
Comprehensive Molecular Dissection of Dermatophilus congolensis Genome and First Observation of tet(Z) Tetracycline Resistance

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Results
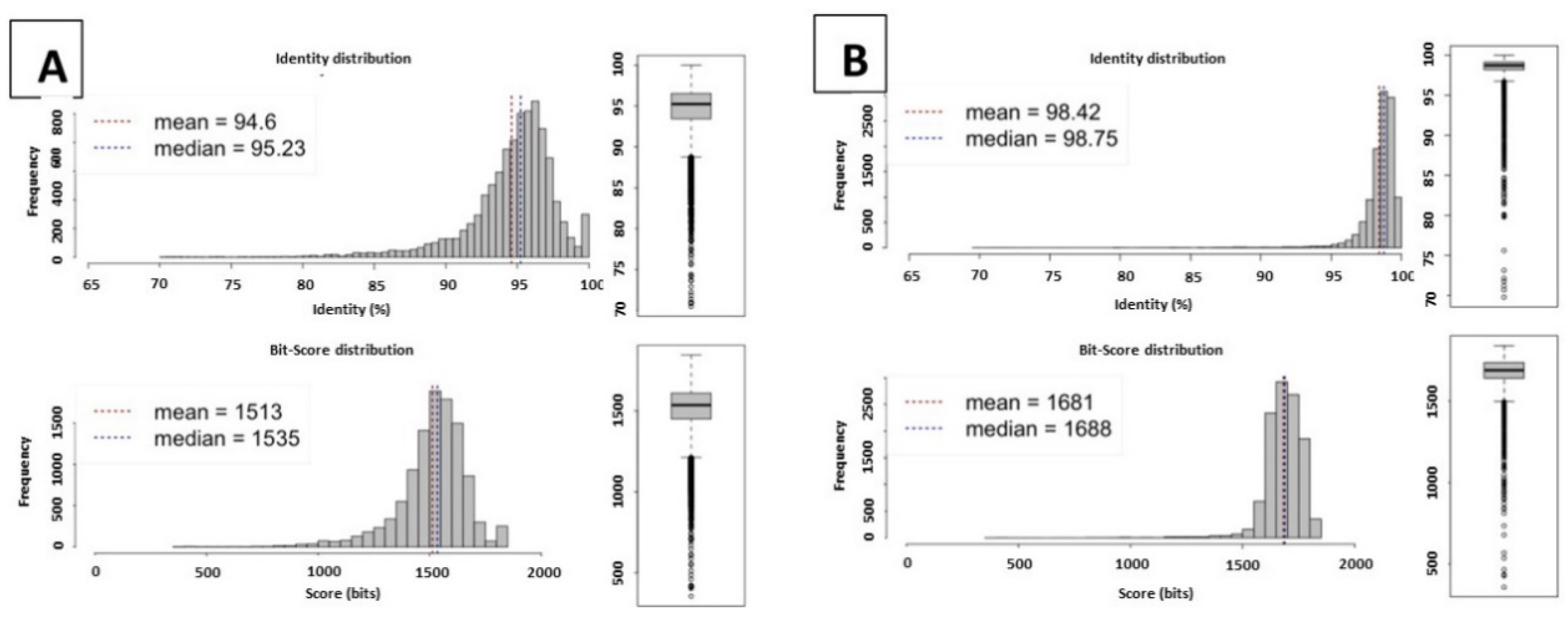
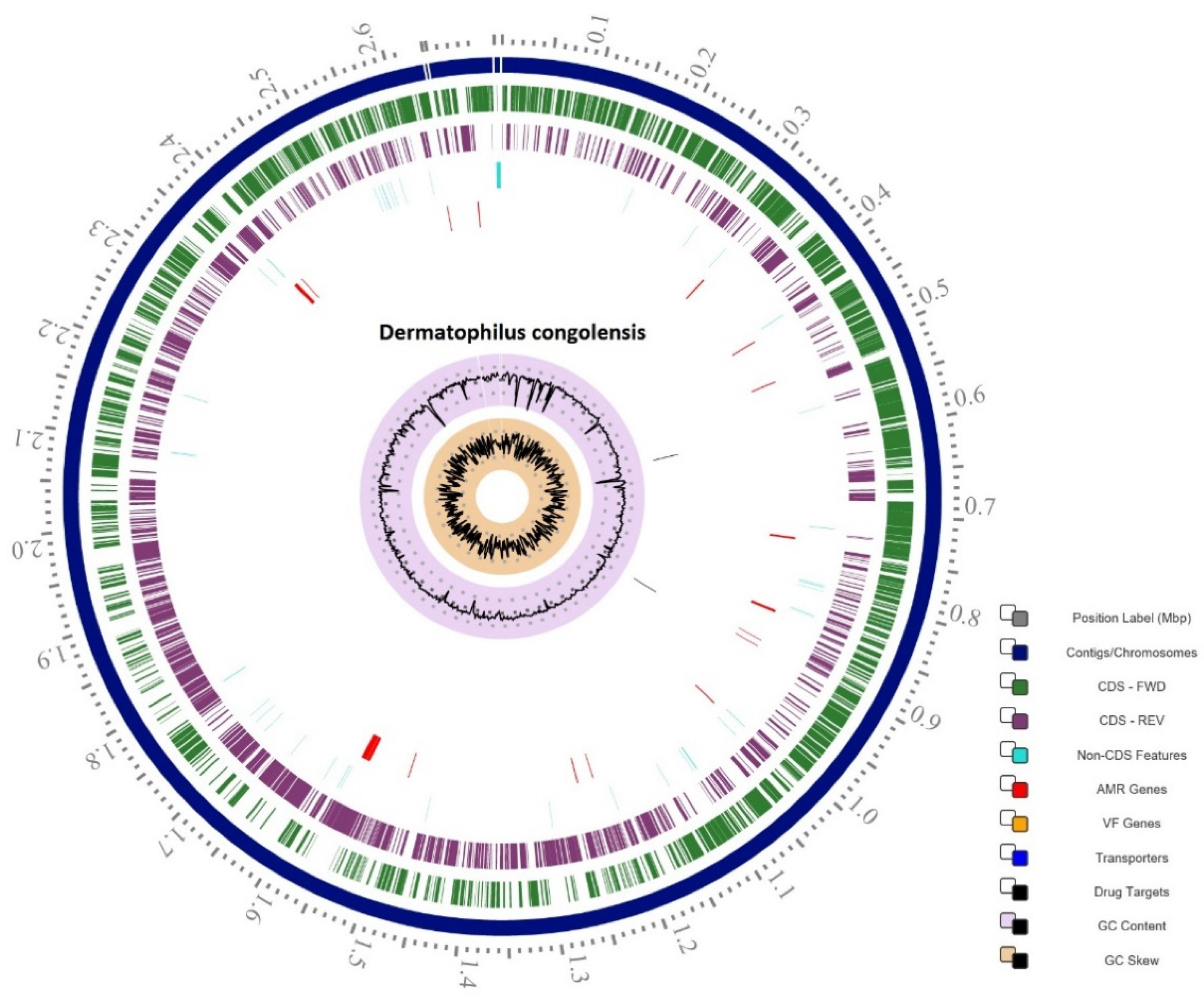
2.1. Genome Statistics
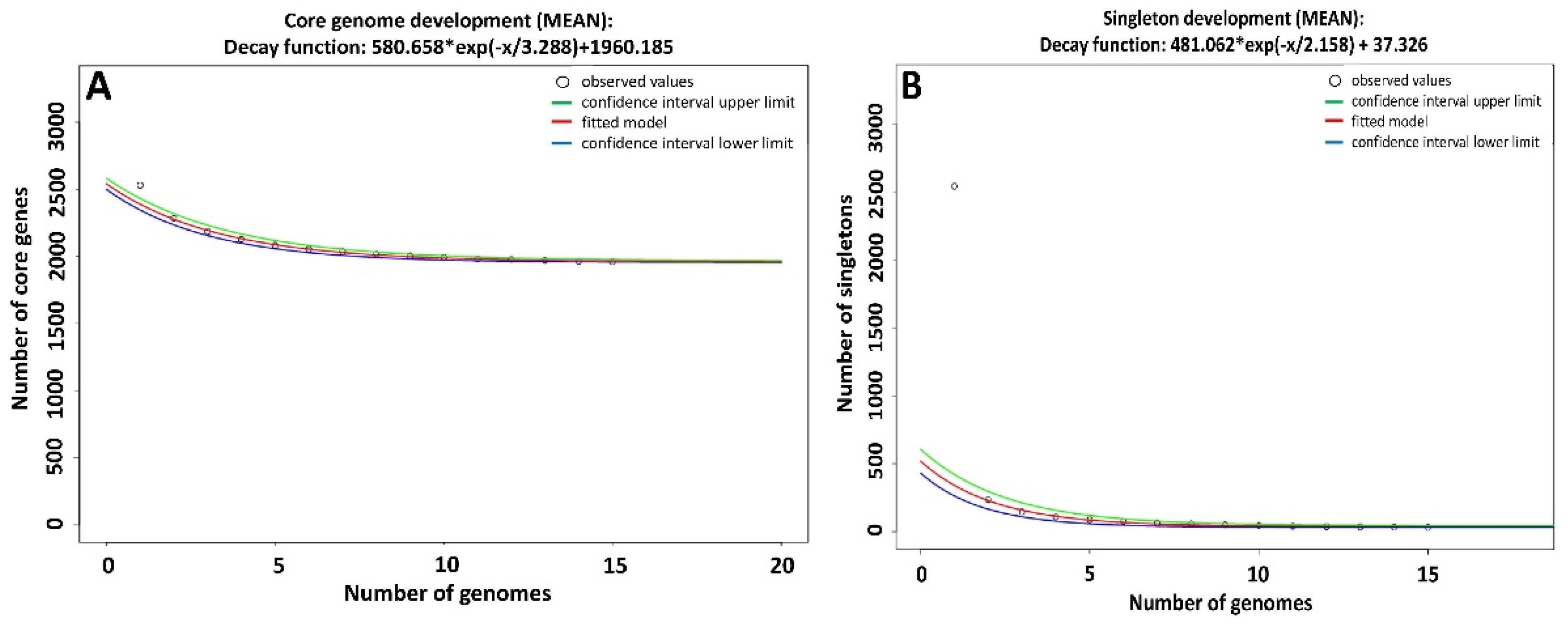
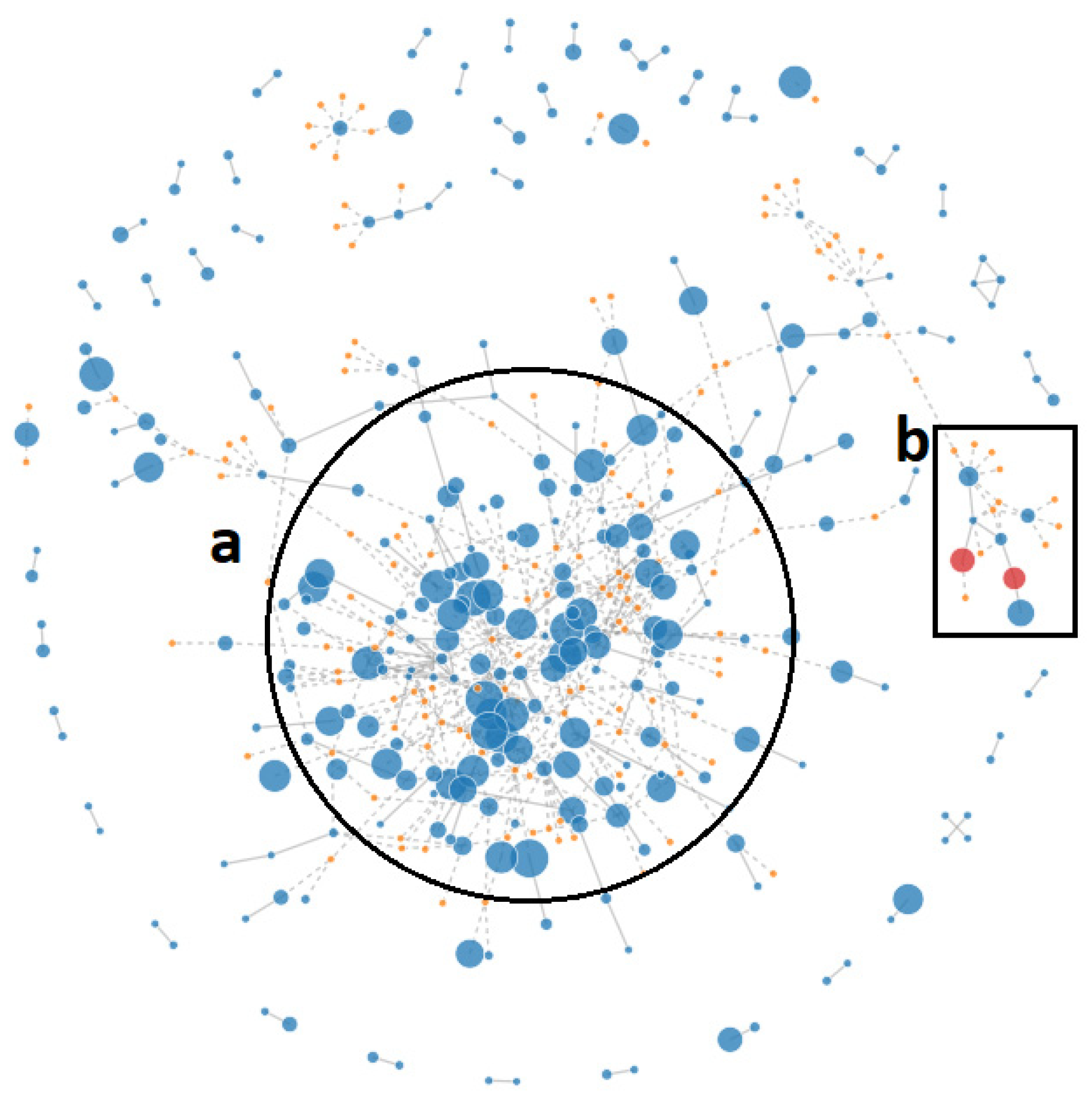
2.2. Pan-Genome Analysis
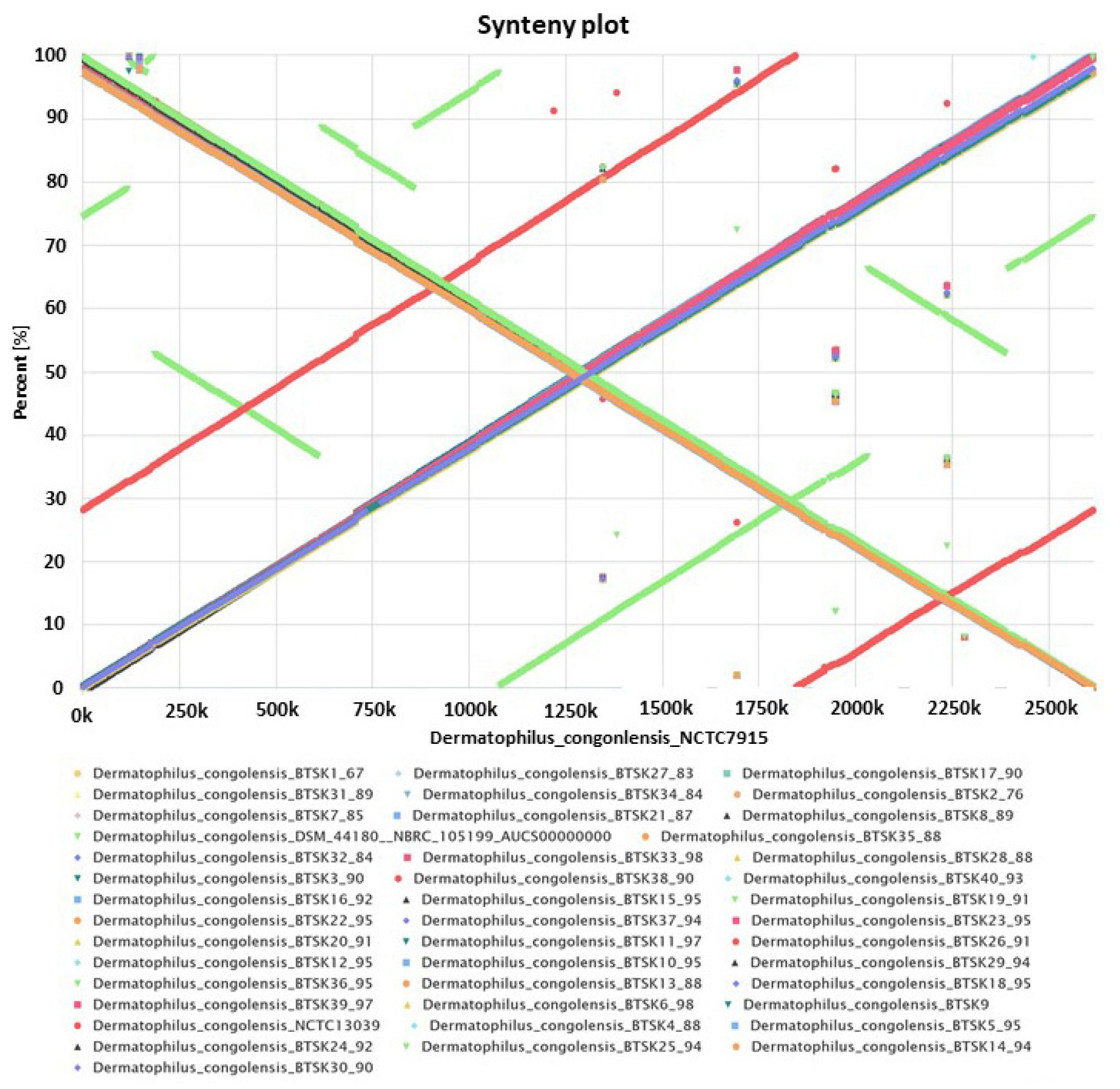
2.3. Synteny Analysis
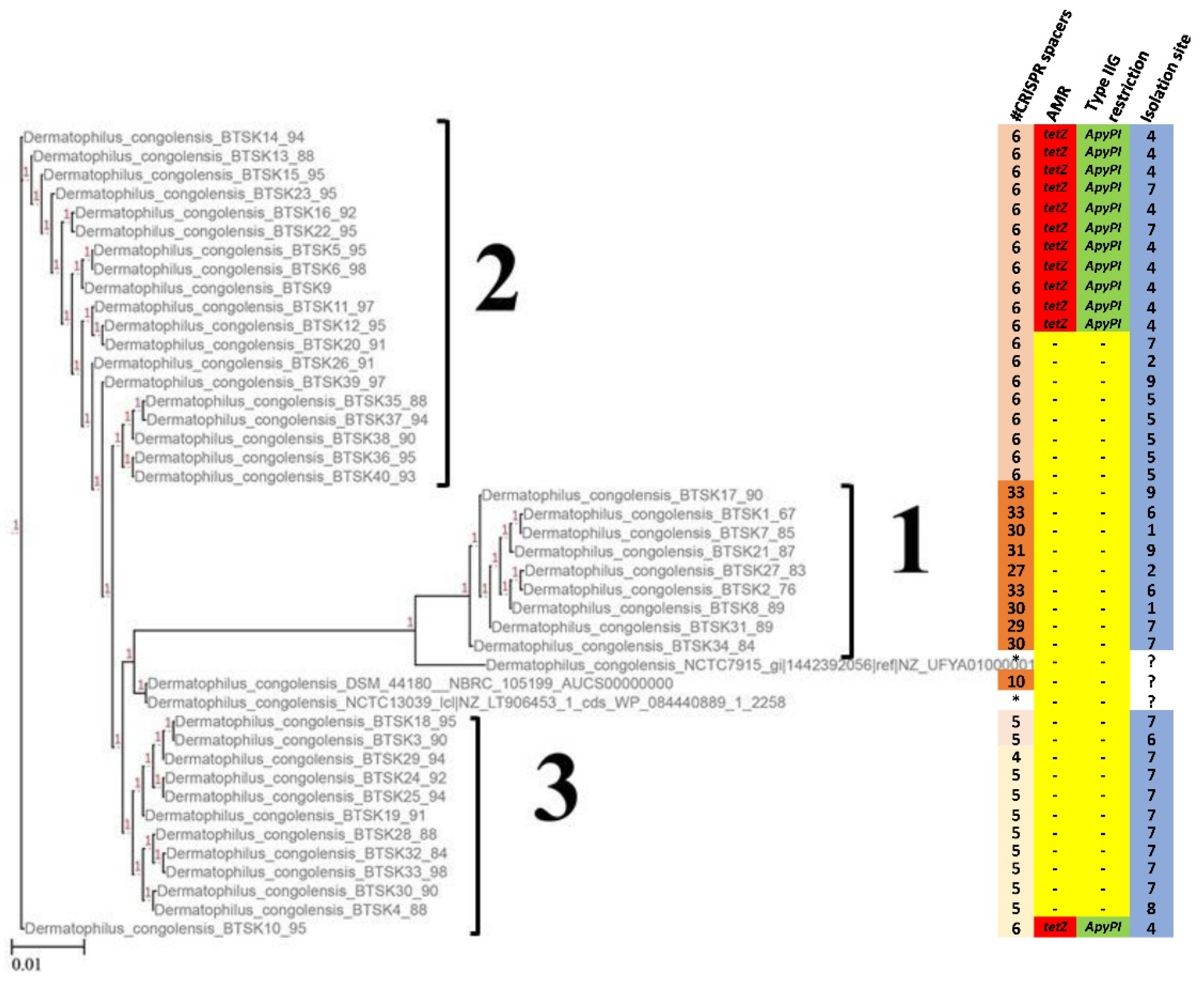
2.4. Phylogenetic Analysis
2.5. Antimicrobial Resistance Mechanisms
2.6. Prophages
2.7. Virulence Factors
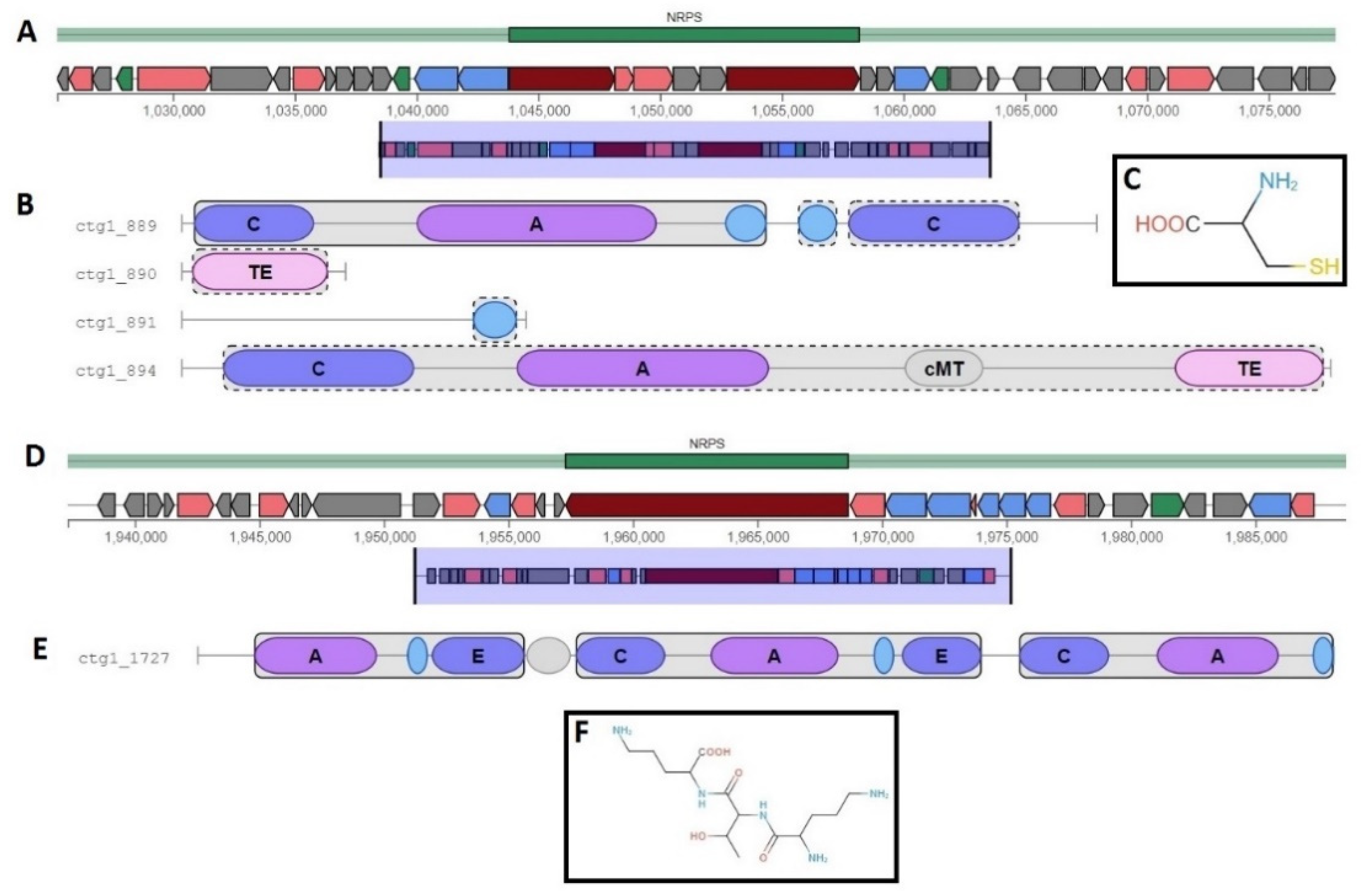
2.8. Metabolism and Secondary Metabolites
3. Discussion
4. Material and Methods
4.1. Ethics Statement
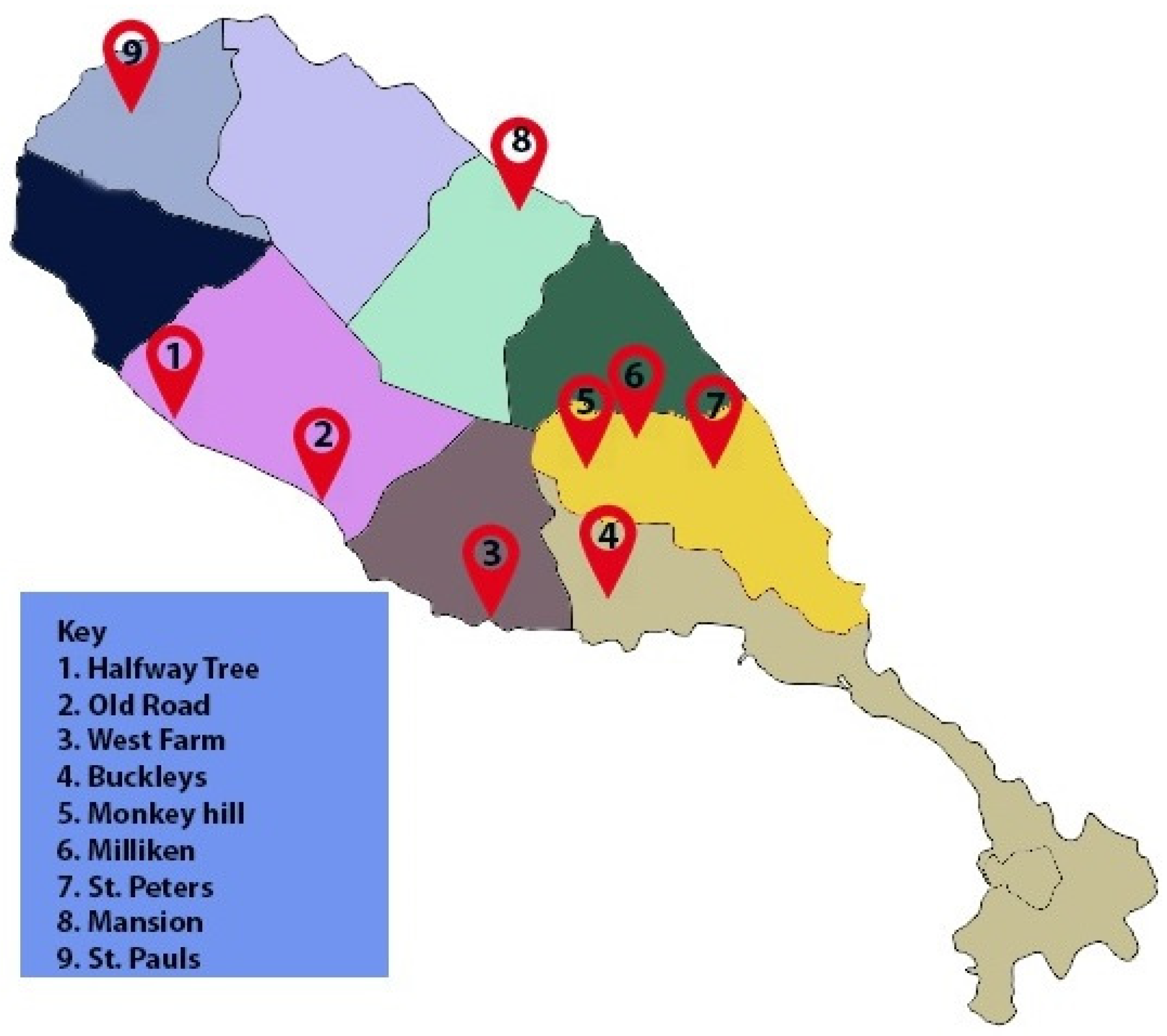
4.2. Isolation of D. congolensis from Field Samples
4.3. DNA Isolation and Sequencing
4.4. Genome Assembly, Annotation, and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amor, A.; Enríquez, A.; Corcuera, M.T.; Toro, C.; Herrero, D.; Baquero, M. Is Infection by Dermatophilus congolensis Underdiagnosed? J. Clin. Microbiol. 2011, 49, 449–451. [Google Scholar] [CrossRef]
