
Identification of 13 Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs to Potentially Inhibit TMPRSS2 for COVID-19 Treatment



Abstract
:
1. Introduction
2. Results
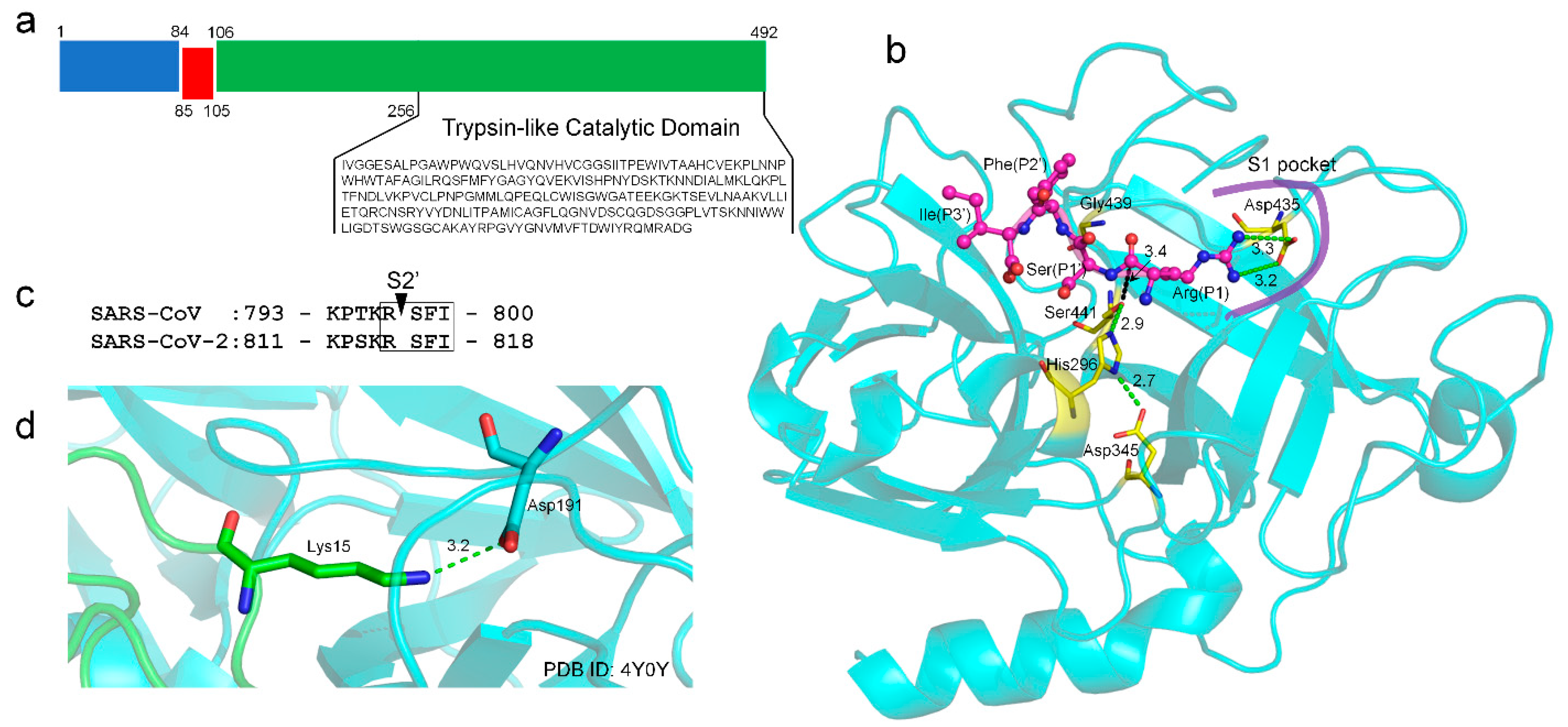
2.1. TMPRSS2 Sequence and Structural Model
2.2. Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs
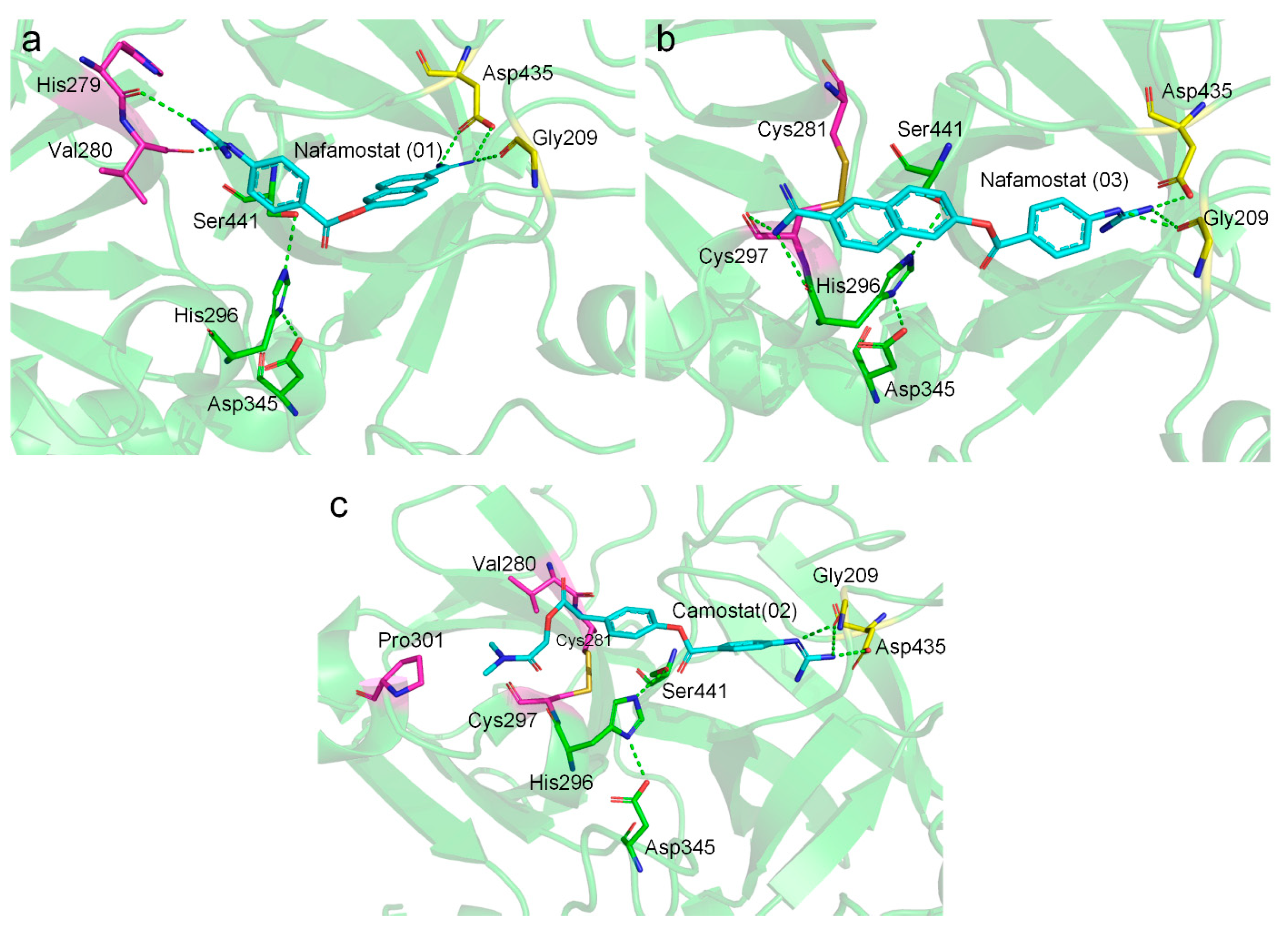
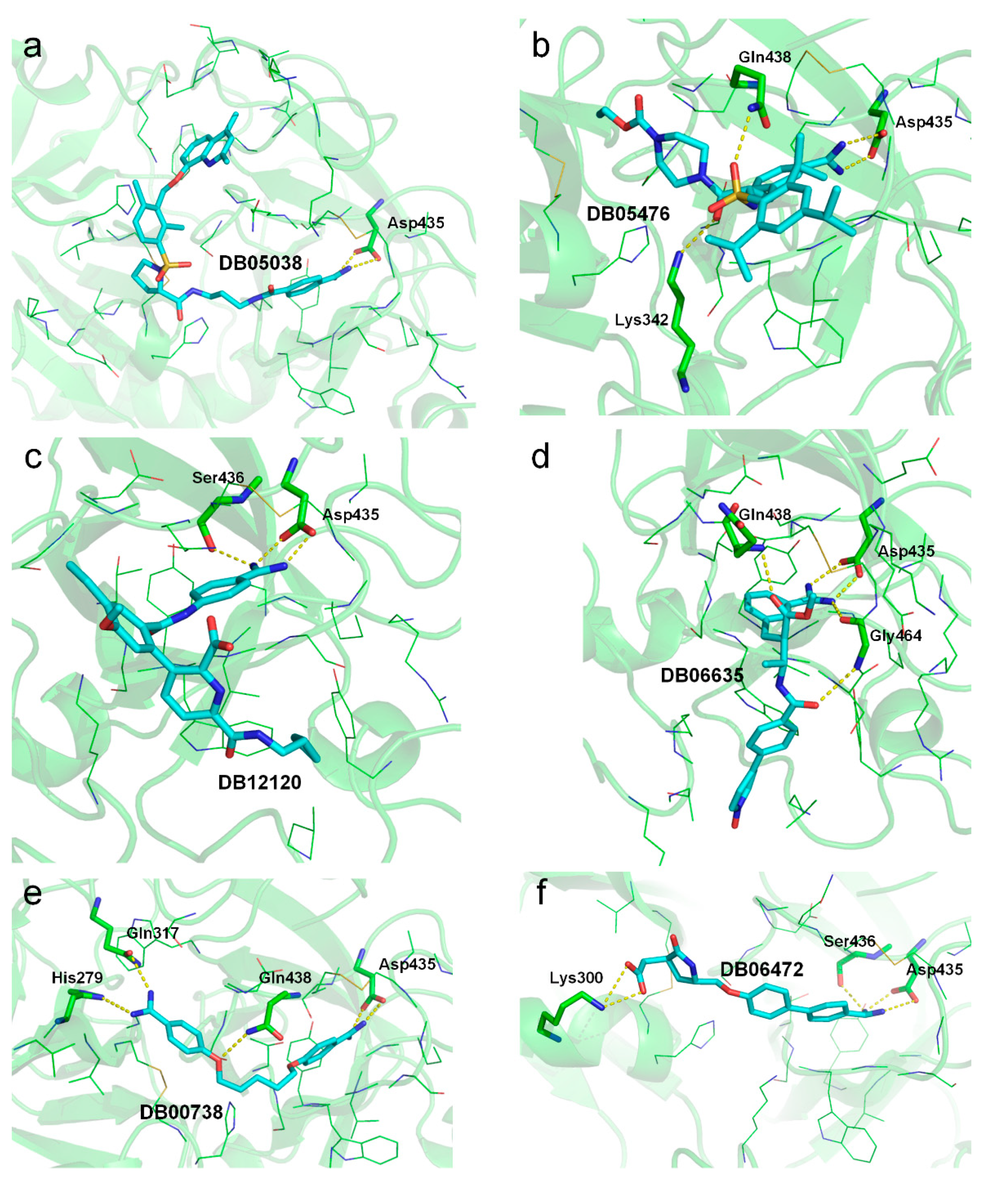
2.3. Molecular Docking Suggests Salt Bridge Interactions between Guanidinobenzoyl or Aminidinobenzoyl and Asp435
2.4. MD Simulations Reveal High Stability of the Putative Salt Bridge Interactions
2.5. MM/GBSA-Binding Free Energy Assessment Suggests High Stability of Binding
3. Discussion
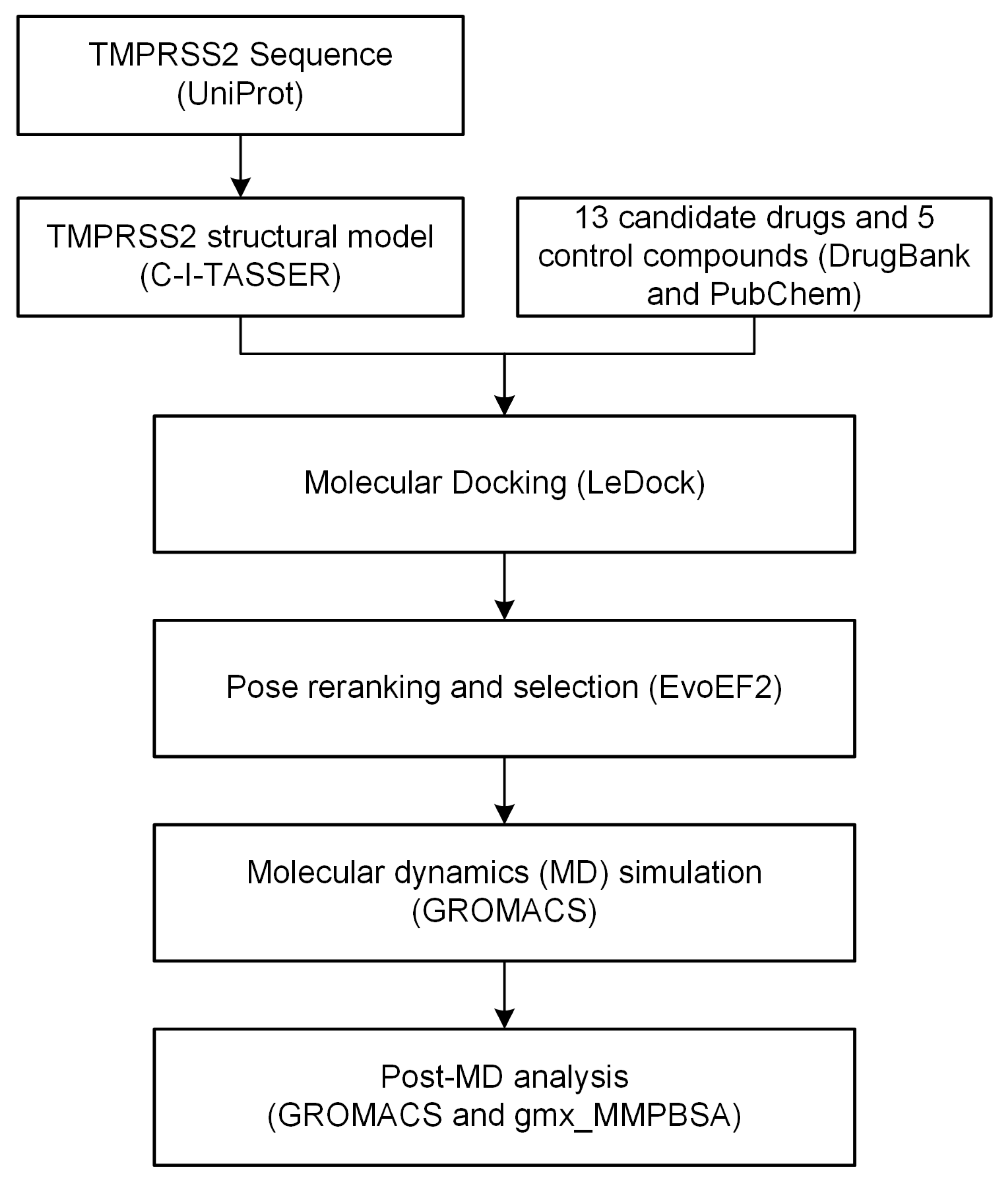
4. Materials and Methods
4.1. TMPRSS2 Structural Modeling and Model Quality Assessment
4.2. Drug Library Construction
4.3. Molecular Docking and Pose Reranking
4.4. MD Simulation
4.5. MM/GBSA-Binding Free Energy Calculation and Per-Residue Energy Decomposition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin-converting enzyme 2 |
| COVID-19 | coronavirus disease 2019 |
| EEU | EvoEF2 energy unit |
| FDA | United States Food and Drug Administration |
| GBPA | 4-(4-guanidinobenzoyloxy)phenylacetic acid |
| MD | molecular dynamics |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MM/GBSA | molecular mechanics/generalized Born surface area |
