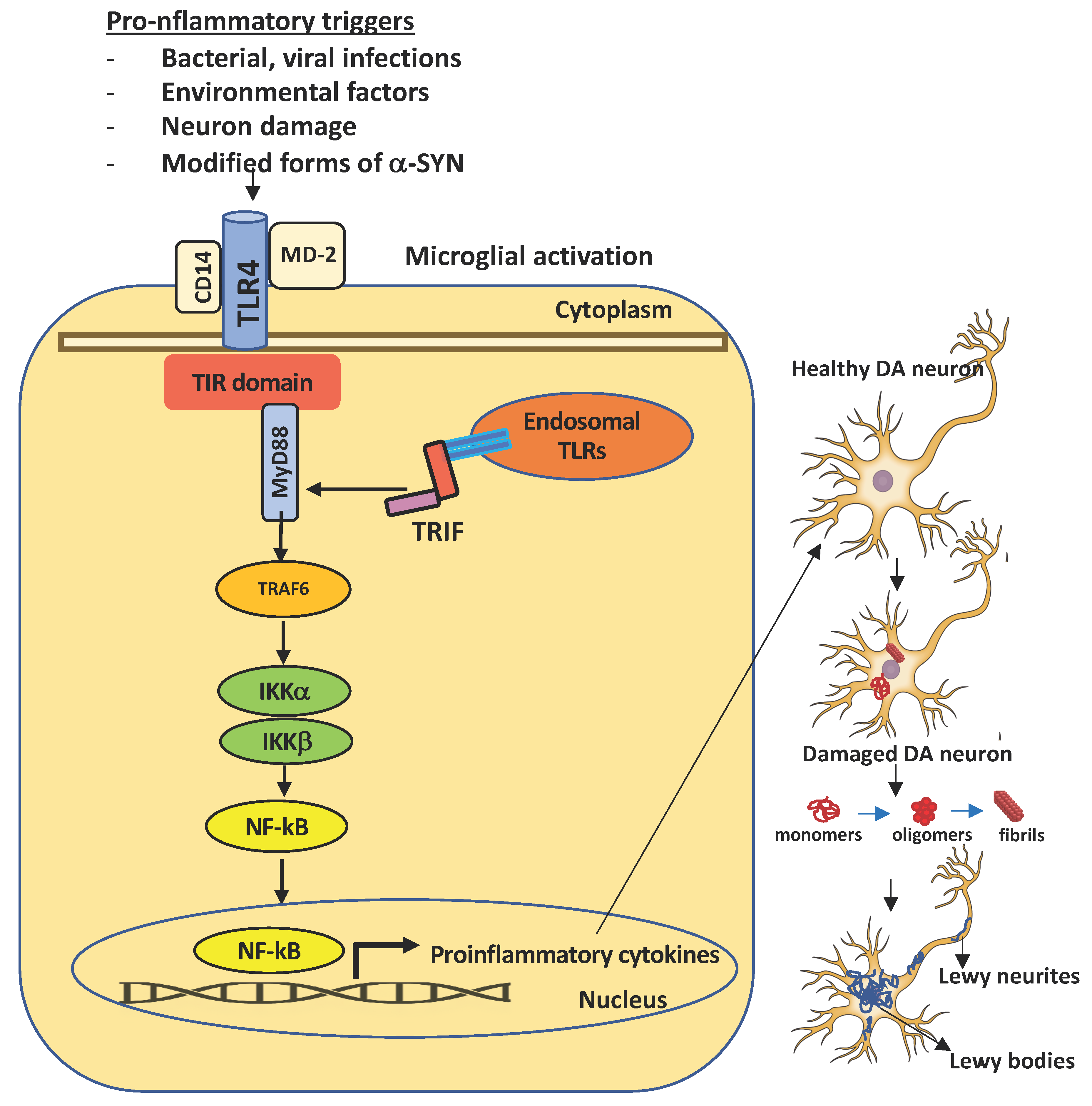
{kind=link}
1. Introduction
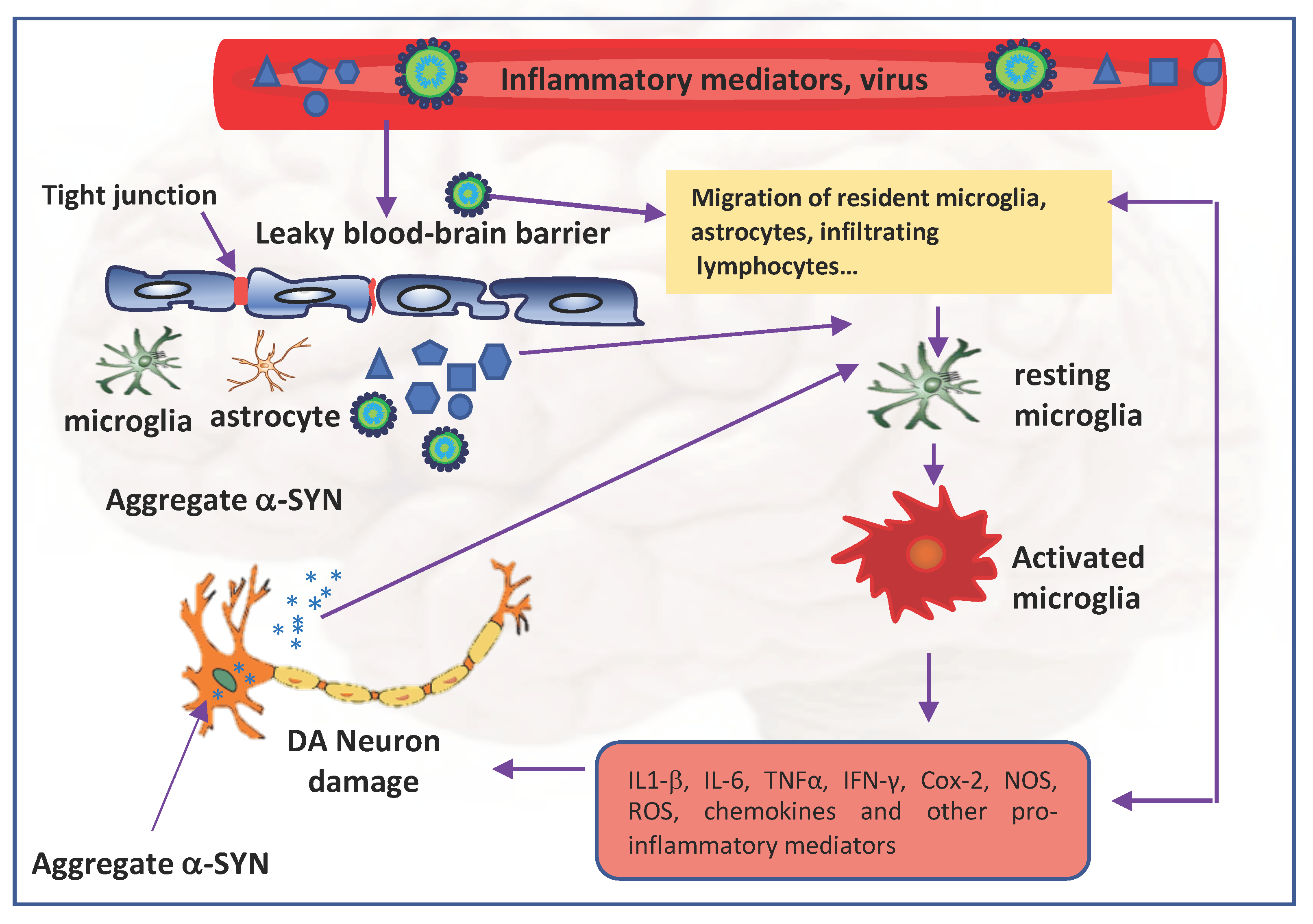
2. Neuroinflammation
3. Toll-Like Receptors
4. TLR4
5. SARS-CoV-2 Inflammatory Response: Role of TLRs
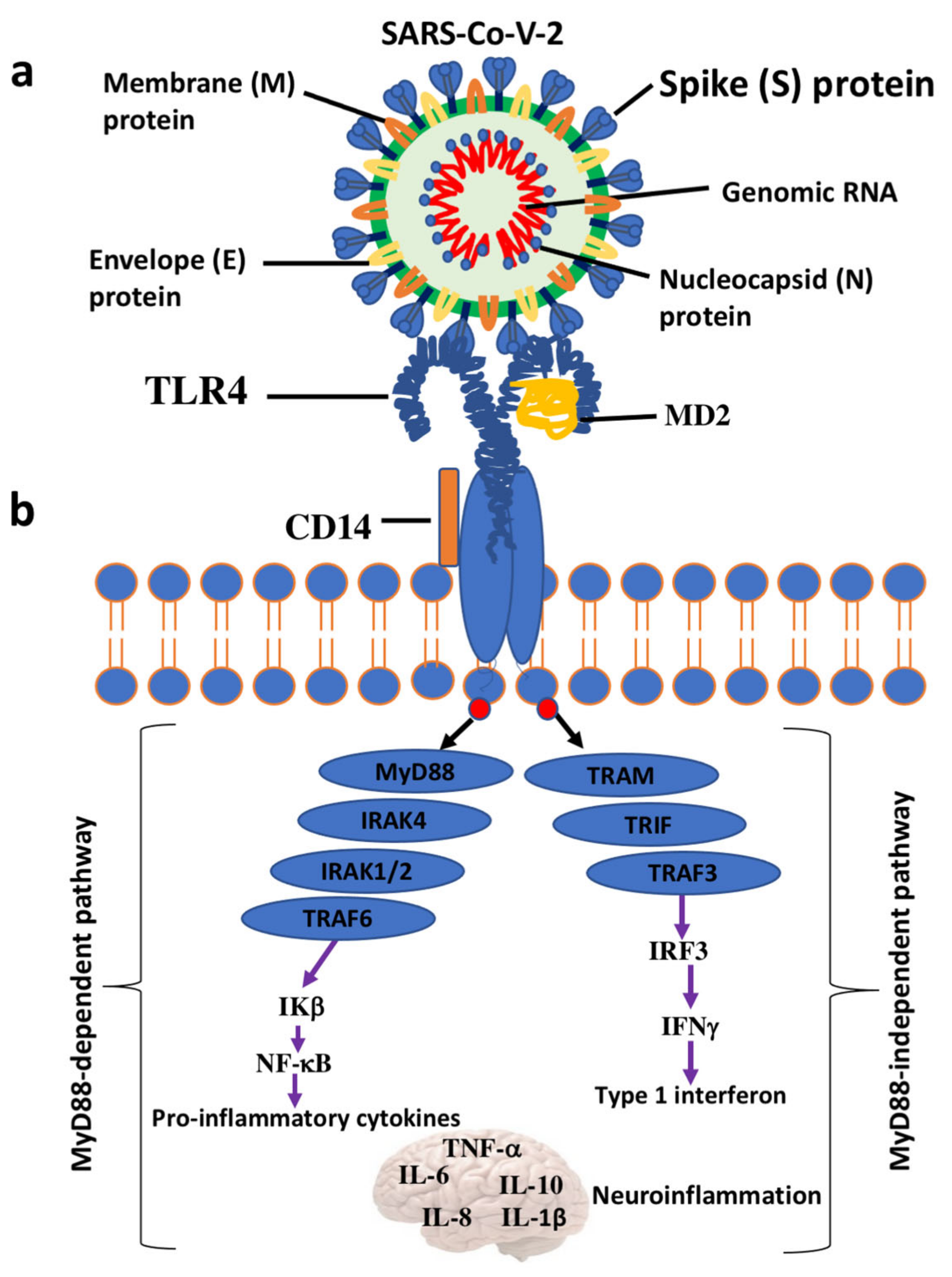
6. Interaction between the SARS-CoV-2 S protein and TLR4
7. TLR4 and PD Pathogenesis
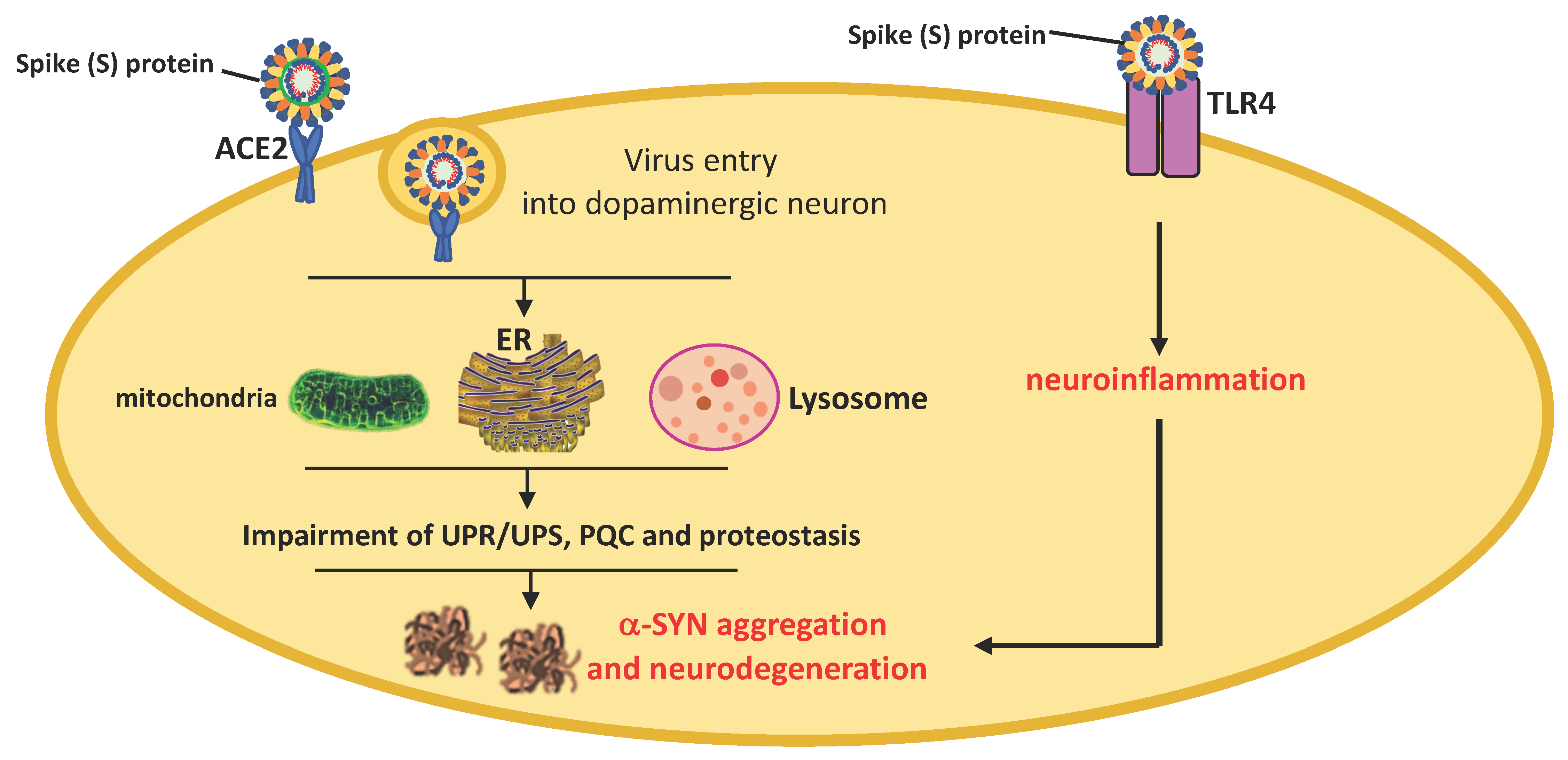
8. Neuroinvasive Potential of SARS-CoV-2 and PD Pathogenesis
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s disease. Subcell Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Bloem, B.R. The Parkinson pandemic—A call to action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Moustafa, A.A.; Chakravarthy, S.; Phillips, J.R.; Gupta, A.; Keri, S.; Polner, B. Motor symptoms in Parkinson’s disease: A unified framework. Neurosci. Biobehav. Rev. 2016, 68, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, W.L.; Stott, K.; He, X.; Wood, T.; Yang, S.; Kwok, J.; Hall, K.; Zhao, Y.; Tietz, O.; Aigbirhio, F.; et al. Systemic α-synuclein injection triggers selective neuronal pathology as seen in patients with Parkinson’s disease. Mol. Psychiatry 2021, 26, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Big, C.; Reineck, L.A.; Aronoff, D.M. Viral infections of the central nervous system: A case-based review. Clin. Med. Res. 2009, 7, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Swanson, P.A.; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Bookstaver, P.B.; Mohorn, P.L.; Shah, A.; Tesh, L.D.; Quidley, A.M.; Kothari, R.; Bland, C.M.; Weissman, S. Management of viral central nervous system infections: A primer for clinicians. J. Cent. Nerv. Syst. Dis. 2017, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Olsen, L.K.; Dowd, E.; McKernan, D.P. A role for viral infections in Parkinson’s etiology? Neuronal. Signal. 2018, 2, NS20170166. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yamada, T.; Nakajima, S.; Nakajima, K.; Yamamoto, T.; Okada, H. The substantia nigra is a major target for neurovirulent influenza A virus. J. Exp. Med. 1995, 181, 2161–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Kobylarek, D.; Kozubski, W. Infectious etiologies of parkinsonism: Pathomechanisms and clinical implications. Front. Neurol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Najjar, S.; Najjar, A.; Chong, D.J.; Pramanik, B.K.; Kirsch, C.; Kuzniecky, R.I.; Pacia, S.V.; Azhar, S. Central nervous system complications associated with SARS-CoV-2 infection: Integrative concepts of pathophysiology and case reports. J. Neuroinflamm. 2020, 17, 231. [Google Scholar] [CrossRef]
- Rocha, N.P.; de Miranda, A.S.; Teixeira, A.L. Insights into neuroinflammation in Parkinson’s disease: From biomarkers to anti-inflammatory based therapies. Biomed. Res. Int. 2015, 628192. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Refolo, V.; Stefanova, N. Neuroinflammation and glial phenotypic changes in alpha-synucleinopathies. Front. Cell Neurosci. 2019, 13, 263. [Google Scholar] [CrossRef] [Green Version]