- Ojong, B.W.; Saccà, E.; Bessong, P.; Piasentier, E. Prevalence of bovine dermatophilosis and disease-associated alleles in zebu Goudali cattle and their Italian Simmental crosses ranching in the western highland plateau savannah of Cameroon. Trop. Anim. Health Prod. 2016, 48, 1329–1335. [Google Scholar] [CrossRef]
- Ambrose, N.C. The pathogenesis of dermatophilosis. Trop. Anim. Health Prod. 1996, 28, 29S–37S. [Google Scholar] [CrossRef]
- Awa, D.N.; Achukwi, M.D. Livestock pathology in the central African region: Some epidemiological considerations and control strategies. Anim. Health Res. Rev. 2010, 11, 235–244. [Google Scholar] [CrossRef]
- Maillard, J.-C.; Berthier, D.; Chantal, I.; Thevenon, S.; Sidibé, I.; Stachurski, F.; Belemsaga, D.; Razafindraïbé, H.; Elsen, J.-M. Selection assisted by a BoLA-DR/DQ haplotype against susceptibility to bovine dermatophilosis. Genet. Evol. GSE 2003, 35, S193–S200. [Google Scholar] [CrossRef]
- Hermoso de Mendoza, J.; Arenas, A.; Rey, J.; Alonso, J.M.; Gil, M.C.; Naranjo, G.; Hermoso de Mendoza, M. In vitro studies of Dermatophilus congolensis antimicrobial susceptibility by determining minimal inhibitory and bacteriocidal concentrations. Br. Vet. J. 1994, 150, 189–196. [Google Scholar] [CrossRef]
