
RAAS: A Convergent Player in Ischemic Heart Failure and Cancer

 , ,
, ,
Abstract
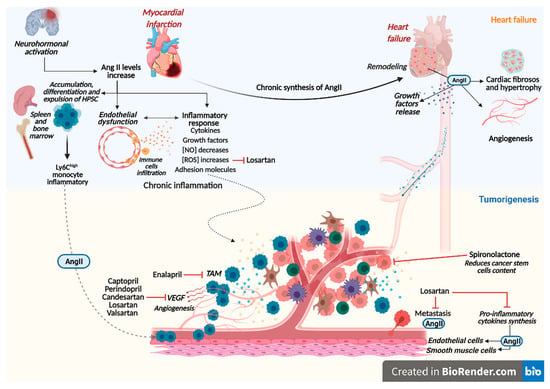
:
1. Introduction
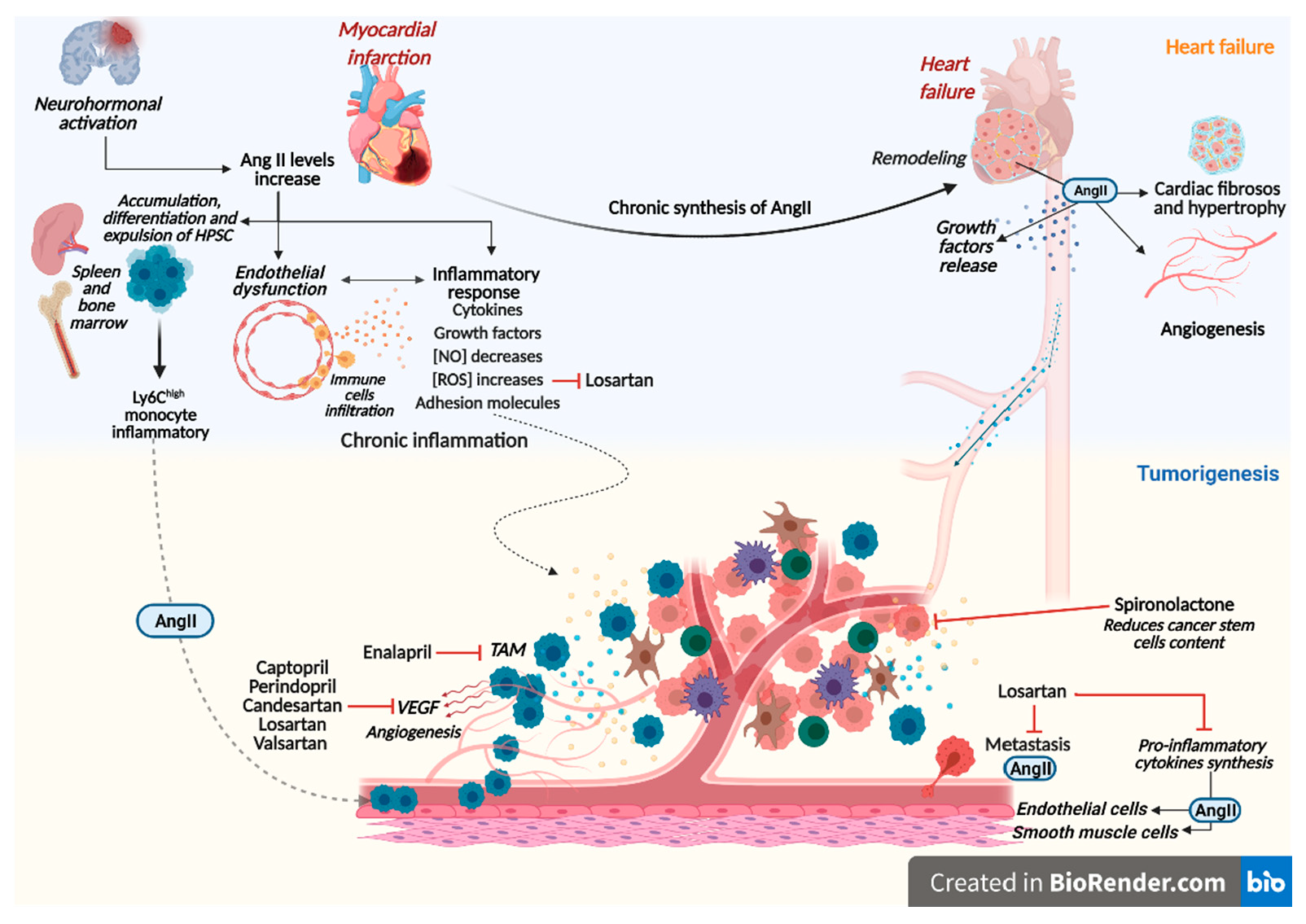
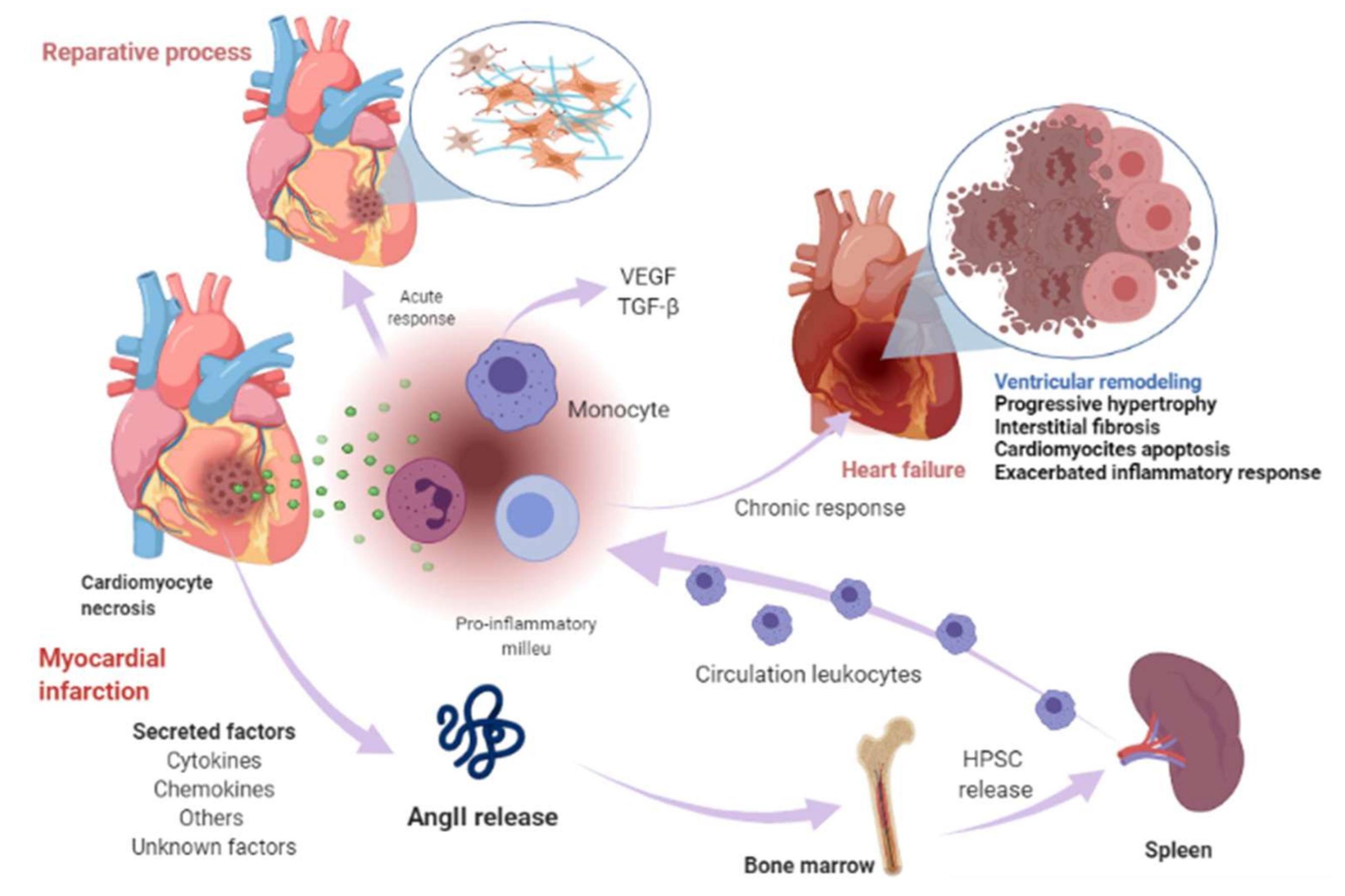
2. Clinical and Preclinical Evidence of Heart Failure Triggering Cancer
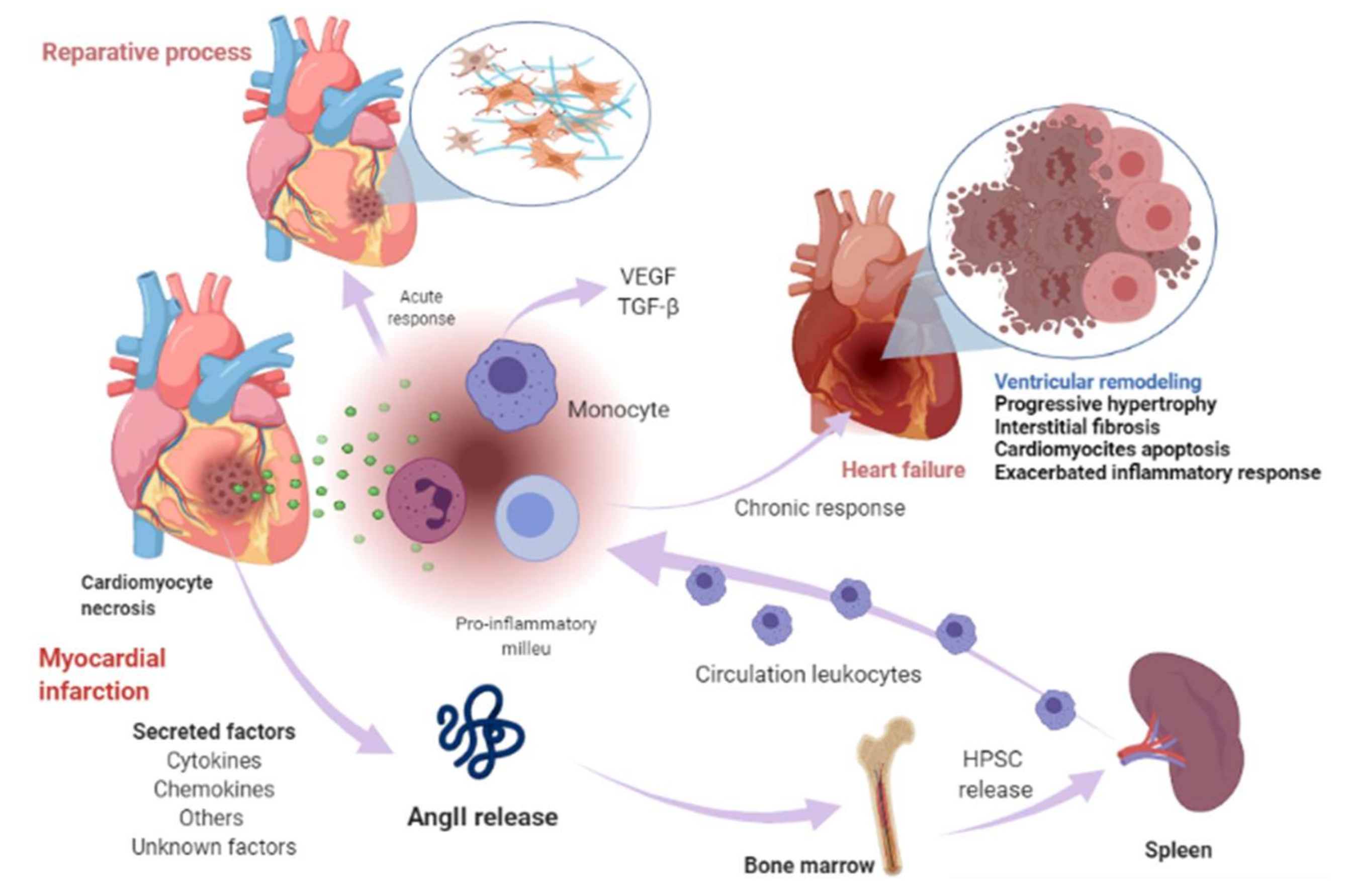
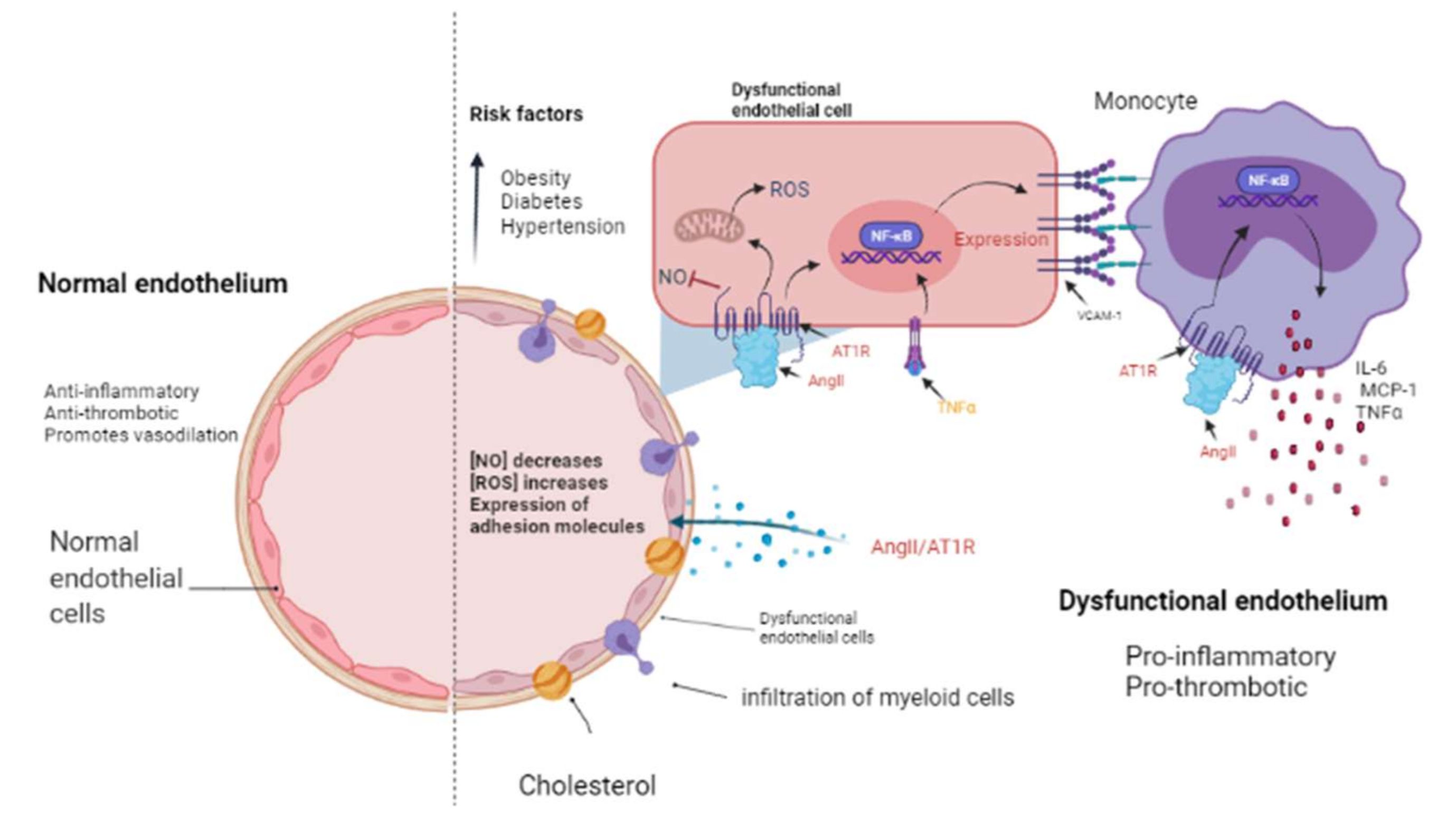
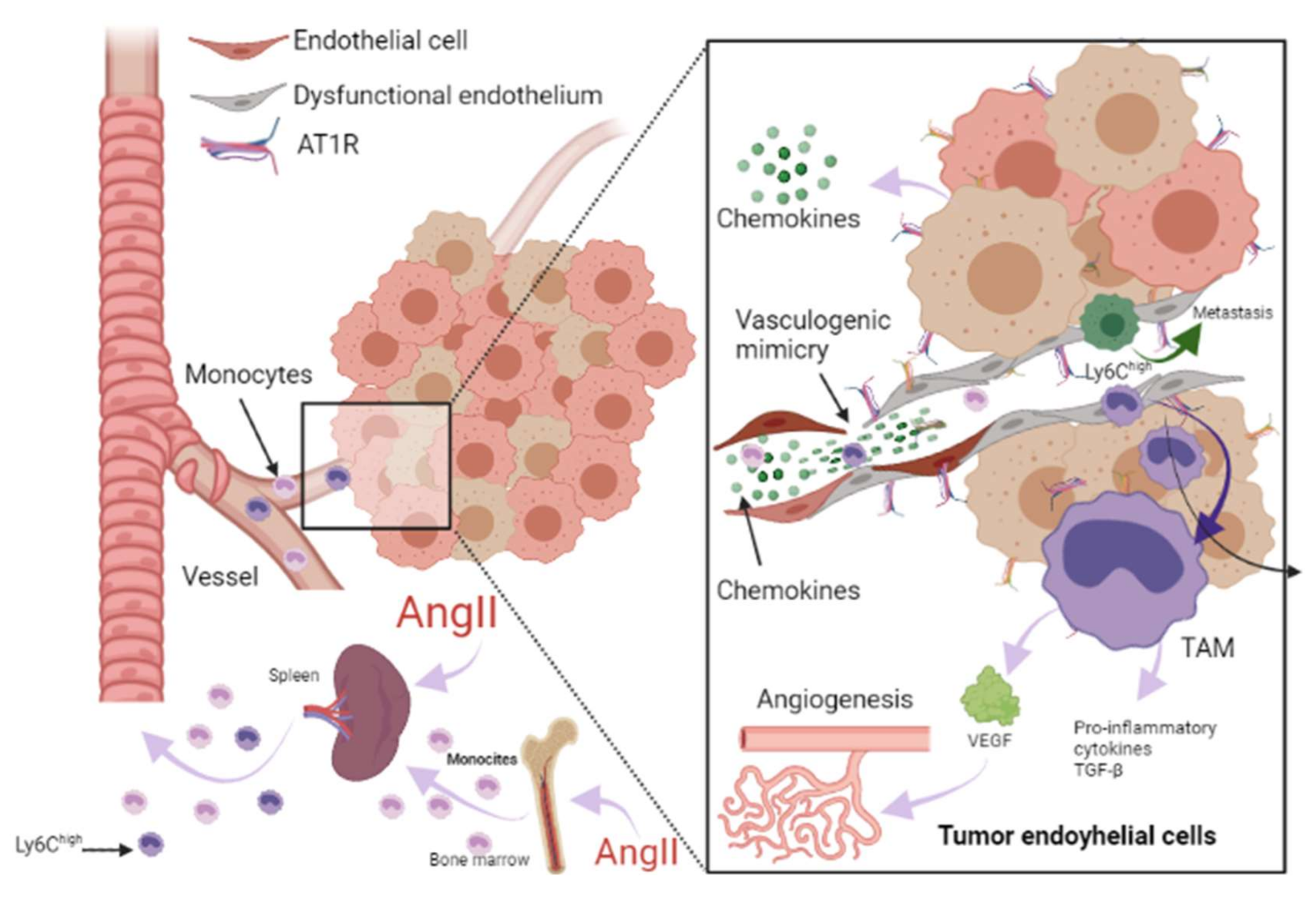
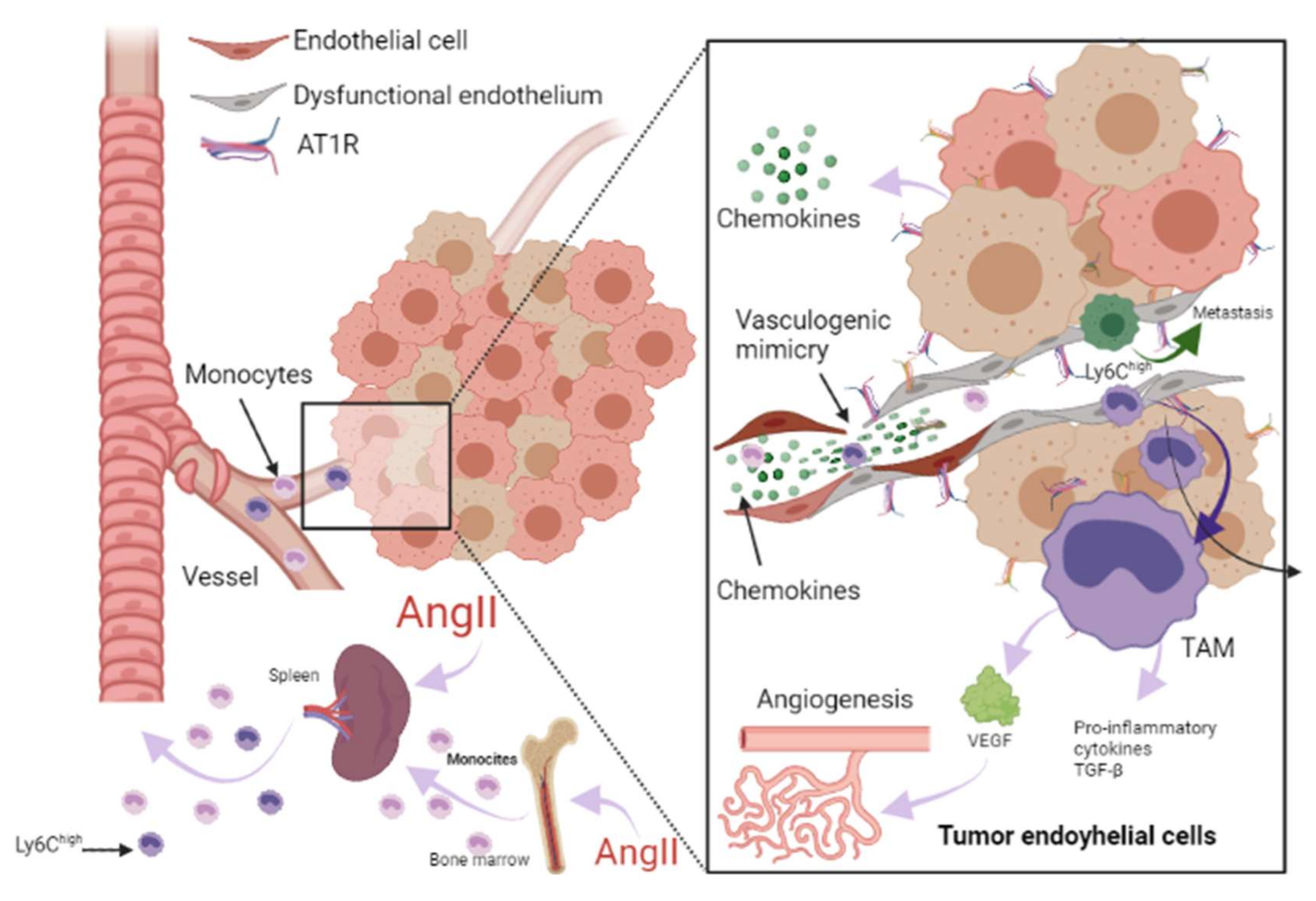
3. Renin-Angiotensin-Aldosterone System Linking Ischemic Heart Failure and Cancer
4. Role of Drug Therapy with RAAS Inhibitors in Heart Failure and Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RAAS Inhibitor | Findings |
|---|---|
| Angiotensin converting enzyme inhibitors (ACEI) | |
| Captopril | Inhibits tumor growth in a gastric cancer model and suppresses the angiogenesis of the tumor by decreasing the expression of vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMP)-7 in a mouse model with human gastric cancer [99]. Attenuates cell migration in a breast cancer model [100]. Inhibits cell growth, decreases c-myc expression, and increases apoptosis on leukemic cell lines [101]. |
| Enalapril | Inhibits tumor progression and reduces number of tumor-associated macrophages (TAMs) [54]. |
| Perindopril | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor by decreasing the expression of VEGF and MMP-7 in a mouse model with human gastric cancer [99]. |
| Ramipril | Decreases systemic inflammation [55]. |
| Trandolapril | Inhibits cell growth, decreases c-myc expression, and increases apoptosis in leukemic cell lines [101]. |
| Angiotensin II type 1 receptor blockers (ARBs) | |
| Telmisartan | Inhibits cell proliferation and tumor growth of esophageal squamous cell carcinoma by inducing s-phase cell cycle arrest [102]. |
| Candesartan | Prevents bladder cancer growth in a mouse model by inhibiting angiogenesis, and combined treatment with candesartan and paclitaxel enhances paclitaxel-induced cytotoxicity [103]. Candesartan treatment significantly sensitizes human lung adenocarcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis [104]. |
| Losartan | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor decreasing the expressions of VEGF [99]. Can exert anti-metastatic activity by inhibiting chemokine receptor type 2 (CCR2) signaling and suppressing monocyte recruitment in a mouse model with tumors and indirectly as anti-inflammatory effect and independently of AT1R [105]. Ameliorates angiogenesis, inflammation and the induction of oxidative stress via type-1 angiotensin-II receptor (AT1R) in a murine model of lung metastasis of colorectal cancer [106]. Inhibits cell growth, decreases c-myc expression and increases apoptosis in leukemic cell lines [101]. |
| Valsartan | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor, decreasing the expressions of VEGF [99]. |
| Aldosterone antagonists | |
| Spironolactone | Inhibits cancerous cell growth and is highly toxic for cancer stem cells; impairs DNA-double-strand breaks repair and induces apoptosis in cancer cells and cancer stem cells (CSCs) while sparing healthy cells. In vivo, this treatment reduces the size and CSC content of tumors [107]. |
5. Future Directions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brancaccio, M.; Pirozzi, F.; Hirsch, E.; Ghigo, A. Mechanisms underlying the cross-talk between heart and cancer. J. Physiol. 2020, 598, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Papazoglou, P.; Peng, L.; Sachinidis, A. Epigenetic Mechanisms Involved in the Cardiovascular Toxicity of Anticancer Drugs. Front. Cardiovasc. Med. 2021, 8, 658900. [Google Scholar] [CrossRef]