- Drouin-Ouellet, J.; St-Amour, I.; Saint-Pierre, M.; Lamontagne-Proulx, J.; Kriz, J.; Barker, R.A.; Cicchetti, F. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 2014, 18, pyu103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.J.; Lucassen, P.J.; van Dam, A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariucci, G.; Pagiotti, R.; Galli, F.; Romani, L.; Conte, C. The potential role of toll-like receptor 4 in mediating dopaminergic cell loss and alpha-synuclein expression in the acute MPTP mouse model of Parkinson’s disease. J. Mol. Neurosci. 2018, 64, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Conte, C.; Roscini, L.; Sardella, R.; Mariucci, G.; Scorzoni, S.; Beccari, T.; Corte, L. Toll like receptor 4 affects the cerebral biochemical changes induced by MPTP treatment. Neurochem. Res. 2017, 42, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Gubernatorova, E.O.; Gorshkova, E.A.; Polinova, A.I.; Drutskaya, M.S. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020, 53, 13–24. [Google Scholar] [CrossRef]
- Alexaki, A.; Wigdahl, B. HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog. 2008, 4, e1000215. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Pino, P.; Kint, J.; Kiseljak, D.; Agnolon, V.; Corradin, G.; Kajava, A.V.; Rovero, P.; Dijkman, R.; den Hartog, G.; McLellan, J.S.; et al. Trimeric SARS-CoV-2 spike proteins produced from CHO cells in bioreactors are high-quality antigens. Processes 2020, 8, 1539. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lazartigues, E. Expression of ACE2 in human neurons supports the neuro-invasive potential of COVID-19 Virus. Cell. Mol. Neurobiol. 2020, 1–5. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 14, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; McCord, K.A.; Bui, D.T.; Bouwman, K.M.; Kitova, E.N.; Kumawat, D.; Daskhan, G.C.; Tomris, I.; Han, L.; Chopra, P.; et al. Sialic acid-dependent binding and viral entry of SARS-CoV-2. BioRxiv 2021. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Geng, X.; Tan, Y.; Li, Q.; Xu, C.; Xu, J.; Hao, L.; Zeng, Z.; Luo, X.; Liu, F.; et al. New understanding of the damage of SARS-CoV-2 infection outside the respiratory system. Biomed. Pharmacother. 2020, 127, 110195. [Google Scholar] [CrossRef]
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; McAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Denis, A.H.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Costa, F.F.; Rosário, W.R.; Ribeiro Farias, A.C.; de Souza, R.G.; Duarte Gondim, R.S.; Barroso, W.A. Metabolic syndrome and COVID-19: An update on the associated comorbidities and proposed therapies. Diabetes Metab. Syndr. 2020, 14, 809–814. [Google Scholar] [CrossRef]
