
Fructose and the Liver


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Deleterious Metabolic Effects of Fructose
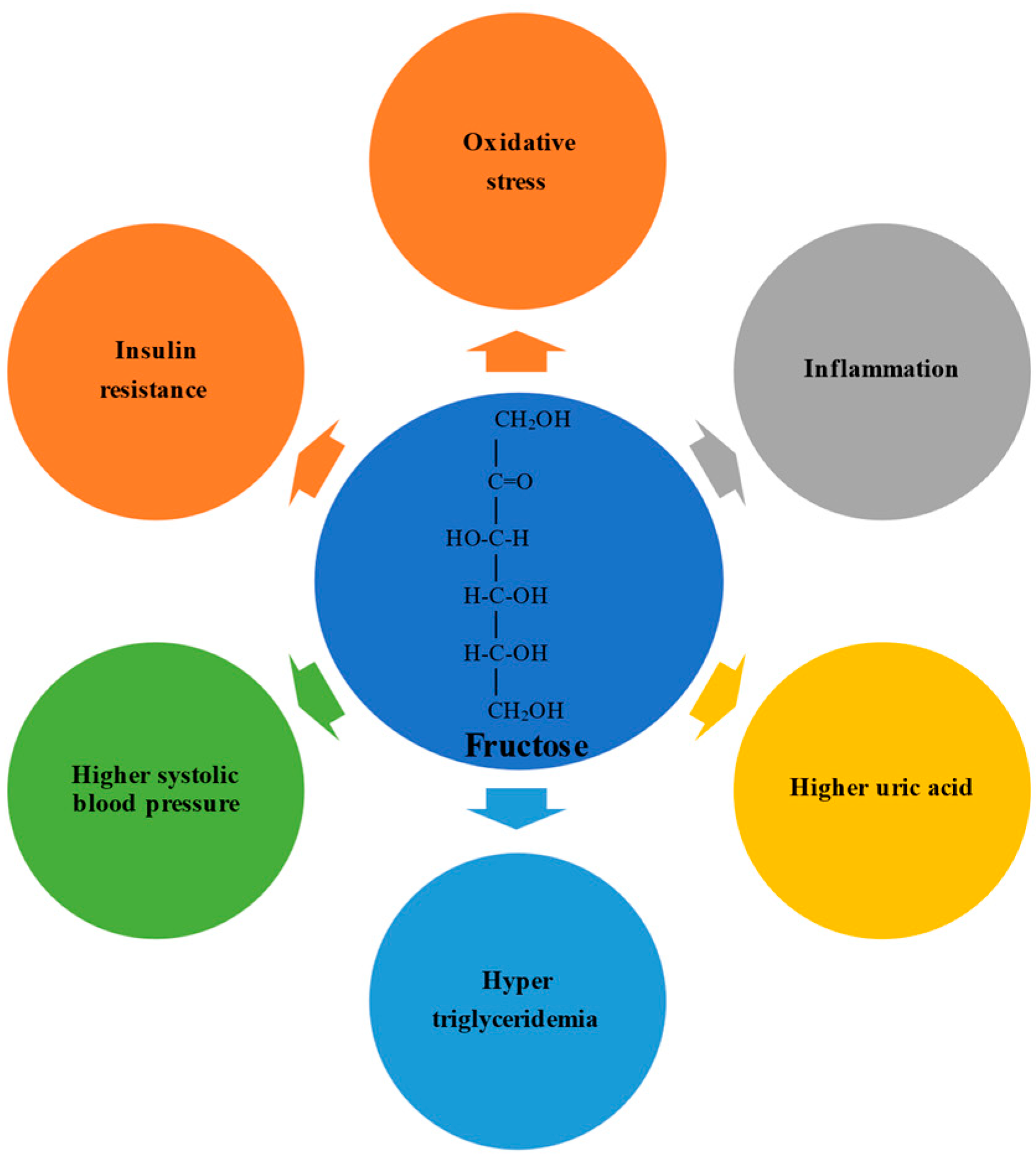
2.1. The Initial Physiological Impact of High Fructose Consumption
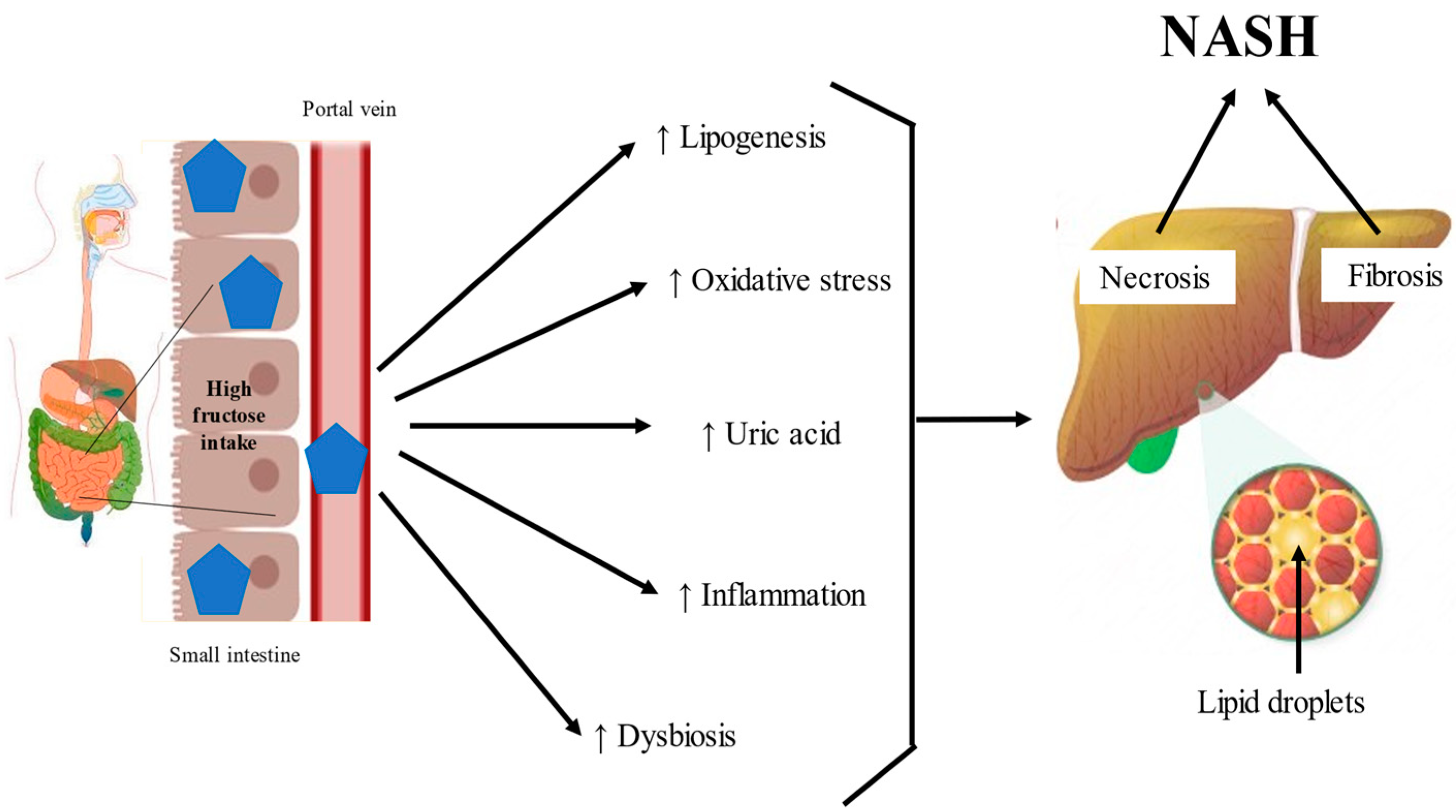
2.2. Fructose in the Intestine
2.2.1. Intestinal Absorption of Fructose
2.2.2. Intestinal Production of Uric Acid by Fructose
2.2.3. Fructose Induces Lipogenesis and Oxidative Stress in the Intestine
2.2.4. Fructose and the Microbiota
2.3. Fructose in the Liver
2.3.1. Ketohexokinase and Fructose
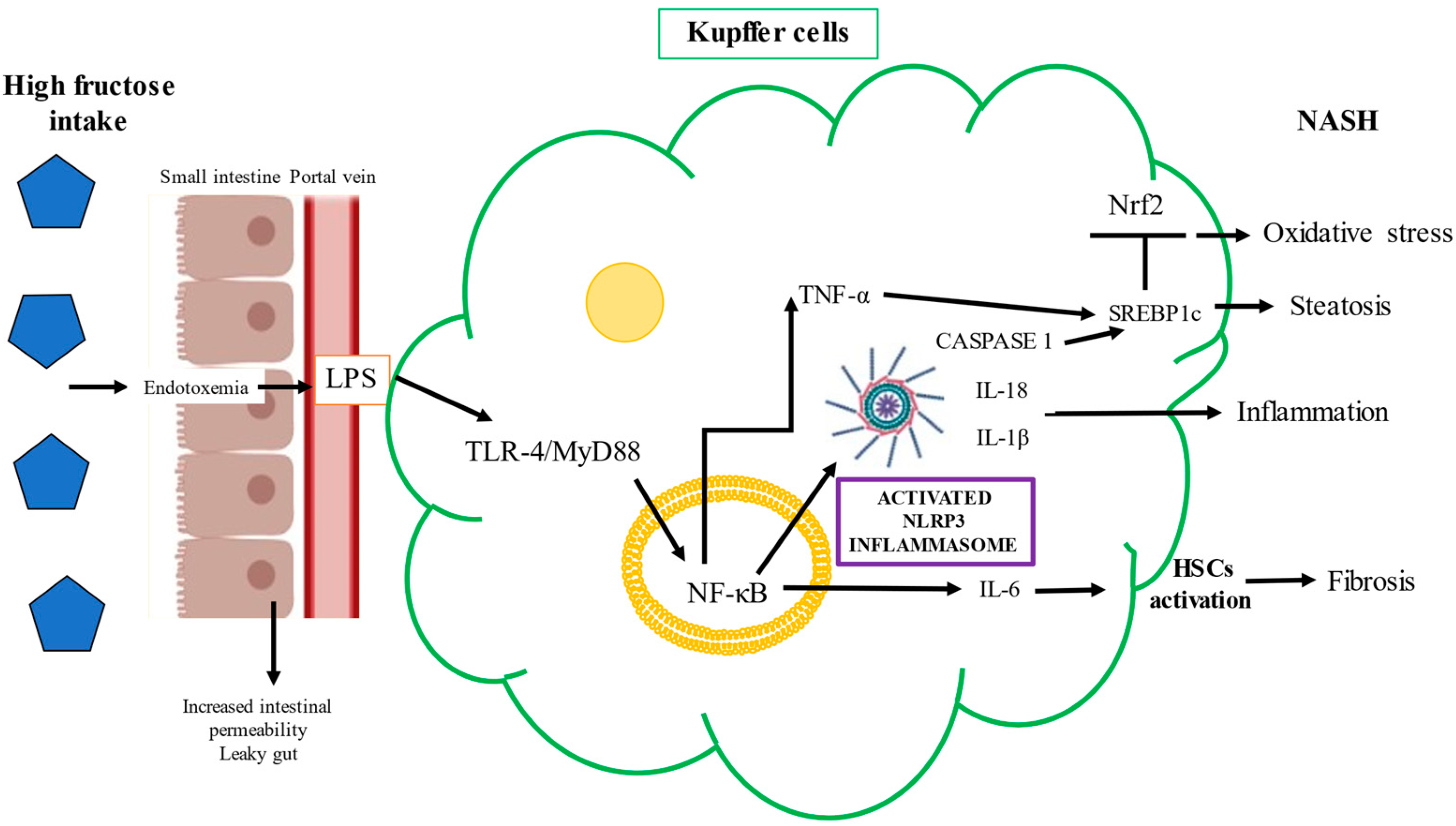
2.3.2. Toll-Like Receptor-4 and Fructose
2.3.3. Nuclear Factor E2-Related Factor 2 and Fructose
2.3.4. Carbohydrate Responsive Element-Binding Protein and Fructose
2.3.5. Sterol-Responsive Element-Binding Protein and Fructose
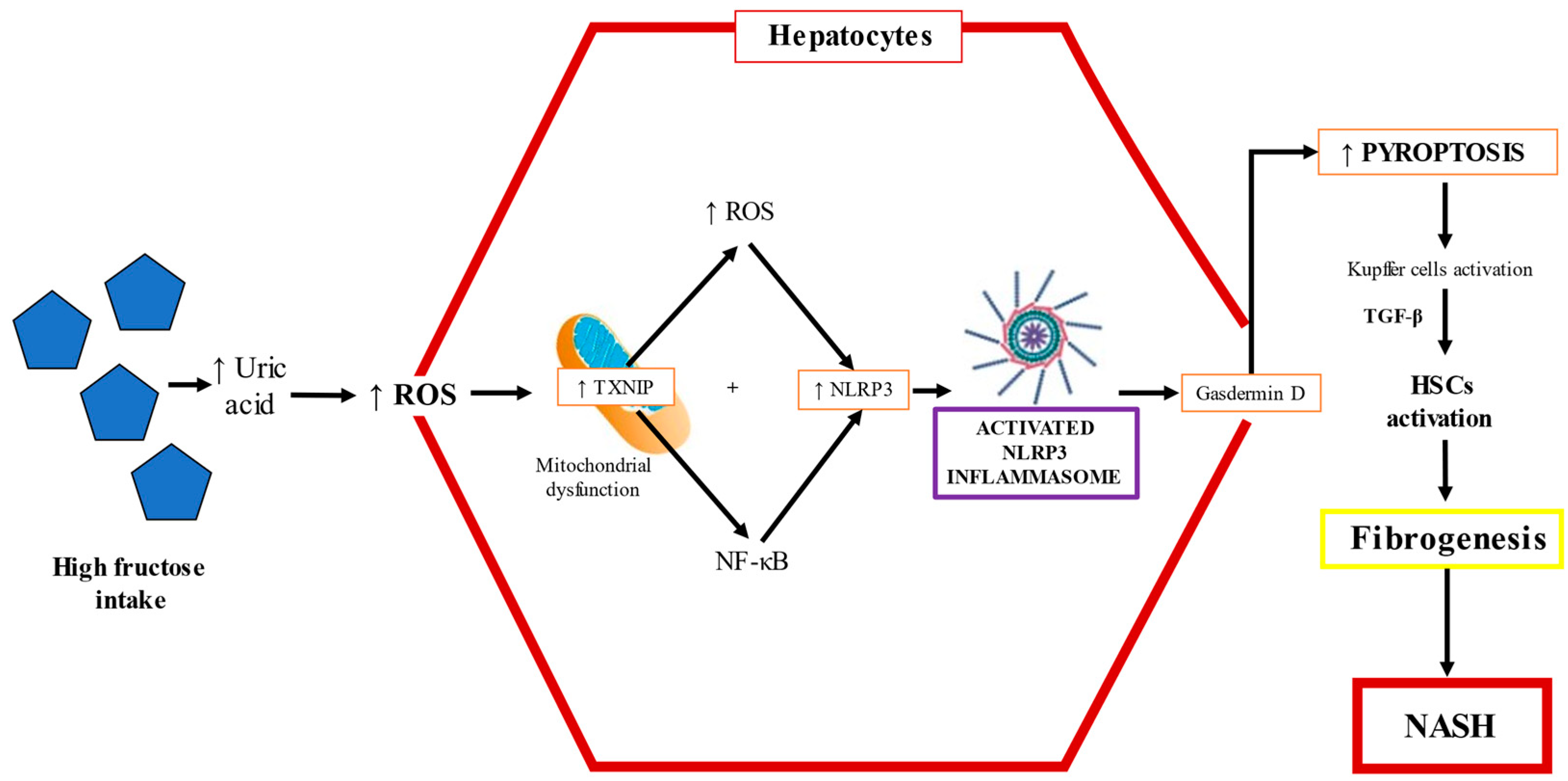
2.3.6. Uric Acid and Fructose
2.3.7. MicroRNAs and Fructose
2.3.8. Cancer and Fructose
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eckel, R.H.; Alberti, K.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Streba, L.A.M. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: An open question. World J. Gastroenterol. 2015, 21, 4103. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.A.; Mohammadnezhad, M. Prevalence of Metabolic Syndrome Among Apparently Healthy Workforce. J. Ayub Med. Coll. 2019, 31, 252–254. [Google Scholar]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.-R.; Morrison, C.D.; Münzberg, H. The obesity epidemic in the face of homeostatic body weight regulation: What went wrong and how can it be fixed? Physiol. Behav. 2020, 222, 112959. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020. [Google Scholar] [CrossRef]