- Domingues, P.F.; Guerra, S.T.; Paula, C.L.D.; Alves, A.C.; Bolanos, C.A.D.; Morais, A.B.C.D.; Risseti, R.M.; Colhado, B.D.S.; Portilho, F.V.R.; Caxito, M.S.; et al. Successful therapy in unusual generalized Dermatophilus congolensis infection in a calf based on modified in vitro disk diffusion test. Arq. Inst. Biológico 2017, 84, e0382017. [Google Scholar] [CrossRef]
- Mannan, M.; Khan, M.; Rahman, M.; Begum, F.; Uddin, U. Isolation and identification of dermatophilus bacteria from the skin Lesions of cattle. Bangladesh J. Vet. Med. 2009, 7, 342–347. [Google Scholar] [CrossRef][Green Version]
- Alejo-Cancho, I.; Bosch, J.; Vergara, A.; Mascaro, J.M.; Marco, F.; Vila, J. Dermatitis by Dermatophilus congolensis. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2015, 21, e73–e74. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Hogg, J.S.; Hu, F.Z.; Janto, B.; Boissy, R.; Hayes, J.; Keefe, R.; Post, J.C.; Ehrlich, G.D. Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains. Genome Biol. 2007, 8, R103. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the Problem of Comparing Whole Bacterial Genomes across Different Sequencing Platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef]
- Antunes, P.; Machado, J.; Sousa, J.C.; Peixe, L. Dissemination of sulfonamide resistance genes (sul1, sul2, and sul3) in Portuguese Salmonella enterica strains and relation with integrons. Antimicrob. Agents Chemother. 2005, 49, 836–839. [Google Scholar] [CrossRef]