| PDB | Protein Data Bank |
| RMSD | root mean square deviation |
| RMSF | root mean square fluctuation |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SMILES | simplified molecular-input line-entry system |
| TMPRSS2 | transmembrane protease serine 2 |
References
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Cannalire, R.; Stefanelli, I.; Cerchia, C.; Beccari, A.R.; Pelliccia, S.; Summa, V. SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. Int. J. Mol. Sci. 2020, 21, 5707. [Google Scholar] [CrossRef]
- Stopsack, K.H.; Mucci, L.A.; Antonarakis, E.S.; Nelson, P.S.; Kantoff, P.W. TMPRSS2 and COVID-19: Serendipity or Opportunity for Intervention? Cancer Discov. 2020, 10, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Kruger, N.; Arora, P.; Sorensen, L.K.; Sogaard, O.S.; Hasselstrom, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Muller, M.A.; Drosten, C.; Pohlmann, S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, e00754-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Shrimp, J.H.; Guo, H.; Zakharov, A.; Jain, S.; Shinn, P.; Simeonov, A.; Hall, M.D.; Shen, M. Discovery of TMPRSS2 inhibitors from virtual screening. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shrimp, J.H.; Kales, S.C.; Sanderson, P.E.; Simeonov, A.; Shen, M.; Hall, M.D. An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef] [PubMed]