- Wai-Sun Wong, V.; Lai-Hung Wong, G.; Woo, J.; Abrigo, J.M.; Ka-Man Chan, C.; She-Ting Shu, S.; Ka-Yu Leung, J.; Mei-Ling Chim, A.; Pik-Shan Kong, A.; Chung-Yan Lui, G.; et al. Impact of the New Definition of Metabolic Associated Fatty Liver Disease on the Epidemiology of the Disease. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Nagaoki, Y.; Hyogo, H.; Aikata, H.; Tanaka, M.; Naeshiro, N.; Nakahara, T.; Honda, Y.; Miyaki, D.; Kawaoka, T.; Takaki, S.; et al. Recent trend of clinical features in patients with hepatocellular carcinoma. Hepatol. Res. 2012, 42, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R. Primary hepatocellular carcinoma and metabolic syndrome: An update. World J. Gastrointest. Oncol. 2013, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.C.; Niederpruem, M.G.; Wallace, J.P.; Lindeman, A.K. Dietary fat, sugar, and fiber predict body fat content. J. Am. Diet. Assoc. 1994, 94, 612–615. [Google Scholar] [CrossRef]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-medicare database. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef]
- Margini, C.; Dufour, J.F. The story of HCC in NAFLD: From epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2016, 36, 317–324. [Google Scholar] [CrossRef]
- Muhidin, S.O.; Magan, A.A.; Osman, K.A.; Syed, S.; Ahmed, M.H. The Relationship between Nonalcoholic Fatty Liver Disease and Colorectal Cancer: The Future Challenges and Outcomes of the Metabolic Syndrome. J. Obes. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Wong, M.C.S.; Jiang, J.Y.; Goggins, W.B.; Liang, M.; Fang, Y.; Fung, F.D.H.; Leung, C.; Wang, H.H.X.; Wong, G.L.H.; Wong, V.W.S.; et al. International incidence and mortality trends of liver cancer: A global profile. Sci. Rep. 2017, 7, 45846. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Li, L.; Dai, J.; Natarajan, Y.; Yu, X.; Asch, S.M.; El-Serag, H.B. Effect of Metabolic Traits on the Risk of Cirrhosis and Hepatocellular Cancer in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 808–819. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Kucera, O. Experimental models of non-alcoholic fatty liver disease in rats. World J. Gastroenterol. 2014, 20, 8364. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef]
- Kmietowicz, Z. Countries that use large amounts of high fructose corn syrup have higher rates of type 2 diabetes. BMJ 2012, 345, e7994. [Google Scholar] [CrossRef]
- Rosas-Villegas, A.; Sánchez-Tapia, M.; Avila-Nava, A.; Ramírez, V.; Tovar, A.; Torres, N. Differential Effect of Sucrose and Fructose in Combination with a High Fat Diet on Intestinal Microbiota and Kidney Oxidative Stress. Nutrients 2017, 9, 393. [Google Scholar] [CrossRef]
- Johnson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J. Dietary Sugars Intake and Cardiovascular Health. Circulation 2009, 120, 1011–1020. [Google Scholar] [CrossRef]
- Basto-Abreu, A.; Braverman-Bronstein, A.; Camacho-García-Formentí, D.; Zepeda-Tello, R.; Popkin, B.M.; Rivera-Dommarco, J.; Hernández-Ávila, M.; Barrientos-Gutiérrez, T. Expected changes in obesity after reformulation to reduce added sugars in beverages: A modeling study. PLoS Med. 2018, 15, e1002664. [Google Scholar] [CrossRef]
- Powell, E.S.; Smith-Taillie, L.P.; Popkin, B.M. Added Sugars Intake Across the Distribution of US Children and Adult Consumers: 1977-2012. J. Acad. Nutr. Diet. 2016, 116, 1543–1550.e1. [Google Scholar] [CrossRef]
- Santhekadur, P.K. The dark face of fructose as a tumor promoter. Genes Dis. 2020, 7, 163–165. [Google Scholar] [CrossRef]
- WHO Guideline: Sugars Intake for Adult and Children; WHO: Geneva, Switzerland, 2015.