- Stoltzfus, K.C.; Zhang, Y.; Sturgeon, K.; Sinoway, L.I.; Trifiletti, D.M.; Chinchilli, V.M.; Zaorsky, N.G. Fatal heart disease among cancer patients. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Banke, A.; Schou, M.; Videbaek, L.; Møller, J.E.; Torp-Pedersen, C.; Gustafsson, F.; Dahl, J.S.; Køber, L.; Hildebrandt, P.R.; Gislason, G.H. Incidence of cancer in patients with chronic heart failure: A long-term follow-up study. Eur. J. Heart Fail. 2016, 18, 260–266. [Google Scholar] [CrossRef]
- Kwak, S.; Kwon, S.; Lee, S.Y.; Yang, S.; Lee, H.J.; Lee, H.; Park, J.B.; Han, K.; Kim, Y.J.; Kim, H.K. Differential risk of incident cancer in patients with heart failure: A nationwide population-based cohort study. J. Cardiol. 2021, 77, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Hasin, T.; Gerber, Y.; McNallan, S.M.; Weston, S.A.; Kushwaha, S.S.; Nelson, T.J.; Cerhan, J.R.; Roger, V.L. Patients with heart failure have an increased risk of incident cancer. J. Am. Coll. Cardiol. 2013, 62, 881–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasin, T.; Gerber, Y.; Weston, S.A.; Jiang, R.; Killian, J.M.; Manemann, S.M.; Cerhan, J.R.; Roger, V.L. Heart Failure After Myocardial Infarction Is Associated With Increased Risk of Cancer. J. Am. Coll. Cardiol. 2016, 68, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Hasegawa, T.; Asakura, M.; Kanzaki, H.; Takahama, H.; Amaki, M.; Mochizuki, N.; Anzai, T.; Hamasaki, T.; Kitakaze, M. Does the pathophysiology of heart failure prime the incidence of cancer? Hypertens. Res. 2017, 40, 831–836. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Wang, L.; Tian, L.; An, N.; Yang, X.; Li, X.; Tian, C.; Yuan, M.; Xiong, X.; et al. Does heart failure increase the risk of incident cancer? A meta-analysis and systematic review. Heart Fail. Rev. 2020, 25, 949–955. [Google Scholar] [CrossRef]
- Bertero, E.; Ameri, P.; Maack, C. Bidirectional Relationship Between Cancer and Heart Failure: Old and New Issues in Cardio-oncology. Card. Fail. Rev. 2019, 5, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsborne, C.; Chaggar, P.S.; Shaw, S.M.; Williams, S.G. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: The past, the present and the future. Postgrad. Med. J. 2016, 93, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The renin-angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Wegman-Ostrosky, T.; Soto-Reyes, E.; Vidal-Millán, S.; Sánchez-Corona, J. The renin-angiotensin system meets the hallmarks of cancer. JRAAS J. Renin Angiotensin Aldosterone Syst. 2015, 16, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation 2013, 16, e147–e239. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Sanchis-Gomar, F. Global epidemiology and future trends of heart failure. AME Med. J. 2020, 5, 15. [Google Scholar] [CrossRef]
- Elgendy, I.Y.; Mahtta, D.; Pepine, C.J. Medical Therapy for Heart Failure Caused by Ischemic Heart Disease. Circ. Res. 2019, 124, 1520–1535. [Google Scholar] [CrossRef]
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef]
- Khan, M.A.; Hashim, M.J.; Mustafa, H.; Baniyas, M.Y.; Al Suwaidi, S.K.B.M.; AlKatheeri, R.; Alblooshi, F.M.K.; Almatrooshi, M.E.A.H.; Alzaabi, M.E.H.; Al Darmaki, R.S.; et al. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 2020, 12, e9349. [Google Scholar] [CrossRef]