- Del Turco, S.; Vianello, A.; Ragusa, R.; Caselli, C.; Basta, G. COVID-19 and cardiovascular consequences: Is the endothelial dysfunction the hardest challenge? Thromb. Res. 2020, 196, 143–151. [Google Scholar] [CrossRef]
- Roberts, K.A.; Colley, L.; Agbaedeng, T.A.; Ellison-Hughes, G.M.; Ross, M.D. Vascular Manifestations of COVID-19-Thromboembolism and Microvascular Dysfunction. Front. Cardiovasc. Med. 2020, 7, 598400. [Google Scholar] [CrossRef]
- The Severe Covid-19 GWAS Group. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Joecks, S.; Burkart, S.S.; Zander, D.Y. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kB. BioRxiv 2020. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef]
- Chavan, S.S.; Pavlov, V.A.; Tracey, K.J. Mechanisms and therapeutic relevance of neuro-immune communication. Immunity 2017, 46, 927–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Standaert, D.G. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef]
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.; Classen, J.; Dresel, C.; Jost, W.; Krüger, R.; Müller, T.; Reichmann, H.; et al. α-Synuclein in Parkinson’s disease: Causal or bystander? J. Neural Transm. 2019, 126, 815–840. [Google Scholar] [CrossRef]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2019, 126, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Riederer, P. The nigral coup in Parkinson’s disease by α-synuclein and its associated rebels. Cells 2021, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Llerena, C.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 235–263. [Google Scholar] [CrossRef]
- Spargo, E.; Everall, I.P.; Lantos, P.L. Neuronal loss in the hippocampus in Huntington’s disease: A comparison with HIV infection. J. Neurol. Neurosurg. Psychiatry 1993, 56, 487–491. [Google Scholar] [CrossRef]
- Giralt, A.; Saavedra, A.; Alberch, J.; Pérez-Navarro, E. Cognitive dysfunction in Huntington’s disease: Humans, mouse models and molecular mechanisms. J. Huntingt. Dis. 2012, 1, 155–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef] [Green Version]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T cells get ready to tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Sevenich, L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front. Immunol. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Carrasco, J.; Hong, S. The jekyll and hyde of TREM2. Trends Neurosci. 2020, 43, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Augusto-Oliveira, M.; Arrifano, G.P.; Lopes-Araújo, A.; Santos-Sacramento, L.; Takeda, P.Y.; Anthony, D.C.; Malva, J.O.; Crespo-Lopez, M.E. What do microglia really do in healthy adult brain? Cells 2019, 8, 1293. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Leak, R.K.; Hu, X. Neurotransmitter receptors on microglia. Stroke Vasc. Neurol. 2016, 1, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Ann. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Chagas, L.; Sandre, P.C.; Ribeiro, E.; Ribeiro, N.; Marcondes, H.; Oliveira Silva, P. environmental signals on microglial function during brain development, neuroplasticity, and disease. Int. J. Mol. Sci. 2020, 21, 2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Robinton, D.; Stevens, B. Microglia and the brain: Complementary partners in development and disease. Ann. Rev. Cell Dev. Biol. 2018, 34, 523–544. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Med. Inflamm. 2013, 480739. [Google Scholar] [CrossRef] [Green Version]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Di Paolo, T.; Tremblay, M.E. Microglial implication in Parkinson’s disease: Loss of beneficial physiological roles or gain of inflammatory functions? Front. Cell Neurosci. 2018, 12, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.Y.; Chao, Y.X.; Rötzschke, O.; Tan, E.K. New insights into immune-mediated mechanisms in Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 9302. [Google Scholar] [CrossRef]
- Bartels, A.L.; Leenders, K.L. Cyclooxygenase and neuroinflammation in Parkinsons disease neurodegeneration. Curr. Neuropharmacol. 2010, 8, 62. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Artaud, F.; Cormier-Dequaire, F.; Rascol, O.; Durif, F.; Derkinderen, P. Examining the reserve hypothesis in Parkinson’s disease: A longitudinal study. Mov. Disord. 2019, 34, 1663–1671. [Google Scholar] [CrossRef]
- Pajares, M.I.; Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation—An Interesting interplay in Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Kouli, A.; Camacho, M.; Allinson, K.; Williams-Gray, C.H. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol. Commun. 2020, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.V. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 2000, 12, 13–19. [Google Scholar] [CrossRef]
- May, M.J.; Ghosh, S. Signal transduction through NF-kappa B. Immunol. Today 1998, 19, 80–88. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological role of toll-like receptor-4 in the brain. J. Neuroimmunol. 2014, 268, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Molteni, M.; Gemma, S.; Rossetti, C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.D.; Choi, M.L.; Ryten, M.; Hopkins, L.; Drews, A.; Botía, J.A.; Iljina, M.; Rodrigues, M.; Gagliano, S.A.; Gandhi, S.; et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2019, 137, 103–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.H.; Jeon, M.T.; Leem, E.; Won, S.Y.; Jeong, K.H.; Park, S.J.; McLean, C.; Lee, S.J.; Jin, B.K.; Jung, U.J.; et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: A potential pathogenic mechanism in Parkinson’s disease. Sci. Rep. 2015, 5, 14764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [Green Version]