- Caliceti, C.; Calabria, D.; Roda, A.; Cicero, A. Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients 2017, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef]
- Tappy, L. Fructose-containing caloric sweeteners as a cause of obesity and metabolic disorders. J. Exp. Biol. 2018, 221, jeb164202. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Ferrocino, I.; Liberto, E.; Chiazza, F.; Cento, A.S.; Collotta, D.; Querio, G.; Nigro, D.; Bitonto, V.; Cutrin, J.C.; et al. Fructose liquid and solid formulations differently affect gut integrity, microbiota composition and related liver toxicity: A comparative in vivo study. J. Nutr. Biochem. 2018, 55, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Roglans, N.; Baena, M.; Sánchez, R.M.; Merlos, M.; Alegret, M.; Laguna, J.C. Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 514–524. [Google Scholar] [CrossRef]
- Glendinning, J.I.; Frim, Y.G.; Hochman, A.; Lubitz, G.S.; Basile, A.J.; Sclafani, A. Glucose elicits cephalic-phase insulin release in mice by activating K ATP channels in taste cells. Am. J. Physiol. Integr. Comp. Physiol. 2017, 312, R597–R610. [Google Scholar] [CrossRef]
- Bodnaruc, A.M.; Prud’homme, D.; Blanchet, R.; Giroux, I. Nutritional modulation of endogenous glucagon-like peptide-1 secretion: A review. Nutr. Metab. 2016, 13, 92. [Google Scholar] [CrossRef]
- Koliaki, C.; Kokkinos, A.; Tentolouris, N.; Katsilambros, N. The Effect of Ingested Macronutrients on Postprandial Ghrelin Response: A Critical Review of Existing Literature Data. Int. J. Pept. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yau, A.M.W.; McLaughlin, J.; Maughan, R.J.; Gilmore, W.; Evans, G.H. Short-term dietary supplementation with fructose accelerates gastric emptying of a fructose but not a glucose solution. Nutrition 2014, 30, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, M.; Lallès, J.-P.; Malbert, C.-H.; Val-Laillet, D. Dietary sugars: Their detection by the gut–brain axis and their peripheral and central effects in health and diseases. Eur. J. Nutr. 2015, 54, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H. Fructose: Metabolic, Hedonic, and Societal Parallels with Ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, R.P.; Choe, J.; Patel, C.R. Intestinal Absorption of Fructose. Annu. Rev. Nutr. 2018, 38, 41–67. [Google Scholar] [CrossRef] [PubMed]
- Lowette, K.; Roosen, L.; Tack, J.; Vanden Berghe, P. Effects of High-Fructose Diets on Central Appetite Signaling and Cognitive Function. Front. Nutr. 2015, 2. [Google Scholar] [CrossRef]
- Kishida, K.; Pearce, S.C.; Yu, S.; Gao, N.; Ferraris, R.P. Nutrient sensing by absorptive and secretory progenies of small intestinal stem cells. Am. J. Physiol. Liver Physiol. 2017, 312, G592–G605. [Google Scholar] [CrossRef]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) Is Essential for the Absorption of Fructose in the Intestine and Generation of Fructose-induced Hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef]
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef]
- Patel, C.; Douard, V.; Yu, S.; Tharabenjasin, P.; Gao, N.; Ferraris, R.P. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am. J. Physiol. Integr. Comp. Physiol. 2015, 309, R499–R509. [Google Scholar] [CrossRef]
- Kellett, G.L.; Brot-Laroche, E.; Mace, O.J.; Leturque, A. Sugar Absorption in the Intestine: The Role of GLUT2. Annu. Rev. Nutr. 2008, 28, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Packard, C.J.; Borén, J. Dietary Fructose and the Metabolic Syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef]
- Zwarts, I.; van Zutphen, T.; Kruit, J.K.; Liu, W.; Oosterveer, M.H.; Verkade, H.J.; Uhlenhaut, N.H.; Jonker, J.W. Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR. Sci. Rep. 2019, 9, 9299. [Google Scholar] [CrossRef]
- Jones, H.F.; Butler, R.N.; Brooks, D.A. Intestinal fructose transport and malabsorption in humans. Am. J. Physiol. Liver Physiol. 2011, 300, G202–G206. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.M. Role of amino acid transporters in amino acid sensing. Am. J. Clin. Nutr. 2014, 99, 223S–230S. [Google Scholar] [CrossRef]
- Shah, A.; Dagdeviren, S.; Lewandowski, J.P.; Schmider, A.B.; Ricci-Blair, E.M.; Natarajan, N.; Hundal, H.; Noh, H.L.; Friedline, R.H.; Vidoudez, C.; et al. Thioredoxin Interacting Protein Is Required for a Chronic Energy-Rich Diet to Promote Intestinal Fructose Absorption. iScience 2020, 23, 101521. [Google Scholar] [CrossRef]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin Interacting Protein: Redox Dependent and Independent Regulatory Mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Jang, C.; Wada, S.; Yang, S.; Gosis, B.; Zeng, X.; Zhang, Z.; Shen, Y.; Lee, G.; Arany, Z.; Rabinowitz, J.D. The small intestine shields the liver from fructose-induced steatosis. Nat. Metab. 2020, 2, 586–593. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Sadidi, M.; Feldman, E.L. Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Rev. Endocr. Metab. Disord. 2008, 9, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and Localization of the Principal Fructose-metabolizing Enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef]
- Afzali, A.; Weiss, N.S.; Boyko, E.J.; Ioannou, G.N. Association between serum uric acid level and chronic liver disease in the United States. Hepatology 2010, 52, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, W.; Zhang, J.; Gobejishvili, L.; Barve, S.S.; McClain, C.J.; Joshi-Barve, S. Elevated Fructose and Uric Acid Through Aldose Reductase Contribute to Experimental and Human Alcoholic Liver Disease. Hepatology 2020, 72, 1617–1637. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Yin, H.; Gao, Z.; Li, Y.; Gao, T.; Duan, J.; Yang, R.; Dong, X.; Zhang, L.; Duan, W. Intestinal tract is an important organ for lowering serum uric acid in rats. PLoS ONE 2017, 12, e0190194. [Google Scholar] [CrossRef]