- Sulo, G.; Igland, J.; Vollset, S.E.; Nygård, O.; Ebbing, M.; Sulo, E.; Egeland, G.M.; Tell, G.S. Heart failure complicating acute myocardial infarction; burden and timing of occurrence: A nation-wide analysis including 86 771 patients from the cardiovascular disease in norway (CVDNOR) project. J. Am. Heart Assoc. 2016, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cahill, T.J.; Kharbanda, R.K. Heart failure after myocardial infarction in the era of primary percutaneous coronary intervention: Mechanisms, incidence and identification of patients at risk. World J. Cardiol. 2017, 9, 407. [Google Scholar] [CrossRef] [PubMed]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237. [Google Scholar] [CrossRef]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 23, 1342–1356. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017, 136, e137–e161. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts and Figures 2021. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2021.html (accessed on 27 June 2021).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameri, P.; Canepa, M.; Anker, M.S.; Belenkov, Y.; Bergler-Klein, J.; Cohen-Solal, A.; Farmakis, D.; López-Fernández, T.; Lainscak, M.; Pudil, R.; et al. Cancer diagnosis in patients with heart failure: Epidemiology, clinical implications and gaps in knowledge. Eur. J. Heart Fail. 2018, 20, 879–887. [Google Scholar] [CrossRef]
- Selvaraj, S.; Bhatt, D.L.; Claggett, B.; Djoussé, L.; Shah, S.J.; Chen, J.; Imran, T.F.; Qazi, S.; Sesso, H.D.; Gaziano, J.M.; et al. Lack of Association Between Heart Failure and Incident Cancer. J. Am. Coll. Cardiol. 2018, 71, 1501–1510. [Google Scholar] [CrossRef]
- Mathieu, S.; El Khoury, N.; Rivard, K.; Paradis, P.; Nemer, M.; Fiset, C. Angiotensin II Overstimulation Leads to an Increased Susceptibility to Dilated Cardiomyopathy and Higher Mortality in Female Mice. Sci. Rep. 2018, 8, 7242. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.K.; Wruck, L.; Quibrera, M.; Matsushita, K.; Loehr, L.R.; Chang, P.P.; Rosamond, W.D.; Wright, J.; Heiss, G.; Coresh, J. Temporal trends in hospitalization for acute decompensated heart failure in the United States, 1998–2011. Am. J. Epidemiol. 2016, 183, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020, 26, 1452–1458. [Google Scholar] [CrossRef]
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; De Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; Van Der Vegt, B.; et al. Heart failure stimulates tumor growth by circulating factors. Circulation 2018, 138, 678–691. [Google Scholar] [CrossRef]
- Thygesen, K.; Mair, J.; Giannitsis, E.; Mueller, C.; Lindahl, B.; Blankenberg, S.; Huber, K.; Plebani, M.; Biasucci, L.M.; Tubaro, M.; et al. How to use high-sensitivity cardiac troponins in acute cardiac care. Eur. Heart J. 2012, 33, 2252–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugliese, N.R.; Masi, S.; Taddei, S. The renin-angiotensin-aldosterone system: A crossroad from arterial hypertension to heart failure. Heart Fail. Rev. 2020, 25, 31–42. [Google Scholar] [CrossRef]
- Ziaja, M.; Urbanek, K.A.; Kowalska, K.; Piastowska-Ciesielska, A.W. Angiotensin II and Angiotensin Receptors 1 and 2—Multifunctional System in Cells Biology, What Do We Know? Cells 2021, 10, 381. [Google Scholar] [CrossRef]
- Steckelings, U.M.; Paulis, L.; Namsolleck, P.; Unger, T. AT2 receptor agonists: Hypertension and beyond. Curr. Opin. Nephrol. Hypertens. 2012, 21, 142–146. [Google Scholar] [CrossRef]
- Wang, Y.; Del Borgo, M.; Lee, H.W.; Baraldi, D.; Hirmiz, B.; Gaspari, T.A.; Denton, K.M.; Aguilar, M.I.; Samuel, C.S.; Widdop, R.E. Anti-fibrotic potential of AT2 receptor agonists. Front. Pharmacol. 2017, 8, 564. [Google Scholar] [CrossRef] [Green Version]