- Panaro, M.A.; Lofrumento, D.D.; Saponaro, C.; De Nuccio, F.; Cianciulli, A.; Mitolo, V.; Nicolardi, G. Expression of TLR4 and CD14 in the central nervous system (CNS) in a MPTP mouse model of Parkinson’s-like disease. Immunopharmacol. Immunotoxicol. 2008, 30, 729–740. [Google Scholar] [CrossRef]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Han, X.; Xue, L.; Zhu, K.; Liu, H.; Xie, A. Association of TLR4 gene polymorphisms with sporadic Parkinson’s disease in a Han Chinese population. Neurol. Sci. 2015, 36, 1659–1665. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [Green Version]
- Viceconte, N.; Burguillos, M.A.; Herrera, A.J.; De Pablos, R.M.; Joseph, B.; Venero, J.L. Neuromelanin activates proinflammatory microglia through a caspase-8-dependent mechanism. J. Neuroinflamm. 2015, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouli, A.; Horne, C.B.; Williams-Gray, C.H. Toll-like receptors and their therapeutic potential in Parkinson’s disease and α-synucleinopathies. Brain Behav. Immun. 2019, 81, 41–51. [Google Scholar] [CrossRef]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targ. 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Campolo, C.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef]
- Loiarro, M.; Ruggiero, V.; Sette, C. Targeting TLR/IL-1R signalling in human diseases. Mediat. Inflamm. 2010, 674363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef]
- Rajaiah, R.; Perkins, D.J.; Ireland, D.D.C.; Vogel, S.N. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, 8391–8396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Deng, S.L.; Lian, Z.X.; Yu, K. Roles of toll-like receptors in nitroxidative stress in mammals. Cells 2019, 8, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Targ. Ther. 2020, 5. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome in times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. Innate receptor activation patterns Involving TLR and NLR synergisms in COVID-19, ALI/ARDS and sepsis cytokine storms: A review and model making novel predictions and therapeutic suggestions. Int. J. Mol. Sci. 2021, 22, 2108. [Google Scholar] [CrossRef] [PubMed]
- Khanolkar, A.; Hartwig, S.M.; Haag, B.A.; Meyerholz, D.K.; Harty, J.T.; Varga, S.M. Toll-like receptor 4 deficiency increases disease and mortality after mouse hepatitis virus type 1 infection of susceptible C3H mice. J. Virol. 2009, 83, 8946–8956. [Google Scholar] [CrossRef] [Green Version]
- Petruk, G.; Puthia, M.; Petrlova, J.; Samsudin, F.; Strömdahl, A.C.; Cerps, S. SARS-CoV-2 Spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell. Biol. 2021, 12, 916–932. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Mallick, B.; Sharma, G.; Lee, S.S.; Chakraborty, C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect. Genet. Evol. 2020, 85, 104587. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Jin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to Increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat. Inflamm. 2021. [Google Scholar] [CrossRef] [PubMed]