- Ndrepepa, G. Uric acid and cardiovascular disease. Clin. Chim. Acta 2018, 484, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, C.; Ogura, J.; Sasaki, S.; Okamoto, K.; Kobayashi, M.; Kuwayama, K.; Narumi, K.; Iseki, K. Fructose suppresses uric acid excretion to the intestinal lumen as a result of the induction of oxidative stress by NADPH oxidase activation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 559–566. [Google Scholar] [CrossRef]
- Al-Jawadi, A.; Patel, C.R.; Shiarella, R.J.; Romelus, E.; Auvinen, M.; Guardia, J.; Pearce, S.C.; Kishida, K.; Yu, S.; Gao, N.; et al. Cell-Type–Specific, Ketohexokinase-Dependent Induction by Fructose of Lipogenic Gene Expression in Mouse Small Intestine. J. Nutr. 2020, 150, 1722–1730. [Google Scholar] [CrossRef]
- Lee, H.-J.; Cha, J.-Y. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Astapova, I.I.; Flier, S.N.; Hannou, S.A.; Doridot, L.; Sargsyan, A.; Kou, H.H.; Fowler, A.J.; Liang, G.; Herman, M.A. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2017, 2, e96703. [Google Scholar] [CrossRef]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features. Hepatol. Res. 2016, 46, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Francis, H.; Alpini, G. Fructose Promotion of Intestinal and Liver Injury: A Sugar by Any Other Name That Isn’t So Sweet. Hepatology 2019, hep.30843. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, D.; Seo, W.; Gao, B.; Yoo, S.; Song, B. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1–Mediated Oxidative and Nitrative Stress. Hepatology 2019, hep.30652. [Google Scholar] [CrossRef] [PubMed]
- Moine, L.; Rivoira, M.; de Barboza, G.D.; Pérez, A.; de Talamoni, N.T. Glutathione depleting drugs, antioxidants and intestinal calcium absorption. World J. Gastroenterol. 2018, 24, 4979–4988. [Google Scholar] [CrossRef]
- Arauz, J.; Ramos-Tovar, E.; Muriel, P. Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Ann. Hepatol. 2016, 15, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef]
- Do, M.; Lee, E.; Oh, M.-J.; Kim, Y.; Park, H.-Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef]
- Softic, S.; Gupta, M.K.; Wang, G.-X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-J.; Akbar, M.; Jo, I.; Hardwick, J.P.; Abdelmegeed, M.A. Translational Implications of the Alcohol-Metabolizing Enzymes, Including Cytochrome P450-2E1, in Alcoholic and Nonalcoholic Liver Disease. Adv. Pharmacol. 2015, 74, 303–372. [Google Scholar]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Dürr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin resistance alters hepatic ethanol metabolism: Studies in mice and children with non-alcoholic fatty liver disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Simeonova, D.; Ivanovska, M.; Murdjeva, M.; Carvalho, A.F.; Maes, M. Recognizing the Leaky Gut as a Trans-diagnostic Target for Neuroimmune Disorders Using Clinical Chemistry and Molecular Immunology Assays. Curr. Top. Med. Chem. 2018, 18, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Francey, C.; Cros, J.; Rosset, R.; Crézé, C.; Rey, V.; Stefanoni, N.; Schneiter, P.; Tappy, L.; Seyssel, K. The extra-splanchnic fructose escape after ingestion of a fructose–glucose drink: An exploratory study in healthy humans using a dual fructose isotope method. Clin. Nutr. ESPEN 2019, 29, 125–132. [Google Scholar] [CrossRef]
- Nergiz-Unal, R.; Ulug, E.; Kisioglu, B.; Tamer, F.; Bodur, M.; Yalcimin, H.; Yuruk, A.A. Hepatic cholesterol synthesis and lipoprotein levels impaired by dietary fructose and saturated fatty acids in mice: Insight on PCSK9 and CD36. Nutrition 2020, 110954. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2021, 3, 100217. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, L.; Xu, S.; Shen, A. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog. Lipid Res. 2020, 77, 101006. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.; Vijayakumar, A.; Ghoshal, S.; Marchand, B.; Yi, S.; Kornyeyev, D.; Zagorska, A.; Hollenback, D.; Walker, K.; Liu, K.; et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J. Hepatol. 2020, 73, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Volynets, V.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Toll-like receptors 1–9 are elevated in livers with fructose-induced hepatic steatosis. Br. J. Nutr. 2012, 107, 1727–1738. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear factor-Κb—A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 1. [Google Scholar] [CrossRef]
- Afonina, I.S.; Zhong, Z.; Karin, M.; Beyaert, R. Limiting inflammation—The negative regulation of NF-κB and the NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. [Google Scholar] [CrossRef]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef]
- Paschos, P.; Athyros, V.G.; Tsimperidis, A.; Katsoula, A.; Grammatikos, N.; Giouleme, O. Can Serum Uric Acid Lowering Therapy Contribute to the Prevention or Treatment of Nonalcoholic Fatty Liver Disease? Curr. Vasc. Pharmacol. 2018, 16, 269–275. [Google Scholar] [CrossRef]
- Wan, X.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; Sang, J.; He, H.; Liu, X.; Li, Y.; Yu, C. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J. Hepatol. 2016, 64, 925–932. [Google Scholar] [CrossRef]