- Rompe, F.; Artuc, M.; Hallberg, A.; Alterman, M.; Ströder, K.; Thöne-Reineke, C.; Reichenbach, A.; Schacherl, J.; Dahlöf, B.; Bader, M.; et al. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor κb. Hypertension 2010, 55, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, S.P. Angiotensin Type 2 Receptor. In Encyclopedia of Signaling Molecules; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 320–327. [Google Scholar]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; He, W.; Guo, L.; Zhang, Y.; Li, H.; Han, S.; Shen, D. The ACE2-Ang (1-7)-Mas receptor axis attenuates cardiac remodeling and fibrosis in post-myocardial infarction. Mol. Med. Rep. 2017, 16, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, K.D.; Coelho, F.M.; Vieira, A.T.; Sachs, D.; Barroso, L.C.; Costa, V.V.; Bretas, T.L.B.; Bader, M.; de Sousa, L.P.; da Silva, T.A.; et al. Anti-Inflammatory Effects of the Activation of the Angiotensin-(1–7) Receptor, Mas, in Experimental Models of Arthritis. J. Immunol. 2010, 185, 5569–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, M.C.; Al Zayadneh, E.M. Angiotensin-(1-7) and the Regulation of Anti-Fibrotic Signaling Pathways. J. Cell Signal. 2017, 2, 2. [Google Scholar] [CrossRef]
- Park, B.M.; Cha, S.A.; Lee, S.H.; Kim, S.H. Angiotensin IV protects cardiac reperfusion injury by inhibiting apoptosis and inflammation via AT4R in rats. Peptides 2016, 79, 66–74. [Google Scholar] [CrossRef]
- de Paula Gonzaga, A.L.A.C.; Palmeira, V.A.; Ribeiro, T.F.S.; Costa, L.B.; de Sá Rodrigues, K.E.; Simões-e-Silva, A.C. ACE2/Angiotensin-(1-7)/Mas Receptor Axis in Human Cancer: Potential Role for Pediatric Tumors. Curr. Drug Targets 2020, 21, 892–901. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Emdin, M.; Fatini, C.; Mirizzi, G.; Poletti, R.; Borrelli, C.; Prontera, C.; Latini, R.; Passino, C.; Clerico, A.; Vergaro, G. Biomarkers of activation of renin-angiotensin-aldosterone system in heart failure: How useful, how feasible? Clin. Chim. Acta 2015, 443, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial infarction accelerates atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Kyaw, T.; Loveland, P.; Kanellakis, P.; Cao, A.; Kallies, A.; Huang, A.L.; Peter, K.; Toh, B.-H.; Bobik, A. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur. Heart J. 2021, 42, 938–947. [Google Scholar] [CrossRef]
- Rasini, E.; Cosentino, M.; Marino, F.; Legnaro, M.; Ferrari, M.; Guasti, L.; Venco, A.; Lecchini, S. Angiotensin II type 1 receptor expression on human leukocyte subsets: A flow cytometric and RT-PCR study. Regul. Pept. 2006, 134, 69–74. [Google Scholar] [CrossRef]
- Kim, S.; Zingler, M.; Harrison, J.K.; Scott, E.W.; Cogle, C.R.; Luo, D.; Raizada, M.K. Angiotensin II Regulation of Proliferation, Differentiation, and Engraftment of Hematopoietic Stem Cells. Hypertension 2016, 67, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Ryan, R.; Pucci, F.; Sio, S.W.; Kuswanto, W.; Rauch, P.J.; Chudnovskiy, A.; Iwamoto, Y.; et al. Angiotensin II Drives the Production of Tumor-Promoting Macrophages. Immunity 2013, 38, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Rudi, W.-S.; Molitor, M.; Garlapati, V.; Finger, S.; Wild, J.; Münzel, T.; Karbach, S.H.; Wenzel, P. ACE Inhibition Modulates Myeloid Hematopoiesis after Acute Myocardial Infarction and Reduces Cardiac and Vascular Inflammation in Ischemic Heart Failure. Antioxidants 2021, 10, 396. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The immune system and the remodeling infarcted heart: Cell biological insights and therapeutic opportunities. J. Cardiovasc. Pharmacol. 