- Chikhale, R.V.; Gupta, V.K.; Eldesoky, G.E.; Wabaidur, S.M.; Patil, S.A.; Islam, M.A. Identification of potential anti-TMPRSS2 natural products through homology modelling, virtual screening and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- DurdaGi, S. Virtual drug repurposing study against SARS-CoV-2 TMPRSS2 target. Turk. J. Biol. 2020, 44, 185–191. [Google Scholar] [CrossRef]
- Elmezayen, A.D.; Al-Obaidi, A.; Sahin, A.T.; Yelekci, K. Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2020, 39, 2980–2992. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef]
- Singh, N.; Decroly, E.; Khatib, A.M.; Villoutreix, B.O. Structure-based drug repositioning over the human TMPRSS2 protease domain: Search for chemical probes able to repress SARS-CoV-2 Spike protein cleavages. Eur. J. Pharm. Sci. 2020, 153, 105495. [Google Scholar] [CrossRef]
- Hempel, T.; Raich, L.; Olsson, S.; Azouz, N.P.; Klingler, A.M.; Hoffmann, M.; Pöhlmann, S.; Rothenberg, M.E.; Noé, F. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem. Sci. 2021, 12, 983–992. [Google Scholar] [CrossRef]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [Green Version]
- Hörnich, B.F.; Großkopf, A.K.; Schlagowski, S.; Tenbusch, M.; Kleine-Weber, H.; Neipel, F.; Stahl-Hennig, C.; Hahn, A.S. SARS-CoV-2 and SARS-CoV spike-mediated cell-cell fusion differ in the requirements for receptor expression and proteolytic activation. J. Virol. 2021. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Huang, X.; Zhu, Y. Using molecular dynamics simulations to evaluate active designs of cephradine hydrolase by molecular mechanics/Poisson–Boltzmann surface area and molecular mechanics/generalized Born surface area methods. RSC Adv. 2019, 9, 13868–13877. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Li, Y.; Zhang, C.; Pearce, R.; Mortuza, S.; Zhang, Y. Deep-learning contact-map guided protein structure prediction in CASP13. Proteins 2019, 87, 1149–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Huggins, D.J. Structural analysis of experimental drugs binding to the SARS-CoV-2 target TMPRSS2. J. Mol. Graph. Model. 2020, 100, 107710. [Google Scholar] [CrossRef]
- Rensi, S.; Altman, R.B.; Liu, T.; Lo, Y.C.; McInnes, G.; Derry, A.; Keys, A. Homology Modeling of TMPRSS2 Yields Candidate Drugs That May Inhibit Entry of SARS-CoV-2 into Human Cells. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- Ye, S.; Loll, B.; Berger, A.A.; Mülow, U.; Alings, C.; Wahl, M.C.; Koksch, B. Fluorine teams up with water to restore inhibitor activity to mutant BPTI. Chem. Sci. 2015, 6, 5246–5254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalante, D.E.; Ferguson, D.M. Structural