- Tauch, A.; Pühler, A.; Kalinowski, J.; Thierbach, G. TetZ, a new tetracycline resistance determinant discovered in gram-positive bacteria, shows high homology to gram-negative regulated efflux systems. Plasmid 2000, 44, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- McGhee, G.C.; Sundin, G.W. Erwinia amylovora CRISPR elements provide new tools for evaluating strain diversity and for microbial source tracking. PLoS ONE 2012, 7, e41706. [Google Scholar] [CrossRef]
- Rezzonico, F.; Smits, T.H.M.; Duffy, B. Diversity, evolution, and functionality of clustered regularly interspaced short palindromic repeat (CRISPR) regions in the fire blight pathogen Erwinia amylovora. Appl. Environ. Microbiol. 2011, 77, 3819–3829. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.A.; Heidelberg, J.F.; White, O.; Salzberg, S.L. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000, 1, 1–9. [Google Scholar] [CrossRef]
- Feßler, A.T.; Schwarz, S. Antimicrobial Resistance in Corynebacterium spp., Arcanobacterium spp., and Trueperella pyogenes. Microbiol. Spectr. 2017, 5, 5–6. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.; Lin, X.; Xu, H.; Li, Y.; Xue, R.; Wang, G.; Sun, S.; Li, J.; Lan, Z.; et al. Genetic characterization, mechanisms and dissemination risk of antibiotic resistance of multidrug-resistant Rothia nasimurium. Infect. Genet. Evol. 2021, 90, 104770. [Google Scholar] [CrossRef]