2014, 63, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Nahrendorf, M.; Swirski, F.K. Leukocytes link local and systemic inflammation in ischemic cardiovascular disease an expanded cardiovascular continuum. J. Am. Coll. Cardiol. 2016, 67, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D. Mechanisms of Heart Failure BT—Management of Heart Failure: Volume 1: Medical; Baliga, R.R., Haas, G.J., Eds.; Springer: London, UK, 2015; pp. 13–30. ISBN 978-1-4471-6657-3. [Google Scholar]
- Dewey, C.M.; Spitler, K.M.; Ponce, J.M.; Hall, D.D.; Grueter, C.E. Cardiac-Secreted Factors as Peripheral Metabolic Regulators and Potential Disease Biomarkers. J. Am. Heart Assoc. 2016, 5, e003101. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [Green Version]
- Kiss, M.; Caro, A.A.; Raes, G.; Laoui, D. Systemic Reprogramming of Monocytes in Cancer. Front. Oncol. 2020, 10, 1399. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Amin, M.N.; Siddiqui, S.A.; Ibrahim, M.; Hakim, M.L.; Ahammed, M.S.; Kabir, A.; Sultana, F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020, 8, 2050312120965752. [Google Scholar] [CrossRef]
- Becher, U.M.; Endtmann, C.; Tiyerili, V.; Nickenig, G.; Werner, N. Endothelial damage and regeneration: The role of the renin-angiotensin- aldosterone system. Curr. Hypertens. Rep. 2011, 13, 86–92. [Google Scholar] [CrossRef]
- Silva, G.M.; França-Falcão, M.S.; Calzerra, N.T.M.; Luz, M.S.; Gadelha, D.D.A.; Balarini, C.M.; Queiroz, T.M. Role of Renin-Angiotensin System Components in Atherosclerosis: Focus on Ang-II, ACE2, and Ang-1–7. Front. Physiol. 2020, 11, 1067. [Google Scholar] [CrossRef]
- Davel, A.P.; Anwar, I.J.; Jaffe, I.Z. The endothelial mineralocorticoid receptor: Mediator of the switch from vascular health to disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Zelada, N.; Zuñiga-Cuevas, U.; Ramirez-Reyes, A.; Lavandero, S.; Riquelme, J.A. Targeting the Endothelium to Achieve Cardioprotection. Front. Pharmacol. 2021, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Toya, T.; Sara, J.D.; Corban, M.T.; Taher, R.; Godo, S.; Herrmann, J.; Lerman, L.O.; Lerman, A. Assessment of peripheral endothelial function predicts future risk of solid-tumor cancer. Eur. J. Prev. Cardiol. 2020, 27, 608–618. [Google Scholar] [CrossRef]
- Franses, J.W.; Drosu, N.C.; Gibson, W.J.; Chitalia, V.C.; Edelman, E.R. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int. J. Cancer 2013, 133, 1334–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molitor, M.; Rudi, W.S.; Garlapati, V.; Finger, S.; Schüler, R.; Kossmann, S.; Lagrange, J.; Nguyen, T.S.; Wild, J.; Knopp, T.; et al. Nox2+myeloid cells drive vascular inflammation and endothelial dysfunction in heart failure after myocardial infarction via angiotensin II receptor type 1. Cardiovasc. Res. 2021, 117, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Catarata, M.J.; Ribeiro, R.; Oliveira, M.J.; Cordeiro, C.R.; Medeiros, R. Renin-angiotensin system in lung tumor and microenvironment interactions. Cancers 2020, 12, 1457. [Google Scholar] [CrossRef]
- Ishikane, S.; Takahashi-Yanaga, F. The role of angiotensin II in cancer metastasis: Potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochem. Pharmacol. 2018, 151, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.-H.; Sun, H.-C.; Zhu, X.-D.; Zhang, S.-Z.; Li, X.-L.; Li, K.-S.; Liu, X.-F.; Lei, M.; Li, Y.; Tang, Z.-Y. Irbesartan inhibits metastasis by interrupting the adherence of tumor cell to endothelial cell induced by angiotensin II in hepatocellular carcinoma. Ann. Transl. Med. 2021, 9, 207. [Google Scholar] [CrossRef]