modeling and analysis of the SARS-CoV-2 cell entry inhibitor camostat bound to the trypsin-like protease TMPRSS2. Med. Chem. Res. 2021, 30, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Chiarparin, E.; Packer, M.J.; Wilson, D.M. Experimental free ligand conformations: A missing link in structure-based drug discovery. Future Med. Chem. 2019, 11, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Lawson, A.D.G.; MacCoss, M.; Heer, J.P. Importance of Rigidity in Designing Small Molecule Drugs To Tackle Protein-Protein Interactions (PPIs) through Stabilization of Desired Conformers. J. Med. Chem. 2018, 61, 4283–4289. [Google Scholar] [CrossRef]
- Verlinde, C.L.; Hol, W.G. Structure-based drug design: Progress, results and challenges. Structure 1994, 2, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, H. Enriching screening libraries with bioactive fragment space. Bioorg. Med. Chem. Lett. 2016, 26, 3594–3597. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Pearce, R.; Zhang, Y. EvoEF2: Accurate and fast energy function for computational protein design. Bioinformatics 2020, 36, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tresanco, M.S.V.; Tresanco, M.E.V.; Valiente, P.A.; Frías, E.M. gmx_MMPBSA (Version v1.4.2). Zenodo. 2021. Available online: http://doi.org/10.5281/zenodo.4569307 (accessed on 18 June 2021).
