
Intestinal Permeability Is a Mechanical Rheostat in the Pathogenesis of Liver Cirrhosis

 , , , ,
, , , ,  , ,
, , 
Abstract
:1. Introduction
2. Mechanisms of Endotoxemia Derived from Gut Microbiota
2.1. Dysbiosis
2.2. Small Intestinal Bacterial Overgrowth (SIBO)
2.3. Disruptions in Intestinal Barrier Function
3. The Role of Endotoxemia in the Progression of Liver Pathogenesis
3.1. Alcoholic Liver Disease
3.2. NASH
3.3. Viral Hepatitis
3.3.1. Hepatitis B Viral Infection
3.3.2. Hepatitis C Viral Infection
3.4. Autoimmune Liver Diseases
3.5. Liver Cirrhosis and Its Complications
3.5.1. Ascites and SBP
3.5.2. Portal Hypertension
3.5.3. Hepatic Encephalopathy
3.6. HCC
4. Conclusions
Funding
Conflicts of Interest
References
- Fukui, H. Gut-liver axis in liver cirrhosis: How to manage leaky gut and endotoxemia. World J. Hepatol. 2015, 7, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenes Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol. 2012, 590, 447–458. [Google Scholar] [CrossRef]
- Seo, Y.S.; Shah, V.H. The role of gut-liver axis in the pathogenesis of liver cirrhosis and portal hypertension. Clin. Mol. Hepatol. 2012, 18, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017, 25, e1055. [Google Scholar] [CrossRef]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Xu, M.; Wang, B.; Fu, Y.; Chen, Y.; Yang, F.; Lu, H.; Chen, Y.; Xu, J.; Li, L. Changes of fecal Bifidobacterium species in adult patients with hepatitis B virus-induced chronic liverdisease. Microb. Ecol. 2012, 63, 304–313. [Google Scholar] [CrossRef]
- Wu, Z.-W.; Lu, H.-F.; Wu, J.; Zuo, J.; Chen, P.; Sheng, J.-F.; Zheng, S.-S.; Li, L.-J. Assessment of the Fecal Lactobacilli Population in Patients with Hepatitis B Virus-Related Decompensated Cirrhosis and Hepatitis B Cirrhosis Treated with Liver Transplant. Microb. Ecol. 2012, 63, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Ridlon, J.M.; Hylemon, P.B.; Thacker, L.R.; Heuman, D.M.; Smith, S.; Sikaroodi, M.; Gillevet, P.M. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Liver Physiol. 2012, 302, G168–G175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborti, C.K. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Heuman, D.M.; Kang, D.J.; Takei, H.; Nittono, H.; Ridlon, J.M.; Fuchs, M.; et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am. J. Physiol. Liver Physiol. 2014, 306, G929–G937. [Google Scholar] [CrossRef] [Green Version]
- Drasar, B.S.; Shiner, M. Studies on the intestinal flora: Part II Bacterial flora of the small intestine in patients with gastrointestinal disorders. Gut 1969, 10, 812–819. [Google Scholar] [CrossRef] [Green Version]
- Unno, N.; Wang, H.; Menconi, M.J.; Tytgat, S.H.; Larkin, V.; Smith, M.; Morin, M.J.; Chavez, A.; Hodin, R.A.; Fink, M.P. Inhibition of inducible nitric oxide synthase ameliorates endotoxin-induced gut mucosal barrier dysfunction in rats. Gastroenterology 1997, 113, 1246–1257. [Google Scholar] [CrossRef]
- Zolotarevsky, Y.; Hecht, G.; Koutsouris, A.; Gonzalez, D.E.; Quan, C.; Tom, J.; Mrsny, R.J.; Turner, J.R. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002, 123, 163–172. [Google Scholar] [CrossRef]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Grąt, M.; Wronka, K.M.; Krasnodębski, M.; Masior, Ł.; Lewandowski, Z.; Kosińska, I.; Grąt, K.; Stypułkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis. Transplant. Proc. 2016, 48, 1687–1691. [Google Scholar] [CrossRef]
- Van Thiel, D.H.; Fagiuoli, S.; Wright, H.I.; Chien, M.C.; Gavaler, J.S. Gastrointestinal transit in cirrhotic patients: Effect of hepatic encephalopathy and its treatment. Hepatology 1994, 19, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Chesta, J.; Defilippi, C. Abnormalities in proximal small bowel motility in patients with cirrhosis. Hepatology 1993, 17, 828–832. [Google Scholar] [PubMed]