- Ojha, A.; Anand, M.; Bhatt, A.; Kremer, L.; Jacobs, W.R., Jr.; Hatfull, G.F. GroEL1: A dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 2005, 123, 861–873. [Google Scholar] [CrossRef]
- Hu, Y.; Henderson, B.; Lund, P.A.; Tormay, P.; Ahmed, M.T.; Gurcha, S.S.; Besra, G.S.; Coates, A.R. A Mycobacterium tuberculosis mutant lacking the groEL homologue cpn60.1 is viable but fails to induce an inflammatory response in animal models of infection. Infect. Immun. 2008, 76, 1535–1546. [Google Scholar] [CrossRef]
- Hu, Y.; Morichaud, Z.; Chen, S.; Leonetti, J.P.; Brodolin, K. Mycobacterium tuberculosis RbpA protein is a new type of transcriptional activator that stabilizes the σ A-containing RNA polymerase holoenzyme. Nucleic Acids Res. 2012, 40, 6547–6557. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Wu, S.; Howard, S.T.; Lakey, D.L.; Kipnis, A.; Samten, B.; Safi, H.; Gruppo, V.; Wizel, B.; Shams, H.; Basaraba, R.J.; et al. The principal sigma factor sigA mediates enhanced growth of Mycobacterium tuberculosis in vivo. Mol. Microbiol. 2004, 51, 1551–1562. [Google Scholar] [CrossRef]