- Armstrong, D.R.; Berrisford, J.M.; Conroy, M.J.; Gutmanas, A.; Anyango, S.; Choudhary, P.; Clark, A.R.; Dana, J.M.; Deshpande, M.; Dunlop, R.; et al. PDBe: Improved findability of macromolecular structure data in the PDB. Nucleic Acids Res. 2020, 48, D335–D343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D. XSEDE: Accelerating scientific discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DrugBank ID | Drug Name | Status with FDA | Primary Indication |
|---|---|---|---|
| DB00738 | Pentamidine | Approved | For the treatment of pneumocystis pneumonia |
| DB03808 | Hexamidine | Approved | For the treatment of acanthamoebiasis |
| DB05038 | Anatibant | Investigational | For the treatment of traumatic brain injuries |
| DB05476 | WX-UK1 | Investigational | For the treatment in solid tumors |
| DB06472 | Fradafiban | Investigational | For the treatment in angina |
| DB06635 | Otamixaban | Investigational | For the treatment of thrombosis |
| DB12120 | Avoralstat | Investigational | For the prevention of hereditary angioedema |
| DB12598 | Nafamostat | Investigational | Used as an anticoagulant in patients with disseminative blood vessel coagulation, hemorrhagic lesions, and hemorrhagic tendencies |
| DB13000 | PCI-27483 | Investigational | For the treatment of pancreatic cancer, ductal adrenocarcinoma, and exocrine pancreatic cancer |
| DB13296 | Propamidine | Investigational | For the treatment of Acanthamoeba infection |
| DB13729 | Camostat | Investigational | For the treatment of chronic pancreatitis and drug-induced lung injury |
| DB14726 | Dabigatran | Investigational | For the treatment and prevention of blood clots and prevention of stroke in people with atrial fibrillation |
| DB14753 | Hydroxystilbamidine | Approved | For the treatment of nonprogressive blastomycosis of the skin and other mycoses |
| DrugBank ID | Drug Name | Number of Poses by LeDock | Best Score | |
|---|---|---|---|---|
| LeDock (kcal/mol) | EvoEF2 (EEU) | |||
| DB00738 | Pentamidine | 12 | −8.73 | −37.36 |
| DB03808 | Hexamidine | 15 | −8.47 | −35.08 |
| DB05038 | Anatibant | 18 | −8.60 | −34.21 |
| DB05476 | WX-UK1 | 17 | −8.57 | −39.29 |
| DB06472 | Fradafiban | 8 | −7.32 | −30.48 |
| DB06635 | Otamixaban | 17 | −8.50 | −40.43 |
| DB12120 | Avoralstat | 7 | −9.35 | −46.30 |
| DB12598 | Nafamostat | 6 | −8.61 | −34.70 |
| DB13000 | PCI-27483 | 16 | −9.44 | −36.84 |
| DB13296 | Propamidine | 14 | −7.93 | −32.48 |
| DB13729 | Camostat | 13 | −8.02 | −27.02 |
| DB14726 | Dabigatran | 15 | −9.51 | −42.58 |
| DB14753 | Hydroxystilbamidine | 4 | −7.56 | −32.56 |
| Drug Name | DrugBank ID_pose | Ligand RMSD (Å) | |
|---|---|---|---|
| Mean ± std | Median | ||
| Pentamidine | DB00738_05 | 3.2 ± 1.0 | 3.1 |
| DB00738_10 | 2.7 ± 1.6 | 2.2 | |
| Hexamidine | DB03808_03 | 3.3 ± 1.4 | 2.7 |
| Anatibant | DB05038_10 | 3.3 ± 0.6 | 3.1 |
| WX-UK1 | DB05476_01 | 2.7 ± 0.3 | 2.6 |
| DB05476_15 | 2.8 ± 0.3 | 2.7 | |
| Fradafiban | DB06472_03 | 3.2 ± 1.4 | 2.9 |
| Otamixaban | DB06635_02 | 3.1 ± 0.4 | 3.1 |
| DB06635_03 | 2.5 ± 1.6 | 2.1 | |
| DB06635_04 | 2.9 ± 0.7 | 2.7 | |
| DB06635_05 | 2.6 ± 0.5 | 2.7 | |
| Avoralstat | DB12120_04 | 2.9 ± 0.6 | 2.8 |