- Patel, R.B.; Colangelo, L.A.; Bielinski, S.J.; Larson, N.B.; Ding, J.; Allen, N.B.; Michos, E.D.; Shah, S.J.; Lloyd-Jones, D.M. Circulating Vascular Cell Adhesion Molecule-1 and Incident Heart Failure: The Multi-Ethnic Study of Atherosclerosis (MESA). J. Am. Heart Assoc. 2020, 9, e019390. [Google Scholar] [CrossRef]
- Carbajo-Lozoya, J.; Lutz, S.; Feng, Y.; Kroll, J.; Hammes, H.P.; Wieland, T. Angiotensin II modulates VEGF-driven angiogenesis by opposing effects of type 1 and type 2 receptor stimulation in the microvascular endothelium. Cell. Signal. 2012, 24, 1261–1269. [Google Scholar] [CrossRef]
- Cespón-Fernández, M.; Raposeiras-Roubín, S.; Abu-Assi, E.; Manzano-Fernández, S.; Flores-Blanco, P.; Barreiro-Pardal, C.; Castiñeira-Busto, M.; Muñoz-Pousa, I.; López-Rodríguez, E.; Caneiro-Queija, B.; et al. Renin–Angiotensin System Blockade and Risk of Heart Failure After Myocardial Infarction Based on Left Ventricular Ejection Fraction: A Retrospective Cohort Study. Am. J. Cardiovasc. Drugs 2019, 19, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Raposeiras-Roubín, S.; Abu-Assi, E.; Cespón-Fernández, M.; Ibáñez, B.; García-Ruiz, J.M.; D’Ascenzo, F.; Simao Henriques, J.P.; Saucedo, J.; Caneiro-Queija, B.; Cobas-Paz, R.; et al. Impact of renin-angiotensin system blockade on the prognosis of acute coronary syndrome based on left ventricular ejection fraction. Rev. Esp. Cardiol. 2020, 73, 114–122. [Google Scholar] [CrossRef]
- Sim, H.W.; Zheng, H.; Richards, A.M.; Chen, R.W.; Sahlen, A.; Yeo, K.K.; Tan, J.W.; Chua, T.; Tan, H.C.; Yeo, T.C.; et al. Beta-blockers and renin-angiotensin system inhibitors in acute myocardial infarction managed with inhospital coronary revascularization. Sci. Rep. 2020, 10, 15184. [Google Scholar] [CrossRef]
- Ranjbar, R.; Shafiee, M.; Hesari, A.R.; Ferns, G.A.; Ghasemi, F.; Avan, A. The potential therapeutic use of renin–angiotensin system inhibitors in the treatment of inflammatory diseases. J. Cell. Physiol. 2019, 234, 2277–2295. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, Y.; Liao, X.; Yao, Y.; Huang, C.; Liu, L. The Relationship Between Anti-Hypertensive Drugs and Cancer: Anxiety to be Resolved in Urgent. Front. Pharmacol. 2020, 11, 2122. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Braunwald, E.; Moyé, L.A.; Basta, L.; Brown, E.J.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C.; et al. Effect of Captopril on Mortality and Morbidity in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 1992, 327, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Aronow, W.S.; Kronzon, I. Effect of enalapril on congestive heart failure treated with diuretics in elderly patients with prior myocardial infarction and normal left ventricular ejection fraction. Am. J. Cardiol. 1993, 71, 602–604. [Google Scholar] [CrossRef]
- Cleland, J.G.F.; Tendera, M.; Adamus, J.; Freemantle, N.; Gray, C.S.; Lye, M.; O’Mahony, D.; Polonski, L.; Taylor, J. Perindopril for elderly people with chronic heart failure: The PEP-CHF study. Eur. J. Heart Fail. 1999, 1, 211–217. [Google Scholar] [CrossRef]
- Ferrari, R. Effects of angiotensin-converting enzyme inhibition with perindopril on left ventricular remodeling and clinical outcome: Results of the randomized Perindopril and Remodeling in Elderly with Acute Myocardial Infarction (PREAMI) study. Arch. Intern. Med. 2006, 166, 659–666. [Google Scholar] [PubMed] [Green Version]
- The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet 1993, 342, 821–828.