- Gibson, J.A.; Thomas, R.J.; Domjahn, R.L. Subcutaneous and lymph node granulomas due to Dermatophilus congolensis in a steer. Vet. Pathol. 1983, 20, 120–122. [Google Scholar] [CrossRef]
- Corretto, E.; Antonielli, L.; Sessitsch, A.; Höfer, C.; Puschenreiter, M.; Widhalm, S.; Swarnalakshmi, K.; Brader, G. Comparative Genomics of Microbacterium Species to Reveal Diversity, Potential for Secondary Metabolites and Heavy Metal Resistance. Front. Microbiol. 2020, 11, 1869. [Google Scholar] [CrossRef]
- Quin, P.J.; Markey, B.K.; Carter, M.E.; Donnelly, W.J.; Leonard, F.C. Dermatophilus Congolensis; Blackwell Science Limited: Oxford, UK, 2002; pp. 69–70. [Google Scholar]
- García, A.; Martínez, R.; Benitez-Medina, J.M.; Risco, D.; García, W.L.; Rey, J.; Alonso, J.M.; de Mendoza, J.H. Development of a real-time SYBR Green PCR assay for the rapid detection of Dermatophilus congolensis. J. Vet. Sci. 2013, 14, 491–494. [Google Scholar] [CrossRef][Green Version]
- Van Driessche, L.; Vanneste, K.; Bogaerts, B.; De Keersmaecker, S.C.J.; Roosens, N.H.; Haesebrouck, F.; De Cremer, L.; Deprez, P.; Pardon, B.; Boyen, F. Isolation of Drug-Resistant Gallibacterium anatis from Calves with Unresponsive Bronchopneumonia, Belgium. Emerg. Infect. Dis. 2020, 26, 721–730. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 11 December 2020).