| DB12120_05 | 2.9 ± 0.7 | 2.7 | |
| Nafamostat | DB12598_01 | 3.3 ± 0.7 | 3.2 |
| DB12598_04 | 2.1 ± 0.5 | 2.1 | |
| DB12598_05 | 2.8 ± 0.7 | 2.6 | |
| DB12598_06 | 3.3 ± 0.7 | 3.4 | |
| Propamidine | DB13296_01 | 2.3 ± 0.7 | 2.1 |
| DB13296_06 | 2.3 ± 0.7 | 2.2 | |
| DB13296_09 | 2.5 ± 0.6 | 2.4 | |
| DB13296_13 | 3.5 ± 2.4 | 2.3 | |
| Camostat | DB13729_04 | 3.1 ± 0.6 | 2.9 |
| Hydroxystilbamidine | DB14753_02 | 2.9 ± 1.0 | 3.0 |
| DB14753_04 | 2.6 ± 0.7 | 2.4 | |
| DrugBank ID_pose | (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) |
|---|---|---|---|---|---|---|
| DB00738_10 | −39.95 ± 3.97 | −45.66 ± 3.95 | 43.74 ± 3.42 | −5.63 ± 0.34 | 12.64 | −34.86 ± 3.96 |
| DB03808_03 | −39.57 ± 3.60 | −37.65 ± 4.17 | 38.21 ± 3.81 | −5.25 ± 0.28 | 13.40 | −30.86 ± 3.38 |
| DB05038_10 | −56.66 ± 4.21 | −25.38 ± 7.13 | 34.16 ± 6.26 | −6.52 ± 0.41 | 13.62 | −40.77 ± 4.06 |
| DB05476_01 | −45.72 ± 5.58 | −61.61 ± 4.81 | 64.33 ± 3.68 | −5.97 ± 0.60 | 9.73 | −39.23 ± 4.77 |
| DB06472_03 | −35.72 ± 5.38 | −24.09 ± 6.50 | 25.59 ± 5.83 | −4.21 ± 0.51 | 12.23 | −26.20 ± 5.19 |
| DB06635_05 | −44.42 ± 3.83 | −53.98 ± 5.26 | 59.27 ± 4.07 | −5.52 ± 0.34 | 9.35 | −35.30 ± 2.95 |
| DB12120_04 | −41.16 ± 4.80 | −23.86 ± 3.92 | 25.42 ± 2.90 | −5.23 ± 0.36 | 8.11 | −36.71 ± 3.97 |
| DB12598_04 | −37.96 ± 3.40 | −39.78 ± 4.44 | 39.03 ± 4.10 | −4.72 ± 0.25 | 12.61 | −30.83 ± 3.33 |
| DB13000_04 | −43.08 ± 6.34 | −35.35 ± 7.21 | 34.19 ± 6.28 | −5.21 ± 0.60 | 19.63 | −29.82 ± 6.29 |
| DB13296_06 | −37.30 ± 3.13 | −40.16 ± 3.87 | 39.34 ± 3.42 | −5.12 ± 0.21 | 10.75 | −32.49 ± 2.77 |
| DB13729_04 | −41.84 ± 3.21 | −49.83 ± 6.21 | 54.96 ± 5.21 | −5.27 ± 0.35 | 11.56 | −30.42 ± 3.02 |
| DB14726_08 | −37.90 ± 4.14 | −31.49 ± 4.79 | 31.87 ± 3.85 | −4.81 ± 0.42 | 11.68 | −30.65 ± 3.61 |
| DB14753_04 | −25.85 ± 4.42 | −42.78 ± 4.90 | 39.32 ± 4.67 | −3.67 ± 0.35 | 18.06 | −14.92 ± 3.54 |
| DrugBank ID | Residues (Per-Residue Energy in kcal/mol) |
|---|---|
| DB00738 | His279 (−0.95), Val280 (−0.82), Cys281 (−0.58), His296 (−1.21), Cys297 (−0.77), Thr393 (−0.59), Asp435 (−1.64), Ser436 (−1.45), Cys437 (−1.77), Gln438 (−2.41), Gly439 (−1.60), Asp440 (−0.82), Ser441 (−1.22), Thr459 (−0.59), Ser460 (−0.55), Trp461 (−1.83), Gly462 (−0.88), Cys465 (−0.67), Tyr474 (−0.54) |
| DB03808 | His279 (−0.50), Val280 (−1.62), Cys281 (−0.62), His296 (−1.59), Cys297 (−0.97), Asp435 (−1.52), Ser436 (−1.52), Cys437 (−1.72), Gln438 (−2.08), Asp440 (−0.57), Ser441 (−0.97), Thr459 (−0.69), Trp461 (−1.68), Gly462 (−0.94), Ser463 (−0.54), Cys465 (−0.64) |
| DB05038 | His279 (−0.55), Val280 (−0.84), Cys281 (−0.73), His296 (−1.26), Cys297 (−0.70), Lys342 (−0.77), Thr393 (−0.85), Asp435 (−2.33), Ser436 (−2.87), Cys437 (−1.74), Gln438 (−2.55), Gly439 (−1.55), Asp440 (−0.75), Ser441 (−0.94), Trp461 (−1.05), Gly462 (−1.19), Ser463 (−0.86), Cys465 (−0.97) |
| DB05476 | Val280 (−0.61), His296 (−1.08), Asp435 (−2.98), Ser436 (−0.78), Cys437 (−1.44), Gln438 (−2.41), Gly439 (−0.89), Asp440 (−0.65), Ser441 (−1.45), Trp461 (−2.70), Gly462 (−0.97), Ser463 (−2.65), Gly464 (−1.91) |