- Brandão, S.C.S.; Ramos, J.O.X.; Dompieri, L.T.; Godoi, E.T.A.M.; Figueiredo, J.L.; Sarinho, E.S.C.; Chelvanambi, S.; Aikawa, M. Is toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2020, 58, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J. Korean Med. Sci. 2020, 35, e343. [Google Scholar] [CrossRef] [PubMed]
- Anderson, F.L.; von Herrmann, K.M.; Andrew, A.S.; Kuras, Y.I.; Young, A.L.; Scherzer, C.R.; Hickey, W.F.; Lee, S.L.; Havrda, M.C. Plasma-borne indicators of inflammasome activity in Parkinson’s disease patients. NPJ Parkinsons Dis. 2021, 7. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Beraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Neitzke, K.; Ton, V.; Chasovskikh, S.; Mhyre, T.; Maguire-Zeiss, K. α-synuclein alters toll-like receptor expression. Front. Naurosci. 2011, 5, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, L.C.; Finkelstein, D.I.; Bush, A.I.; Evans, A.H.; Barnham, K.J. Parkinsonism as a third wave of the COVID-19 pandemic? J. Parkinsons Dis. 2020, 10, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Ter Meulen, V. Coronaviruses: A challenge of today and a call for extended human postmortem brain analyses. J. Neural Transm. 2020, 127, 1217–1228. [Google Scholar] [CrossRef]
- Matsuda, K.; Park, C.H.; Sunden, Y.; Kimura, T.; Ochiai, K.; Kida, H.; Umemura, T. The vagus nerve is one route of transneural invasion for intranasally inoculated influenza a virus in mice. Vet. Pathol. 2004, 41, 101–107. [Google Scholar] [CrossRef]
- Chasey, D.; Alexander, D.J. Morphogenesis of avian infectious bronchitis virus in primary chick kidney cells. Arch. Virol. 1976, 52, 101–111. [Google Scholar] [CrossRef]
- Guo, Y.; Korteweg, C.; McNutt, M.A.; Gu, J. Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008, 133, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Pesce, M.; Seguella, L.; Sanseverino, W.; Lu, J.; Sarnelli, G. Can the enteric nervous system be an alternative entrance door in SARS-CoV2 neuroinvasion? Brain Behav. Immun. 2020, 87, 93–94. [Google Scholar] [CrossRef]
- Beyrouti, R.; Adams, M.E.; Benjamin, L.; Cohen, H.; Farmer, S.F.; Goh, Y.Y.; Humphries, F.; Jäger, H.R.; Losseff, N.A.; Perry, R.J.; et al. Characteristics of ischaemic stroke associated with COVID-19. J. Neurol. Neurosurg. Psychiatry 2020, 91, 889–891. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef]
- Jones, A.R.; Overly, C.C.; Sunkin, S.M. The allen brain atlas: 5 years and beyond. Nat. Rev. Neurosci. 2019, 10, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Merello, M.; Bhatia, K.P.; Obeso, J.A. SARS-CoV-2 and the risk of Parkinson’s disease: Facts and fantasy. Lancet Neurol. 2021, 20, 94–95. [Google Scholar] [CrossRef]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood–Brain barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Vieites Prado, A.