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.; Konstantinidis, K. Bypassing Cultivation To Identify Bacterial Species. Microbe 2014, 9, 111–118. [Google Scholar] [CrossRef]
- Blom, J.; Albaum, S.P.; Doppmeier, D.; Pühler, A.; Vorhölter, F.J.; Zakrzewski, M.; Goesmann, A. EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 2009, 10, 154. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- de Toro, M.; Lanza, V.F.; Vielva, L.; Redondo-Salvo, S.; de la Cruz, F. Plasmid Reconstruction from Next-Gen Data: A Detailed Protocol for the Use of PLACNETw for the Reconstruction of Plasmids from WGS Datasets. Methods Mol. Biol. 2020, 2075, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Vielva, L.; de Toro, M.; Lanza, V.F.; de la Cruz, F. PLACNETw: A web-based tool for plasmid reconstruction from bacterial genomes. Bioinformatics 2017, 33, 3796–3798. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Branford, I.; Johnson, S.; Zayas, S.; Chapwanya, A.; Boyen, F.; Mielcarska, M.B.; Szulc-Dąbrowska, L.; Butaye, P.; Toka, F.N. Draft Genome Sequences of 40 Dermatophilus congolensis Isolates from Bovine Dermatophilosis Cases in St. Kitts and Nevis. Microbiol. Resour. Announc. 2021, 10, e0033421. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Name | Species | GenBank Accession No. | Genome Size (bp) | CDS | G+C (%) | AMR * | CRISPR-Array | CRISPR-Repeat | CRISPR-Spacer | Repeat-Region |
|---|---|---|---|---|---|---|---|---|---|---|
| BTSK1 | D. congolensis | JAAFOV000000000 | 2,636,036 | 2482 | 58.8 | 22 | 1 | 33 | 32 | 4 |
| BTSK2 | D. congolensis | JAAFOU000000000 | 2,635,845 | 2501 | 58.8 | 22 | 1 | 33 | 32 | 4 |
| BTSK3 | D. congolensis | JAAFOT000000000 | 2,645,506 | 2506 | 59.2 | 22 | 1 | 5 | 4 | 2 |
| BTSK4 | D. congolensis | JAAFOS000000000 | 2,644,574 | 2504 | 59.2 | 22 | 1 | 5 | 4 | 2 |
| BTSK5 | D. congolensis | JAAFOR000000000 | 2,707,787 | 2566 | 59.2 | 23 | 1 | 6 | 5 | 2 |
| BTSK6 | D. congolensis | JAAFOQ000000000 | 2,707,101 | 2580 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK7 | D. congolensis | JAAFOP000000000 | 2,634,952 | 2546 | 58.8 | 22 | 1 | 30 | 29 | 6 |
| BTSK8 | D. congolensis | JAAFOO000000000 | 2,610,909 | 2530 | 58.8 | 21 | 1 | 30 | 29 | 6 |
| BTSK9 | D. congolensis | JAAFON000000000 | 2,705,920 | 2577 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK10 | D. congolensis | JAAFOM000000000 | 2,706,861 | 2581 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK11 | D. congolensis | JAAFOL000000000 | 2,704,542 | 2564 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK12 | D. congolensis | JAAFOK000000000 | 2,706,162 | 2567 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK13 | D.congolemsis | JAAFOJ000000000 | 2,705,629 | 2562 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK14 | D. congolensis | JAAFOI000000000 | 2,706,605 | 2520 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK15 | D. congolensis | JAAFOH000000000 | 2,706,691 | 2553 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK16 | D. congolensis | JAAFOG000000000 | 2,704,123 | 2561 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK17 | D. congolensis | JAAFOF000000000 | 2,635,364 | 2551 | 58.8 | 22 | 1 | 33 | 32 | 4 |
| BTSK18 | D. congolensis | JAAFOE000000000 | 2,643,372 | 2486 | 59.2 | 22 | 1 | 5 | 4 | 2 |
| BTSK19 | D. congolensis | JAAFOD000000000 | 2,646,426 | 2505 | 59.2 | 22 | 1 | 5 | 4 | 2 |
| BTSK20 | D. congolensis | JAAFOC000000000 | 2,643,778 | 2498 | 59.2 | 22 | 1 | 6 | 5 | 2 |
| BTSK21 | D. congolensis | JAAFOB000000000 | 2,609,406 | 2513 | 58.8 | 21 | 1 | 31 | 30 | 4 |
| BTSK22 | D. congolensis | JAAFOA000000000 | 2,705,748 | 2584 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK23 | D. congolensis | JAAFNZ000000000 | 2,707,702 | 2584 | 59.1 | 23 | 1 | 6 | 5 | 2 |
| BTSK24 | D. congolensis | JAAFNY000000000 | 2,644,395 | 2480 | 59.2 | 22 | 1 | 5 | 4 | 2 |
| BTSK25 | D. congolensis | JAAFNX000000000 | 2,644,659 | 2481 | 59.2 | 22 | 1 | 5 | 4 | 2 |
| BTSK26 | D. congolensis | JAAFNW000000000 | 2,690,948 | 2546 | 59.1 | 22 | 1 | 6 | 5 | 2 |
| BTSK27 | D. congolensis | JAAFNV000000000 | 2,635,575 | 2505 | 58.8 | 22 | 1 | 27 | 26 | 4 |
| BTSK28 | D. congolensis | JAAFNU000000000 | 2,644,675 | 2536 | 59.2 | 22 | 1 | 5 | 4 | 2 |
| BTSK29 | D. congolensis | JAAFNT000000000 | 2,641,543 | 2518 | 59.2 | 22 | 1 | 4 | 3 | 2 |
| BTSK30 | D. congolensis | JAAFNS000000000 | 2,692,568 | 2526 | 59.1 | 22 | 1 | 5 | 3 | 2 |
| BTSK31 | D. congolensis | JAAFNR000000000 | 2,635,532 | 2536 | 58.8 | 22 | 1 | 29 | 28 | 4 |
| BTSK32 | D. congolensis | JAAFNQ000000000 | 2,640,714 | 2499 | 59.2 | 22 | 1 | 5 | 4 | 4 |
| BTSK33 | D. congolensis | JAAFNP000000000 | 2,640,994 | 2490 | 59.2 | 22 | 1 | 5 | 4 | 4 |
| BTSK34 | D. congolensis | JAAFNO000000000 | 2,635,305 | 2527 | 58.8 | 22 | 1 | 30 | 29 | 4 |
| BTSK35 | D. congolensis | JAAFNN000000000 | 2,642,663 | 2488 | 59.2 | 22 | 1 | 6 | 5 | 2 |
| BTSK36 | D. congolensis | JAAFNM000000000 | 2,649,493 | 2496 | 59.2 | 22 | 1 | 6 | 5 | 2 |
| BTSK37 | D. congolensis | JAAFNL000000000 | 2,642,605 | 2517 | 59.2 | 22 | 1 | 6 | 5 | 2 |