| DB06472 | Val280 (−0.80), His296 (−1.04), Cys297 (−0.54), Asp435 (−1.31), Ser436 (−1.42), Cys437 (−2.00), Gln438 (−2.35), Gly439 (−0.90), Asp440 (−0.85), Ser441 (−0.94), Thr459 (−0.60), Trp461 (−1.19), Gly462 (−0.90), Ser463 (−0.64), Cys465 (−0.85) |
| DB06635 | Thr341 (−1.06), Lys342 (−0.60), Leu419 (−1.52), Asp435 (−3.64), Cys437 (−1.56), Gln438 (−1.68), Asp440 (−0.64), Thr459 (−0.51), Trp461 (−5.00), Gly462 (−1.53), Ser463 (−1.43), Gly464 (−1.22) |
| DB12120 | Lys342 (−0.65), Tyr416 (−0.60), Asp435 (−1.93), Ser436 (−0.86), Cys437 (−1.82), Gln438 (−3.67), Thr459 (−0.58), Trp461 (−3.70), Gly462 (−2.26), Ser463 (−1.89), Gly464 (−0.53), Cys465 (−0.65) |
| DB12598 | Val280 (−0.90), Cys281 (−0.59), His296 (−1.73), Cys297 (−0.85), Asp435 (−1.73), Ser436 (−1.54), Cys437 (−2.11), Gln438 (−2.48), Asp440 (−0.96), Ser441 (−1.31), Thr459 (−0.60), Trp461 (−1.40), Gly462 (−0.95), Ser463 (−0.91), Cys465 (−0.64) |
| DB13000 | His296 (−0.84), Asp435 (−0.71), Ser436 (−1.22), Cys437 (−2.18), Gln438 (−4.36), Gly439 (−1.32), Asp440 (−0.55), Ser441 (−1.10), Thr459 (−0.58), Trp461 (−1.69), Gly462 (−1.44), Ser463 (−1.58), Gly464 (−0.54), Cys465 (−0.91), Pro471 (−0.60) |
| DB13296 | His279 (−0.93), Val280 (−0.82), Cys281 (−0.80), His296 (−0.64), Asp435 (−1.65), Ser436 (−1.30), Cys437 (−1.99), Gln438 (−2.04), Gly439 (−1.49), Asp440 (−0.99), Ser441 (−1.25), Thr459 (−0.64), Trp461 (−1.58), Gly462 (−0.97), Cys465 (−0.56) |
| DB13729 | Val278 (−0.66), Val280 (−2.04), Cys281 (−0.74), His296 (−1.05), Cys297 (−0.70), Asp435 (−2.58), Ser436 (−0.69), Cys437 (−1.62), Gln438 (−1.40), Gly439 (−1.04), Asp440 (−0.95), Ser441 (−1.50), Thr459 (−0.69), Trp461 (−1.10), Gly462 (−0.72), Gly464 (−0.84) |
| DB14726 | Asp435 (−3.07), Ser436 (−0.76), Cys437 (−2.02), Gln438 (−3.05), Asp440 (−0.57), Thr459 (−0.64), Trp461 (−1.44), Gly462 (−1.63), Ser463 (−1.25), Gly464 (−0.52), Cys465 (−1.24), Val473 (−0.76), Tyr474 (−1.94) |
| DB14753 | His296 (−0.97), Asp435 (−2.48), Ser436 (−1.25), Cys437 (−2.10), Gln438 (−1.98), Gly439 (−0.67), Asp440 (−0.77), Ser441 (−0.69), Thr459 (−0.58), Trp461 (−1.44), Gly462 (−1.13), Gly464 (−0.66), Cys465 (−0.79) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Pearce, R.; Omenn, G.S.; Zhang, Y. Identification of 13 Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs to Potentially Inhibit TMPRSS2 for COVID-19 Treatment. Int. J. Mol. Sci. 2021, 22, 7060. https://doi.org/10.3390/ijms22137060
Huang X, Pearce R, Omenn GS, Zhang Y. Identification of 13 Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs to Potentially Inhibit TMPRSS2 for COVID-19 Treatment. International Journal of Molecular Sciences. 2021; 22(13):7060. https://doi.org/10.3390/ijms22137060
Chicago/Turabian StyleHuang, Xiaoqiang, Robin Pearce, Gilbert S. Omenn, and Yang Zhang. 2021. "Identification of 13 Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs to Potentially Inhibit TMPRSS2 for COVID-19 Treatment" International Journal of Molecular Sciences 22, no. 13: 7060. https://doi.org/10.3390/ijms22137060
APA StyleHuang, X., Pearce, R., Omenn, G. S., & Zhang, Y. (2021). Identification of 13 Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs to Potentially Inhibit TMPRSS2 for COVID-19 Treatment. International Journal of Molecular Sciences, 22(13), 7060. https://doi.org/10.3390/ijms22137060