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Brison, E.; Meessen-Pinard, M.; Talbot, P.J. Neuroinvasive and neurotropic human respiratory coronaviruses: Potential neurovirulent agents in humans. Adv. Exp. Med. Biol. 2014, 807, 75–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bale, J.F., Jr. Virus and immune-mediated encephalitides: Epidemiology, diagnosis, treatment, and prevention. Pediatr. Neurol. 2015, 53, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailles, A.; Stahl, J.P.; Bloch, K.C. Update and new insights in encephalitis. Clin. Microbiol. Infect. 2017, 23, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Bohmwald, K.; Gálvez, N.M.S.; Ríos, M.; Kalergis, A.M. Neurologic alterations due to respiratory virus infections. Front. Cell Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Jiangm, D.; Huangm, J.T. SARS-CoV-2 detected in cerebrospinal fluid by PCR in a case of COVID-19 encephalitis. Brain Behav. Immun. 2020, 87, 149. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Mahajan, S.D. SARS-COV2 alters blood brain barrier integrity contributing to neuro-inflammation. J. Neuroimmune Pharmacol. 2021, 16, 4–6. [Google Scholar] [CrossRef]
- Alquisiras-Burgos, I.; Peralta-Arrieta, I.; Alonso-Palomares, L.A. Neurological complications associated with the blood-brain barrier damage induced by the inflammatory response during SARS-CoV-2 infection. Mol. Neurobiol. 2020, 58, 1–16. [Google Scholar] [CrossRef]
- Kustin, T.; Ling, G.; Sharabi, S.; Ram, D.; Friedman, N.; Zuckerman, N.; Bucris, E.D.; Glatman-Freedman, A.; Stern, A.; Mandelboim, M. A method to identify respiratory virus infections in clinical samples using next-generation sequencing. Sci. Rep. 2019, 9, 2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzini, A.; Padovani, A. Lifting the mask on neurological manifestations of COVID-19. Nat. Rev. Neurol. 2020, 16, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Guerrero, A.; Laespada-García, M.I.; Gómez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rábano-Suárez, P.; Álvarez-Torres, E. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.A. Case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Faber, I.; Brandão, P.R.; Menegatti, F.; de Carvalho Bispo, D.D.; Maluf, F.B.; Cardoso, F. Coronavirus disease 2019 and Parkinsonism: A non-post-encephalitic case. Mov. Disord. 2020, 35, 1721–1722. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.E.; Ho, A.P.; Ho, M.A.; Ho, E.C. Case report of familial COVID-19 cluster associated with high prevalence of anosmia, ageusia, and gastrointestinal symptoms. IDCases 2020, 22, e00975. [Google Scholar] [CrossRef] [PubMed]
- Tarakad, A.; Jankovic, J. Anosmia and ageusia in Parkinson’s disease. Int. Rev. Neurobiol. 2017, 133, 541–556. [Google Scholar] [CrossRef]
- Sepehrinezhad, A.; Shahbazi, A.; Negah, S.S. COVID-19 virus may have neuroinvasive potential and cause neurological complications: A perspective review. J. Neurovirol. 2020, 26, 324–329. [Google Scholar] [CrossRef]