- Merino, B.; Fernández-Díaz, C.M.; Cózar-Castellano, I.; Perdomo, G. Intestinal Fructose and Glucose Metabolism in Health and Disease. Nutrients 2019, 12, 94. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.-H.; Chen, X.-Y.; Hu, Q.-H.; Wang, M.-X.; Jin, R.; Zhang, Q.-Y.; Wang, W.; Wang, R.; Kang, L.-L.; et al. Reactive Oxygen Species-Induced TXNIP Drives Fructose-Mediated Hepatic Inflammation and Lipid Accumulation Through NLRP3 Inflammasome Activation. Antioxid. Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [PubMed]
- Ignat, S.-R.; Dinescu, S.; Hermenean, A.; Costache, M. Cellular Interplay as a Consequence of Inflammatory Signals Leading to Liver Fibrosis Development. Cells 2020, 9, 461. [Google Scholar] [CrossRef]
- Artlett, C.M. The Role of the NLRP3 Inflammasome in Fibrosis. Open Rheumatol. J. 2012, 6, 80–86. [Google Scholar] [CrossRef]
- Wu, X.; Dong, L.; Lin, X.; Li, J. Relevance of the NLRP3 Inflammasome in the Pathogenesis of Chronic Liver Disease. Front. Immunol. 2017, 8, 1728. [Google Scholar] [CrossRef] [PubMed]
- Nigro, D.; Menotti, F.; Cento, A.S.; Serpe, L.; Chiazza, F.; Dal Bello, F.; Romaniello, F.; Medana, C.; Collino, M.; Aragno, M.; et al. Chronic administration of saturated fats and fructose differently affect SREBP activity resulting in different modulation of Nrf2 and Nlrp3 inflammasome pathways in mice liver. J. Nutr. Biochem. 2017, 42, 160–171. [Google Scholar] [CrossRef]
- Kim, S.-K.; Choe, J.-Y.; Park, K.-Y. TXNIP-mediated nuclear factor-κB signaling pathway and intracellular shifting of TXNIP in uric acid-induced NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2019, 511, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Noblet, B.; Benhamed, F.; O-Sullivan, I.; Zhang, W.; Filhoulaud, G.; Montagner, A.; Polizzi, A.; Marmier, S.; Burnol, A.-F.; Guilmeau, S.; et al. Dual regulation of TxNIP by ChREBP and FoxO1 in liver. iScience 2021, 102218. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Free radicals, antioxidants, nuclear factor-E2-related factor-2 and liver damage. J. Appl. Toxicol. 2020, 40, 151–168. [Google Scholar] [CrossRef]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26, 173–179. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2019, 9, 1428. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Yagishita, Y.; Fazzari, M.; Palliyaguru, D.L.; Rao, U.N.M.; Zaravinos, A.; Khoo, N.K.H.; Schopfer, F.J.; Weiss, K.R.; Michalopoulos, G.K.; et al. Nrf2 prevents Notch-induced insulin resistance and tumorigenesis in mice. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Maurya, C.K.; Arha, D.; Avisetti, D.R.; Prathapan, A.; Raj, P.S.; Raghu, K.G.; Kalivendi, S.V.; Tamrakar, A.K. Fructose induces mitochondrial dysfunction and triggers apoptosis in skeletal muscle cells by provoking oxidative stress. Apoptosis 2015, 20, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, S.; Ospina, E.; Shabandri, O.; Lank, D.; Akakpo, J.Y.; Zhao, Z.; Yang, M.; Wu, J.; Jaeschke, H.; et al. Fructose Protects Against Acetaminophen-Induced Hepatotoxicity Mainly by Activating the Carbohydrate-Response Element-Binding Protein α–Fibroblast Growth Factor 21 Axis in Mice. Hepatol. Commun. 2021, hep4.1683. [Google Scholar] [CrossRef]
- Zhao, X.-J.; Yu, H.-W.; Yang, Y.-Z.; Wu, W.-Y.; Chen, T.-Y.; Jia, K.-K.; Kang, L.-L.; Jiao, R.-Q.; Kong, L.-D. Polydatin prevents fructose-induced liver inflammation and lipid deposition through increasing miR-200a to regulate Keap1/Nrf2 pathway. Redox Biol. 2018, 18, 124–137. [Google Scholar] [CrossRef]
- Novelle, M.G.; Bravo, S.B.; Deshons, M.; Iglesias, C.; García-Vence, M.; Annells, R.; da Silva Lima, N.; Nogueiras, R.; Fernández-Rojo, M.A.; Diéguez, C.; et al. Impact of liver-specific GLUT8 silencing on fructose-induced inflammation and omega oxidation. iScience 2021, 24, 102071. [Google Scholar] [CrossRef]
- Ishihara, Y.; Kado, S.Y.; Hoeper, C.; Harel, S.; Vogel, C.F.A. Role of NF-kB RelB in Aryl Hydrocarbon Receptor-Mediated Ligand Specific Effects. Int. J. Mol. Sci. 2019, 20, 2652. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.H.; Tang, J.; Beane, K.E.; Redding, M.C.; Cho, Y.J.; Kim, Y.J.; Zhao, J.; Shin, E.-C.; Lee, J.H.; Kong, B.C.; et al. Hepatic transcriptomics analysis reveals that fructose intervention down-regulated xenobiotics-metabolising enzymes through aryl hydrocarbon receptor signalling suppression in C57BL/6N mice. Br. J. Nutr. 2019, 122, 769–779. [Google Scholar] [CrossRef]
- Pan, J.H.; Tang, J.; Beane, K.; Redding, M.; Zhao, J.; Lee, J.H.; Kong, B.; Kim, J.K. Hepatic Transcriptome Reveals That Fructose Downregulated Xenobiotics Metabolizing Enzymes Through Aryl Hydrocarbon Receptor Signaling Suppression in C57BL/6 N Mice (P15-011-19). Curr. Dev. Nutr. 2019, 3. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Shi, J.-H.; Lu, J.-Y.; Chen, H.-Y.; Wei, C.-C.; Xu, X.; Li, H.; Bai, Q.; Xia, F.-Z.; Lam, S.M.; Zhang, H.; et al. Liver ChREBP Protects Against Fructose-Induced Glycogenic Hepatotoxicity by Regulating L-Type Pyruvate Kinase. Diabetes 2020, 69, 591–602. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.-R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli, A.; Crescenzo, R.; Cigliano, L.; Spagnuolo, M.S.; Cancelliere, R.; Gatto, C.; Iossa, S. Early Hepatic Oxidative Stress and Mitochondrial Changes Following Western Diet in Middle Aged Rats. Nutrients 2019, 11, 2670. [Google Scholar] [CrossRef]
- Softic, S.; Stanhope, K.L.; Boucher, J.; Divanovic, S.; Lanaspa, M.A.; Johnson, R.J.; Kahn, C.R. Fructose and hepatic insulin resistance. Crit. Rev. Clin. Lab. Sci. 2020, 1–15. [Google Scholar] [CrossRef]