- Tepper, D.; Greenberg, S. A clinical trial of the angiotensin-converting enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Cardiovasc. Rev. Rep. 1996, 17, 49. [Google Scholar]
- The Telmisartan Randomised AssessmeNt Study in ACE iNtolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators; Yusuf, S.; Teo, K.; Anderson, C.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: A randomised controlled trial. Lancet 2008, 372, 1174–1183. [Google Scholar]
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.V.; Michelson, E.L.; Olofsson, B.; Östergren, J. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-preserved trial. Lancet 2003, 362, 777–781. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; McMurray, J.J.; Östergren, J.; Granger, C.B.; Yusuf, S.; Pitt, B. Candesartan reduced mortality and hospital admissions in chronic heart failure. Evid. Based. Med. 2004, 9, 44–45. [Google Scholar]
- Konstam, M.A.; Neaton, J.D.; Dickstein, K.; Drexler, H.; Komajda, M.; Martinez, F.A.; Riegger, G.A.; Malbecq, W.; Smith, R.D.; Guptha, S.; et al. Effects of high-dose versus low-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): A randomised, double-blind trial. Lancet 2009, 374, 1840–1848. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; McMurray, J.; Leizorovicz, A.; Maggioni, A.P.; Rouleau, J.L.; Van De Werf, F.; Henis, M.; Neuhart, E.; Gallo, P.; Edwards, S.; et al. Valsartan in acute myocardial infarction trial (VALIANT): Rationale and design. Am. Heart J. 2000, 140, 727–750. [Google Scholar] [CrossRef]
- Bissessor, N.; White, H. Valsartan in the treatment of heart failure or left ventricular dysfunction after myocardial infarction. Vasc. Health Risk Manag. 2007, 3, 425–430. [Google Scholar] [PubMed]
- Hayashi, M.; Tsutamoto, T.; Wada, A.; Tsutsui, T.; Ishii, C.; Ohno, K.; Fujii, M.; Taniguchi, A.; Hamatani, T.; Nozato, Y.; et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003, 107, 2559–2565. [Google Scholar] [PubMed] [Green Version]
- Song, T.; Choi, C.H.; Kim, M.K.; Kim, M.L.; Yun, B.S.; Seong, S.J. The effect of angiotensin system inhibitors (angiotensin-converting enzyme inhibitors or angiotensin receptor blockers) on cancer recurrence and survival: A meta-analysis. Eur. J. Cancer Prev. 2017, 26, 78–85. [Google Scholar] [CrossRef]
- Asgharzadeh, F.; Hashemzehi, M.; Moradi-Marjaneh, R.; Hassanian, S.M.; Ferns, G.A.; Khazaei, M.; Avan, A. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers as therapeutic options in the treatment of renal cancer: A meta-analysis. Life Sci. 2020, 242, 117181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, D.-S.; Xin, L.; Zhou, L.-Q.; Zhang, H.-T.; Liu, L.; Yuan, Y.-W.; Li, S.-H. The renin–angiotensin system blockers and survival in digestive system malignancies. Medicine 2020, 99, e19075. [Google Scholar] [CrossRef]
- Fang, K.; Zhang, Y.; Liu, W.; He, C. Effects of angiotensin-converting enzyme inhibitor/angiotensin receptor blocker use on cancer therapy-related cardiac dysfunction: A meta-analysis of randomized controlled trials. Heart Fail. Rev. 2021, 26, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, S.R.; Zhang, C.H.; He, Y.L.; Zhan, W.H.; Wu, H.; Peng, J.J. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor blockers on lymphangiogenesis of gastric cancer in a nude mouse model. Chin. Med. J. 2008, 121, 2167–2171. [Google Scholar] [CrossRef] [PubMed]
- Rasha, F.; Kahathuduwa, C.; Ramalingam, L.; Hernandez, A.; Moussa, H.; Moustaid-Moussa, N. Combined Effects of Eicosapentaenoic Acid and Adipocyte Renin–Angiotensin System Inhibition on Breast Cancer Cell Inflammation and Migration. Cancers 2020, 12, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Iglesia Iñigo, S.; López-Jorge, C.E.; Gómez-Casares, M.T.; Lemes Castellano, A.; Martín Cabrera, P.; López Brito, J.; Suárez Cabrera, A.; Molero Labarta, T. Induction of apoptosis in leukemic cell lines treated with captopril, trandolapril and losartan: A new role in the treatment of leukaemia for these agents. Leuk. Res. 2009, 33, 810–816. [Google Scholar] [CrossRef]
- Matsui, T.; Chiyo, T.; Kobara, H.; Fujihara, S.; Fujita, K.; Namima, D.; Nakahara, M.; Kobayashi, N.; Nishiyama, N.; Yachida, T.; et al. Telmisartan Inhibits Cell Proliferation and Tumor Growth of Esophageal Squamous Cell Carcinoma by Inducing S-Phase Arrest In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3197. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, M.; Miyajima, A.; Kikuchi, E.; Kosaka, T.; Horiguchi, Y.; Murai, M. Effect of angiotensin II type 1 receptor antagonist on tumor growth and angiogenesis in a xenograft model of human bladder cancer. Hum. Cell 2007, 20, 1–9. [Google Scholar] [CrossRef]
- Rasheduzzaman, M.; Park, S.Y. Antihypertensive drug-candesartan attenuates TRAIL resistance in human lung cancer via AMPK-mediated inhibition of autophagy flux. Exp. Cell Res. 2018, 368, 126–135. [Google Scholar] [CrossRef]
- Regan, D.P.; Coy, J.W.; Chahal, K.K.; Chow, L.; Kurihara, J.N.; Guth, A.M.; Kufareva, I.; Dow, S.W. The Angiotensin Receptor Blocker Losartan Suppresses Growth of Pulmonary Metastases via AT1R-Independent Inhibition of CCR2 Signaling and Monocyte Recruitment. J. Immunol. 2019, 202, 3087–3102. [Google Scholar] [CrossRef] [PubMed]
- Hashemzehi, M.; Naghibzadeh, N.; Asgharzadeh, F.; Mostafapour, A.; Hassanian, S.M.; Ferns, G.A.; Cho, W.C.; Avan, A.; Khazaei, M. The therapeutic potential of losartan in lung metastasis of colorectal cancer. EXCLI J. 2020, 19, 927–935. [Google Scholar]
- Gold, A.; Eini, L.; Nissim-Rafinia, M.; Viner, R.; Ezer, S.; Erez, K.; Aqaqe, N.; Hanania, R.; Milyavsky, M.; Meshorer, E.; et al. Spironolactone inhibits the growth of cancer stem cells by impairing DNA damage response. Oncogene 2019, 38, 3103–3118. [Google Scholar] [CrossRef]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D.M. Cardiotoxicity of anticancer drugs: The need for cardio-oncology and cardio-oncological prevention. J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Kalam, K.; Marwick, T.H. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: A systematic review and meta-analysis. Eur. J. Cancer 2013, 49, 2900–2909. [Google Scholar] [CrossRef]
- Perez, I.E.; Taveras Alam, S.; Hernandez, G.A.; Sancassani, R. Cancer Therapy-Related Cardiac Dysfunction: An Overview for the Clinician. Clin. Med. Insights Cardiol. 2019, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Tini, G.; Bertero, E.; Signori, A.; Sormani, M.P.; Maack, C.; De Boer, R.A.; Canepa, M.; Ameri, P. Cancer Mortality in Trials of Heart Failure With Reduced Ejection Fraction: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2020, 9. [Google Scholar] [CrossRef]