- Espinosa, P.S.; Rizvi, Z.; Sharma, P.; Hindi, F.; Filatov, A. Neurological complications of Coronavirus Disease (COVID-19): Encephalopathy, MRI brain and cerebrospinal fluid findings: Case 2. Cureus 2020, 12, e7930. [Google Scholar] [CrossRef]
- Zhang, J.; Hao, Y.; Ou, W.; Ming, F.; Liang, G.; Cai, Q.; Dong, S.; Hu, S.; Wang, W.; Wei, S. Serum interleukin-6 is an indicator for severity in 901 patients with SARS-CoV-2 infection: A cohort study. J. Transl. Med. 2020, 18, 406–413. [Google Scholar] [CrossRef]
- Pereira, J.R.; Santos, L.V.D.; Santos, R.M.S.; Campos, A.L.F.; Pimenta, A.L.; de Oliveira, M.S. IL-6 serum levels are elevated in Parkinson’s disease patients with fatigue compared to patients without fatigue. J. Neurol. Sci. 2016, 370, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Veselý, B.; Dufek, M.; Thon, V.; Brozman, M.; Királová, S.; Halászová, T.; Koritáková, E.; Rektor, I. Interleukin 6 and complement serum level study in Parkinson’s disease. J. Neural Transm. 2018, 125, 875–881. [Google Scholar] [CrossRef]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-synuclein expression restricts RNA viral infections in the brain. J. Virol. 2015, 90, 2767–2782. [Google Scholar] [CrossRef] [Green Version]
- Ait Wahmane, S.; Achbani, A.; Ouhaz, Z.; Elatiqi, M.; Belmouden, A.; Nejmeddine, M. The possible protective role of α-synuclein against severe acute respiratory syndrome Coronavirus 2 infections in patients with Parkinson’s disease. Mov. Disord. 2020, 35, 1293–1294. [Google Scholar] [CrossRef]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-protein and α-synuclein accelerate amyloid formation. BioRxiv 2021. [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Chaná-Cuevas, P.; Salles-Gándara, P.; Rojas-Fernandez, A.; Salinas-Rebolledo, C.; Milán-Solé, A. The potential role of SARS-COV-2 in the pathogenesis of Parkinson’s disease. Front. Neurol. 2020, 11, 1044. [Google Scholar] [CrossRef] [PubMed]
- Tulisiak, C.T.; Mercado, G.; Peelaerts, W.; Brundin, L.; Brundin, P. Can infections trigger alpha-synucleinopathies? Prog. Mol. Biol. Transl. Sci. 2019, 168, 299–322. [Google Scholar] [CrossRef]
- Labrie, V.; Brundin, P. Alpha-synuclein to the rescue: Immune cell recruitment by alpha-synuclein during gastrointestinal infection. J. Innate Immun. 2017, 9, 80. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. https://doi.org/10.3390/ijms22137135
Conte C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. International Journal of Molecular Sciences. 2021; 22(13):7135. https://doi.org/10.3390/ijms22137135
Chicago/Turabian StyleConte, Carmela. 2021. "Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4" International Journal of Molecular Sciences 22, no. 13: 7135. https://doi.org/10.3390/ijms22137135
APA StyleConte, C. (2021). Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. International Journal of Molecular Sciences, 22(13), 7135. https://doi.org/10.3390/ijms22137135