- Softic, S.; Meyer, J.G.; Wang, G.-X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.P.M.M.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e4. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.; Bruce, W.R.; Dong, Q.; Bruce, J.; Mehta, R.; O’Brien, P.J. Fructose and carbonyl metabolites as endogenous toxins. Chem. Biol. Interact. 2009, 178, 332–339. [Google Scholar] [CrossRef]
- Hieronimus, B.; Griffen, S.C.; Keim, N.L.; Bremer, A.A.; Berglund, L.; Nakajima, K.; Havel, P.J.; Stanhope, K.L. Effects of Fructose or Glucose on Circulating ApoCIII and Triglyceride and Cholesterol Content of Lipoprotein Subfractions in Humans. J. Clin. Med. 2019, 8, 913. [Google Scholar] [CrossRef] [PubMed]
- Ichigo, Y.; Takeshita, A.; Hibino, M.; Nakagawa, T.; Hayakawa, T.; Patel, D.; Field, C.J.; Shimada, M. High-Fructose Diet-Induced Hypertriglyceridemia Is Associated with Enhanced Hepatic Expression of ACAT2 in Rats. Physiol. Res. 2019, 1021–1026. [Google Scholar] [CrossRef]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, M.; Kimura, T.; Li, C.; Tanaka, M.; Matsuhashi, A.; Yoshida, H.; Noda, A.; Xu, P.; Takase, S.; Okazaki, S.; et al. Critical Role of SREBP-1c Large-VLDL Pathway in Environment-Induced Hypertriglyceridemia of Apo AV Deficiency. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 373–386. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Hieronimus, B.; Stanhope, K.L. Dietary fructose and dyslipidemia. Curr. Opin. Lipidol. 2020, 31, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Milutinović, D.V.; Brkljačić, J.; Teofilović, A.; Bursać, B.; Nikolić, M.; Gligorovska, L.; Kovačević, S.; Djordjevic, A.; Preitner, F.; Tappy, L.; et al. Chronic Stress Potentiates High Fructose–Induced Lipogenesis in Rat Liver and Kidney. Mol. Nutr. Food Res. 2020, 64, 1901141. [Google Scholar] [CrossRef] [PubMed]
- Steenson, S.; Shojaee-Moradie, F.; Whyte, M.B.; Jackson, K.G.; Lovegrove, J.A.; Fielding, B.A.; Umpleby, A.M. The Effect of Fructose Feeding on Intestinal Triacylglycerol Production and De Novo Fatty Acid Synthesis in Humans. Nutrients 2020, 12, 1781. [Google Scholar] [CrossRef]
- Jiang, J.X.; Török, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef]
- Lian, C.-Y.; Zhai, Z.-Z.; Li, Z.-F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Biol. Interact. 2020, 330, 109199. [Google Scholar] [CrossRef]
- Pereira, R.; Botezelli, J.; da Cruz Rodrigues, K.; Mekary, R.; Cintra, D.; Pauli, J.; da Silva, A.; Ropelle, E.; de Moura, L. Fructose Consumption in the Development of Obesity and the Effects of Different Protocols of Physical Exercise on the Hepatic Metabolism. Nutrients 2017, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.S.; Petrovsky, N. Calcium Signaling As a Therapeutic Target for Liver Steatosis. Trends Endocrinol. Metab. 2019, 30, 270–281. [Google Scholar] [CrossRef]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [PubMed]
- Mellouk, K.Z.; Louchami, E.; Hupkens, A.; Sener, D.A.; Yahia, W.J.M. The metabolic syndrome of fructose-fed rats: Effects of long-chain polyunsaturated ω3 and ω6 fatty acids. V. Post-mortem findings. Mol. Med. Rep. 2012, 6, 1399–1403. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, L.-C.; Varghese, Z.; Moorhead, J.F.; Lee, C.-T.; Chen, J.-B.; Ruan, X.Z. Cross-talk between TLR4-MyD88-NF-κB and SCAP-SREBP2 pathways mediates macrophage foam cell formation. Am. J. Physiol. Circ. Physiol. 2013, 304, H874–H884. [Google Scholar] [CrossRef]
- Jegatheesan, P.; De Bandt, J. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef]
- Farrell, G.C.; van Rooyen, D. Liver cholesterol: Is it playing possum in NASH? Am. J. Physiol. Liver Physiol. 2012, 303, G9–G11. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Yamazaki, M.; Munetsuna, E.; Yamada, H.; Ando, Y.; Mizuno, G.; Fujii, R.; Nouchi, Y.; Kageyama, I.; Teshigawara, A.; Ishikawa, H.; et al. Maternal fructose consumption down-regulates Lxra expression via miR-206-mediated regulation. J. Nutr. Biochem. 2020, 82, 108386. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Munetsuna, E.; Yamada, H.; Ando, Y.; Yamazaki, M.; Taromaru, N.; Nagura, A.; Ishikawa, H.; Suzuki, K.; Teradaira, R.; et al. High fructose consumption induces DNA methylation at PPARα and CPT1A promoter regions in the rat liver. Biochem. Biophys. Res. Commun. 2015, 468, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Johnson, R.J.; Lanaspa, M.A. Endogenous fructose production. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Boulestin, H.; Campbell, F.M.; Williams, L.M. The Role of Dietary Advanced Glycation End Products in Metabolic Dysfunction. Mol. Nutr. Food Res. 2020, 1900934. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Zhang, Y.; Zeng, C. Recent advances in fructose intake and risk of hyperuricemia. Biomed. Pharmacother. 2020, 131, 110795. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Vicente, A.; Cabral, P.D.; Hong, N.J.; Asirwatham, J.; Saez, F.; Garvin, J.L. Fructose reabsorption by rat proximal tubules: Role of Na + -linked cotransporters and the effect of dietary fructose. Am. J. Physiol. Physiol. 2019, 316, F473–F480. [Google Scholar] [CrossRef]
- Bjornstad, P.; Lanaspa, M.A.; Ishimoto, T.; Kosugi, T.; Kume, S.; Jalal, D.; Maahs, D.M.; Snell-Bergeon, J.K.; Johnson, R.J.; Nakagawa, T. Fructose and uric acid in diabetic nephropathy. Diabetologia 2015, 58, 1993–2002. [Google Scholar] [CrossRef]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef]