- Singh, S.; Hassan, D.; Aldawsari, H.M.; Molugulu, N.; Shukla, R.; Kesharwani, P. Immune checkpoint inhibitors: A promising anticancer therapy. Drug Discov. Today 2020, 25, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Cheng, T.; Lin, J.; Zhang, L.; Zheng, J.; Liu, Y.; Xie, G.; Wang, B.; Yuan, Y. Local angiotensin II contributes to tumor resistance to checkpoint immunotherapy. J. Immunother. Cancer 2018, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tomczak, K.; Li, J.; Ochieng, J.K.; Lee, Y.; Haymaker, C. Next-Generation Immunotherapies to Improve Anticancer Immunity. Front. Pharmacol. 2021, 11, 1947. [Google Scholar] [CrossRef] [PubMed]

| Clinical Study/Population | Type of Malignancy (Percentage) | HR, 95% IC, p Value | Findings |
|---|---|---|---|
| Hasin et al. 2013 [6]. Subjects newly diagnosed with HF and without cancer at index | Digestive system (19%) Male reproductive (19%) Hematologic (16%) Breast a (10%) Respiratory (8%) Urinary (8%) Female reproductive (3%) Skin (3%) Other cancers c (14%) | Not provided | HF patients had a 68% higher risk of developing cancer (HR: 1.68; 95% CI: 1.13–2.50) adjusted for body mass index, smoking, and comorbidities in the HF group. |
| Banke et al. 2016 [4]. HF patients without a prior diagnosis of cancer. | Lung (15.7%) | 1.81, 1.54–2.12, p < 0.0001 | Risk of any type of cancer increased (IRR: 1.24; 95% CI: 1.15–1.33, c < 0.0001), except for prostate cancer. |
| Skin (16.3%) | 1.84, 1.57–2.15, p < 0.0001 | ||
| Kidney and urinary system (8.2%) | 1.75, 1.41–2.18, p < 0.0001 | ||
| Liver/biliary system (4.7%) | 1.60, 1.20–2.13, p = 0.0015 | ||
| Lymph/blood (6.8%) | 1.45, 1.14–1.85, p = 0.0027 | ||
| Colon/rectal (12.3%) | 1.24, 1.04–1.49, p = 0.0180 | ||
| Breast a (4.8%) | 1.36, 1.02–1.81, p < 0.038 | ||
| Prostate b (13%) Other c (18%) | 1.04, 0.88–1.24, p < 0.6345- | ||
| Hasin et al. 2016 [7]. Survivors of a first MI who developed HF | Respiratory system: 29% Digestive system: 29% Hematologic: 14% Skin: 7% Male reproductive: 4% Breast: 4% Urinary: 4% Female reproductive: 4% | Not provided | Patients who develop HF after MI have an increased risk of cancer (HR: 2.16, 95% CI: 1.39–3.35). |
| Sakamoto et al. 2017 [8]. Chronic HF patients without a prior diagnosis of cancer. | Stomach (0.41%) | 95% IC: 0.25–0.61, p < 0.0001 * | The incidence of cancer in chronic HF patients was approximately four times higher contrasting with control patients (2.27% vs. 0.59%, 95% CI: 1.89–2.71, p < 0.0001). |
| Lung (0.22%) | 0.12–0.40, p = 0.0002 | ||
| Prostate (0.24%) | 0.09–0.46, p < 0.0001 | ||
| Breast (0.51%) | 0.26–0.93, p < 0.0001 | ||
| Colon (0.21%) | 0.10–0.38, p = 0.006 | ||
| Others d | 1.89–2.71, p < 0.0001 | ||
| Overall cancer (2,27%) | |||
| Kwak et al. 2021 [5]. Patients with HF and aged ≥20 years | Gastrointestinal (3.3%) | (1.49, 1.44–1.54, p < 0.0001) | Patients with HF presented a higher risk for cancer development compared to controls (HR: 1.64, 95% CI: 1.61–1.68) and the increased risk was consistent for all site-specific cancers. |
| Liver/Biliary/Pancreas (2.2%) | (1.80, 1.72–1.88), p < 0.0001) | ||
| Lung (2%) | (2.22, 2.12–2.32, p< 0.0001) | ||
| Prostate c (1.7%) | (1.40, 1.31–1.49, p < 0.0001) | ||
| Hematology (0.7%) | (2.77, 2.55–3.00, p < 0.0001) | ||
| Genitourinary (0.6%) | (1.55, 1.43–1.69, p < 0.0001) | ||
| Thyroid (0.4%) | 1.30, 1.18–1.43, p < 0.0001) | ||
| Breast b (0.6%) | (1.36, 1.21–1.52, p < 0.0001) | ||
| Female reproductive (0.6%) | (1.90, 1.68–2.15, p < 0.0001) | ||
| Head and neck (0.2%) | 1.62, 1.41–1.87, p < 0.0001 | ||
| Skin (0.04%) | (1.53, 1.11–2.11, p = 0.0081) | ||
| Overall cancer (9.2%) | (1.64, 1.61–1.68, p < 0.0001) |
| RAAS Inhibitor | Observations |
|---|---|
| Angiotensin converting enzyme inhibitors (ACEI) | |
| Captopril | Long-term administration was associated with an improvement in survival and reduced morbidity and mortality due to major cardiovascular events in patients with asymptomatic left ventricular (LV) dysfunction after myocardial infarction (MI) [82]. |
| Enalapril | Increased exercise time and left ventricular ejection fraction (LVEF) [83]. |
| Perindopril | Increased 6 min walk distance but did not decrease mortality [84]. After 1-year treatment reduced progressive LV remodeling but it was not associated with better clinical outcomes [85]. |
| Ramipril | Administration to patients with clinical evidence of either transient or ongoing heart failure (HF) after MI resulted in a substantial reduction in premature death from all causes [86]. |
| Trandolapril | Long-term treatment in patients with reduced LV function soon after MI significantly reduced the risk of overall mortality, mortality from cardiovascular causes, sudden death, and the development of severe HF [87]. |
| Angiotensin II type 1 receptor blockers (ARBs) | |
| Telmisartan | Telmisartan was well tolerated in patients unable to tolerate ACEI. Although the drug had no significant effect on hospitalizations for HF, it modestly reduced the risk of the composite outcome of cardiovascular death, MI, or stroke [88]. |
| Candesartan | Slightly decreased hospitalizations but did not decrease mortality [89]. Reduced cardiovascular mortality and hospital admissions for worsening chronic HF. Patients with reduced ejection fraction were the most benefited [90]. |
| Losartan | Reduced the rate of death or admission for HF in patients with HF, reduced LVEF, and intolerance to ACEI [91]. |
| Valsartan | In patients with MI associated with HF and/or LV dysfunction, valsartan administration in the immediate post MI period demonstrated equal efficacy than captopril [92,93]. |
| Aldosterone antagonists | |
| Spironolactone | Prevented LV fibrosis and remodeling after MI [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Garduño, T.C.; Padilla-Gutierrez, J.R.; Cambrón-Mora, D.; Valle, Y. RAAS: A Convergent Player in Ischemic Heart Failure and Cancer. Int. J. Mol. Sci. 2021, 22, 7106. https://doi.org/10.3390/ijms22137106
Garcia-Garduño TC, Padilla-Gutierrez JR, Cambrón-Mora D, Valle Y. RAAS: A Convergent Player in Ischemic Heart Failure and Cancer. International Journal of Molecular Sciences. 2021; 22(13):7106. https://doi.org/10.3390/ijms22137106
Chicago/Turabian StyleGarcia-Garduño, Texali C., Jorge R. Padilla-Gutierrez, Diego Cambrón-Mora, and Yeminia Valle. 2021. "RAAS: A Convergent Player in Ischemic Heart Failure and Cancer" International Journal of Molecular Sciences 22, no. 13: 7106. https://doi.org/10.3390/ijms22137106
APA StyleGarcia-Garduño, T. C., Padilla-Gutierrez, J. R., Cambrón-Mora, D., & Valle, Y. (2021). RAAS: A Convergent Player in Ischemic Heart Failure and Cancer. International Journal of Molecular Sciences, 22(13), 7106. https://doi.org/10.3390/ijms22137106