| BTSK38 | D. congolensis | JAAFNK000000000 | 2,640,313 | 2503 | 59.2 | 22 | 1 | 6 | 5 | 2 |
| BTSK39 | D. congolensis | JAAFNJ000000000 | 2,640,884 | 2540 | 59.2 | 22 | 1 | 6 | 5 | 2 |
| BTSK40 | D. congolensis | JAAFNI000000000 | 2,640,858 | 2510 | 59.2 | 22 | 1 | 6 | 5 | 2 |
| Gene | Product | |
|---|---|---|
| rpoB | DNA-directed RNA polymerase beta subunit (EC 2.7.7.6) | Antibiotic target in susceptible species. Rifamycins, Peptide antibiotics |
| MtrA | Two component system response regulator MtrA | Regulator modulating expression of antibiotic resistance genes. (azithromycin, erythromycin, penicillin) |
| Ddl | D-alanine--D-alanine ligase (EC 6.3.2.4) | Antibiotic target in susceptible species. Cycloserine |
| S12p | SSU ribosomal protein S12p (S23e) | Antibiotic target in susceptible species. Aminoglycosides (streptomycin) |
| EF-Tu | Translation elongation factor Tu | Antibiotic-resistant gene variant or mutant, elfamycin resistance gene |
| rpoC | DNA-directed RNA polymerase beta’ subunit (EC 2.7.7.6) | Antibiotic target in susceptible species. daptomycin |
| Alr | Alanine racemase (EC 5.1.1.1) | Antibiotic target in susceptible species. D-cycloserine |
| OxyR | Hydrogen peroxide-inducible genes activator = OxyR | Regulator modulating expression of antibiotic resistance genes. Isoniazid |
| rho | Transcription termination factor Rho | Antibiotic target in susceptible species. Bicyclomycins |
| folA, Dfr | Dihydrofolate reductase (EC 1.5.1.3) | Antibiotic target in susceptible species. Diaminopyrimidines: trimethoprim, brodimoprim, tetroxoprim, iclaprim |
| PgsA | CDP-diacylglycerol--glycerol-3-phosphate 3-phosphatidyltransferase (EC 2.7.8.5) | Protein altering cell wall charge conferring antibiotic resistance. Peptide antibiotics: daptomycin |
| Iso-tRNA | Isoleucyl-tRNA synthetase (EC 6.1.1.5) | Antibiotic target in susceptible species. Mupirocin |
| gyrB | DNA gyrase subunit B (EC 5.99.1.3) | Antibiotic target in susceptible species. Antibiotics Class: Fluoroquinolones Quinolones Quinolines, Aminocoumarin antibiotics |
| folP | Dihydropteroate synthase (EC 2.5.1.15) | Antibiotic target in susceptible species. Antibiotics Class: Sulfonamides |
| gyrA | DNA gyrase subunit A (EC 5.99.1.3) | Antibiotic target in susceptible species Antibiotics Class: Fluoroquinolones Quinolones Quinolines |
| S10p | SSU ribosomal protein S10p (S20e) | Antibiotic target in susceptible species Antibiotics Class: Tetracyclines, Glycylcyclines |
| inhA, fabI | Enoyl-[acyl-carrier-protein] reductase [NADH] (EC 1.3.1.9) | Antibiotic target in susceptible species Antibiotics Class: Isoniazid, Ethionamide, Triclosan |
| dxr | 1-deoxy-D-xylulose 5-phosphate reductoisomerase (EC 1.1.1.267) | Antibiotic target in susceptible species Antibiotics Class: Fosmidomycin |
| MtrB | Two component system sensor histidine kinase MtrB | Regulator modulating expression of antibiotic resistance genes Antibiotics Class: Macrolides, Penams |
| tet(Z) | Tetracycline resistance, MFS efflux pump = tet(Z) | Efflux pump conferring antibiotic resistance Antibiotics Class: Tetracyclines |
| gidB | 16S rRNA (guanine(527)-N(7))-methyltransferase (EC 2.1.1.170) | Gene conferring resistance via absence Antibiotics Class: Aminoglycosides |
| EF-G | Translation elongation factor G | Antibiotic target in susceptible species. Antibiotics Class: Fusidic acid |
| kasA | 3-oxoacyl-[acyl-carrier-protein] synthase, KASII (EC 2.3.1.179) | Antibiotic target in susceptible species Antibiotics Class: Isoniazid, Triclosan |
| rpsL | SSU ribosomal protein S12p (S23e) | Aminoglycoside resistance gene, antibiotic-resistant gene variant or mutant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branford, I.; Johnson, S.; Chapwanya, A.; Zayas, S.; Boyen, F.; Mielcarska, M.B.; Szulc-Dąbrowska, L.; Butaye, P.; Toka, F.N. Comprehensive Molecular Dissection of Dermatophilus congolensis Genome and First Observation of tet(Z) Tetracycline Resistance. Int. J. Mol. Sci. 2021, 22, 7128. https://doi.org/10.3390/ijms22137128
Branford I, Johnson S, Chapwanya A, Zayas S, Boyen F, Mielcarska MB, Szulc-Dąbrowska L, Butaye P, Toka FN. Comprehensive Molecular Dissection of Dermatophilus congolensis Genome and First Observation of tet(Z) Tetracycline Resistance. International Journal of Molecular Sciences. 2021; 22(13):7128. https://doi.org/10.3390/ijms22137128
Chicago/Turabian StyleBranford, Ian, Shevaun Johnson, Aspinas Chapwanya, Samantha Zayas, Filip Boyen, Matylda Barbara Mielcarska, Lidia Szulc-Dąbrowska, Patrick Butaye, and Felix Ngosa Toka. 2021. "Comprehensive Molecular Dissection of Dermatophilus congolensis Genome and First Observation of tet(Z) Tetracycline Resistance" International Journal of Molecular Sciences 22, no. 13: 7128. https://doi.org/10.3390/ijms22137128
APA StyleBranford, I., Johnson, S., Chapwanya, A., Zayas, S., Boyen, F., Mielcarska, M. B., Szulc-Dąbrowska, L., Butaye, P., & Toka, F. N. (2021). Comprehensive Molecular Dissection of Dermatophilus congolensis Genome and First Observation of tet(Z) Tetracycline Resistance. International Journal of Molecular Sciences, 22(13), 7128. https://doi.org/10.3390/ijms22137128