- Aldaba-Muruato, L.R.; Moreno Gil, M.; Shibayama, M.; Tsutsumi, V.; Muriel, P. Protective effects of allopurinol against acute liver damage and cirrhosis induced by carbon tetrachloride: Modulation of NF-κB, cytokine production and oxidative stress. Biochim. Biophys. Acta 2012, 1820, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Albano, E.; Mottaran, G.; Occhino, E.; Reale, M.V. Review article: Role of oxidative stress in the progression of non-alcoholic steatosis. Aliment. Pharmacol. Ther. 2005, 22, 71–73. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, L.; Jiang, L.; Wen, P.; Cao, H.; He, W.; Dai, C.; Yang, J. Uric Acid Induces Renal Inflammation via Activating Tubular NF-κB Signaling Pathway. PLoS ONE 2012, 7, e39738. [Google Scholar] [CrossRef]
- Spiga, R.; Marini, M.A.; Mancuso, E.; Di Fatta, C.; Fuoco, A.; Perticone, F.; Andreozzi, F.; Mannino, G.C.; Sesti, G. Uric Acid Is Associated with Inflammatory Biomarkers and Induces Inflammation Via Activating the NF-κB Signaling Pathway in HepG2 Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.; Clare, K.; George, J.; Dillon, J.F. Determining the role for uric acid in non-alcoholic steatohepatitis development and the utility of urate metabolites in diagnosis: An opinion review. World J. Gastroenterol. 2020, 26, 1683–1690. [Google Scholar] [CrossRef]
- Oral; Sahin; Turker; Kocak Relationship Between Serum Uric Acid Levels and Nonalcoholic Fatty Liver Disease in Non-Obese Patients. Medicina 2019, 55, 600. [CrossRef]
- Yang, Y.; Zhou, Y.; Cheng, S.; Sun, J.L.; Yao, H.; Ma, L. Effect of uric acid on mitochondrial function and oxidative stress in hepatocytes. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Jhang, J.-J.; Cheng, Y.-T.; Ho, C.-Y.; Yen, G.-C. Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell. Mol. Immunol. 2015, 12, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Zarfeshani, A.; Ngo, S.; Sheppard, A. MicroRNA Expression Relating to Dietary-Induced Liver Steatosis and NASH. J. Clin. Med. 2015, 4, 1938–1950. [Google Scholar] [CrossRef] [PubMed]
- Hanousková, B.; Neprašová, B.; Skálová, L.; Maletínská, L.; Zemanová, K.; Ambrož, M.; Matoušková, P. High-fructose drinks affect microRNAs expression differently in lean and obese mice. J. Nutr. Biochem. 2019, 68, 42–50. [Google Scholar] [CrossRef]
- Dong, X.; Liu, H.; Chen, F.; Li, D.; Zhao, Y. MiR-214 Promotes the Alcohol-Induced Oxidative Stress via Down-Regulation of Glutathione Reductase and Cytochrome P450 Oxidoreductase in Liver Cells. Alcohol. Clin. Exp. Res. 2014, 38, 68–77. [Google Scholar] [CrossRef]
- Dai, B.-H.; Geng, L.; Wang, Y.; Sui, C.-J.; Xie, F.; Shen, R.-X.; Shen, W.-F.; Yang, J.-M. microRNA-199a-5p protects hepatocytes from bile acid-induced sustained endoplasmic reticulum stress. Cell Death Dis. 2013, 4, e604. [Google Scholar] [CrossRef]
- Jimenez Calvente, C.; Del Pilar, H.; Tameda, M.; Johnson, C.D.; Feldstein, A.E. MicroRNA 223 3p Negatively Regulates the NLRP3 Inflammasome in Acute and Chronic Liver Injury. Mol. Ther. 2020, 28, 653–663. [Google Scholar] [CrossRef]
- Pan, J.H.; Cha, H.; Tang, J.; Lee, S.; Lee, S.H.; Le, B.; Redding, M.C.; Kim, S.; Batish, M.; Kong, B.C.; et al. The role of microRNA-33 as a key regulator in hepatic lipogenesis signaling and a potential serological biomarker for NAFLD with excessive dietary fructose consumption in C57BL/6N mice. Food Funct. 2021, 12, 656–667. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Huang, Y.-H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lanaspa, M.A.; Millan, I.S.; Fini, M.; Rivard, C.J.; Sanchez-Lozada, L.G.; Andres-Hernando, A.; Tolan, D.R.; Johnson, R.J. Fructose contributes to the Warburg effect for cancer growth. Cancer Metab. 2020, 8, 16. [Google Scholar] [CrossRef]
- Healy, M.E.; Chow, J.D.Y.; Byrne, F.L.; Breen, D.S.; Leitinger, N.; Li, C.; Lackner, C.; Caldwell, S.H.; Hoehn, K.L. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J. Hepatol. 2015, 62, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Healy, M.E.; Lahiri, S.; Hargett, S.R.; Chow, J.D.Y.; Byrne, F.L.; Breen, D.S.; Kenwood, B.M.; Taddeo, E.P.; Lackner, C.; Caldwell, S.H.; et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci. Rep. 2016, 6, 22292. [Google Scholar] [CrossRef]
- Kim, J.; Kang, J.; Kang, Y.-L.; Woo, J.; Kim, Y.; Huh, J.; Park, J.-W. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun. 2020, 11, 5436. [Google Scholar] [CrossRef]
- Krause, N.; Wegner, A. Fructose Metabolism in Cancer. Cells 2020, 9, 2635. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.-Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262. [Google Scholar] [CrossRef]
- Hayashi, M.; Yamada, S.; Tanabe, H.; Takami, H.; Inokawa, Y.; Sonohara, F.; Shimizu, D.; Hattori, N.; Kanda, M.; Tanaka, C.; et al. High Serum Uric Acid Levels Could Be a Risk Factor of Hepatocellular Carcinoma Recurrences. Nutr. Cancer 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seki, K.; Kitade, M.; Nishimura, N.; Kaji, K.; Asada, K.; Namisaki, T.; Moriya, K.; Kawaratani, H.; Okura, Y.; Takaya, H.; et al. Oral administration of fructose exacerbates liver fibrosis and hepatocarcinogenesis via increased intestinal permeability in a rat steatohepatitis model. Oncotarget 2018, 9, 28638–28651. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muriel, P.; López-Sánchez, P.; Ramos-Tovar, E. Fructose and the Liver. Int. J. Mol. Sci. 2021, 22, 6969. https://doi.org/10.3390/ijms22136969
Muriel P, López-Sánchez P, Ramos-Tovar E. Fructose and the Liver. International Journal of Molecular Sciences. 2021; 22(13):6969. https://doi.org/10.3390/ijms22136969
Chicago/Turabian StyleMuriel, Pablo, Pedro López-Sánchez, and Erika Ramos-Tovar. 2021. "Fructose and the Liver" International Journal of Molecular Sciences 22, no. 13: 6969. https://doi.org/10.3390/ijms22136969
APA StyleMuriel, P., López-Sánchez, P., & Ramos-Tovar, E. (2021). Fructose and the Liver. International Journal of Molecular Sciences, 22(13), 6969. https://doi.org/10.3390/ijms22136969