- Lombardo, L.; Foti, M.; Ruggia, O.; Chiecchio, A. Increased Incidence of Small Intestinal Bacterial Overgrowth during Proton Pump Inhibitor Therapy. Clin. Gastroenterol. Hepatol. 2010, 8, 504–508. [Google Scholar] [CrossRef]
- Farquhar, M.G.; Palade, G.E. JUNCTIONAL COMPLEXES IN VARIOUS EPITHELIA. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [Green Version]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.B.; Lu, Y.; Choate, K.A.; Vekazquez, H.; Al-Sabban, E.; Praga, M.; Casari, G.; Bettinelli, A.; Colussi, G.; Rodriguez-Soriano, J.; et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2þ resorption. Science 1999, 285, 103–106. [Google Scholar] [CrossRef]
- Shen, L.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight Junction Pore and Leak Pathways: A Dynamic Duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef] [Green Version]
- Luissint, A.-C.; Parkos, C.A.; Nusrat, A. Inflammation and the Intestinal Barrier: Leukocyte–Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology 2016, 151, 616–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiippala, K.; Jouhten, H.; Ronkainen, A.; Hartikainen, A.; Kainulainen, V.; Jalanka, J.; Satokari, R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients 2018, 10, 988. [Google Scholar] [CrossRef] [Green Version]
- Hendrikx, T.; Schnabl, B. Antimicrobial proteins: Intestinal guards to protect against liver disease. J. Gastroenterol. 2018, 54, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Gao, B.; Zakhari, S.; Nagy, L.E. Inflammation in Alcoholic Liver Disease. Annu. Rev. Nutr. 2012, 32, 343–368. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Bataller, R. Alcoholic Liver Disease: Pathogenesis and New Therapeutic Targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic Liver Disease: The Gut Microbiome and Liver Cross Talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, J.C.; Bode, C.; Heidelbach, R.; Dürr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34. [Google Scholar]
- Morencos, F.C.; de las Heras Castano, G.; Martin Ramos, L.; López Arias, M.J.; Ledesma, F.; Romero, F.P. Small bowel bacterial over-growth in patients with alcoholic cirrhosis. Dig. Dis. Sci. 1995, 40, 1252–1256. [Google Scholar] [CrossRef]
- Hartmann, P.; Chen, P.; Wang, H.J.; Wang, L.; McCole, D.F.; Brandl, K.; Stärkel, P.; Belzer, C.; Hellerbrand, C.; Tsukamoto, H.; et al. De-ficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 2013, 58, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Vassallo, G.; Mirijello, A.; Ferrulli, A.; Antonelli, M.; Landolfi, R.; Gasbarrini, A.; Addolorato, G. Review article: Alcohol and gut microbiota–the possible role of gut microbiota modulation in the treatment of alcoholic liver disease. Aliment. Pharmacol. Ther. 2015, 41, 917–927. [Google Scholar] [CrossRef]
- Fukui, H.; Brauner, B.; Bode, J.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kirpich, I.; Liu, Y.; Ma, Z.; Barve, S.; McClain, C.J.; Feng, W. Lactobacillus rhamnosus GG Treatment Potentiates Intestinal Hypoxia-Inducible Factor, Promotes Intestinal Integrity and Ameliorates Alcohol-Induced Liver Injury. Am. J. Pathol. 2011, 179, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Meta-genomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic mi-crobiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, C.A.; Patel, V.C.; Singanayagam, A.; Shawcross, D.L. Review article: The gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment. Pharmacol. Ther. 2018, 47, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G. Gut–Liver Axis in Alcoholic Liver Disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Ilan, Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2609–2618. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; Nilaweera, K.; Ross, P.R.; Shanahan, F.; O’Toole, P.W.; Cotter, P.D. The gut microbiota and its relationship to diet and obesity: New insights. Gut Microbes 2012, 3, 186–202. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nat. Cell Biol. 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Panasevich, M.R.; Peppler, W.T.; Oerther, D.; Wright, D.C.; Rector, R.S. Microbiome and NAFLD: Potential influence of aerobic fitness and lifestyle modification. Physiol. Genom. 2017, 49, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Alisi, A.; Manco, M.; De Vito, R.; Piemonte, F.; Nobili, V. Endotoxin and Plasminogen Activator Inhibitor-1 Serum Levels Associated With Nonalcoholic Steatohepatitis in Children. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 645–649. [Google Scholar] [CrossRef]
- Farhadi, A.; Gundlapalli, S.; Shaikh, M.; Frantzides, C.; Harrell, L.; Kwasny, M.M.; Keshavarzian, A. Susceptibility to gut leakiness: A possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008, 28, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2015, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Clemens, M.; Diehl, A.M. Obesity increases sensitivity to endotoxin liver injury: Implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. USA 1997, 94, 2557–2562. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Gaborit, B.; Dutour, A.; Clement, K. Gut microbiota and non-alcoholic fatty liver disease: New insights. Clin. Microbiol. Infect. 2013, 19, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Douhara, A.; Moriya, K.; Yoshiji, H.; Noguchi, R.; Namisaki, T.; Kitade, M.; Kaji, K.; Aihara, Y.; Nishimura, N.; Takeda, K.; et al. Reduction of en-dotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol. Med. Rep. 2015, 11, 1693–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzer, S.; Machnowska, P.; Aßmus, J.; Sieber, M.; Pieper, R.; Schmidt, M.F.; Brockmann, G.A.; Scharek-Tedin, L.; Johne, R. Feeding of the probiotic bacterium Enterococcus faecium NCIMB 10415 differentially affects shedding of enteric viruses in pigs. Veter Res. 2012, 43, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.K.; Yan, P.; Chung, R.T.; Butt, A.A.; Abou-Samra, A.-B. Proton pump inhibitors are associated with accelerated development of cirrhosis, hepatic decompensation and hepatocellular carcinoma in noncirrhotic patients with chronic hepatitis C infection: Results from ERCHIVES. Aliment. Pharmacol. Ther. 2017, 47, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci. Rep. 2016, 6, 34055. [Google Scholar] [CrossRef]
- Mohamadkhani, A. On the potential role of intestinal microbial community in hepatocarcinogenesis in chronic hepatitis B. Cancer Med. 2018, 7, 3095–3100. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Wu, Z.; Xu, W.; Yang, J.; Chen, Y.; Li, L. Intestinal Microbiota Was Assessed in Cirrhotic Patients with Hepatitis B Virus Infection. Microb. Ecol. 2011, 61, 693–703. [Google Scholar] [CrossRef]
- Zhao, Y.; Mao, Y.-F.; Tang, Y.-S.; Ni, M.-Z.; Liu, Q.-H.; Wang, Y.; Feng, Q.; Peng, J.-H.; Hu, Y.-Y. Altered oral microbiota in chronic hepatitis B patients with different tongue coatings. World J. Gastroenterol. 2018, 24, 3448–3461. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Norkina, O.; Kodys, K.; Catalano, D.; Bakis, G.; Marshall, C.; Mandrekar, P.; Szabo, G. Viral and Host Factors Induce Macrophage Activation and Loss of Toll-Like Receptor Tolerance in Chronic HCV Infection. Gastroenterology 2007, 133, 1627–1636. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Nakayama, J.; Moriya, K.; Kawaratani, H.; Momoda, R.; Ito, K.; Lio, E.; Nojiri, S.; Fujiwara, K.; Yoneda, M.; et al. Gut dysbiosis associated with hepatitis C virus in-fection. Clin. Infect. Dis. 2018, 67, 869–877. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Putignani, L.; Sterbini, F.P.; Petito, V.; Picca, A.; Del Chierico, F.; Reddel, S.; Calvani, R.; Marzetti, E.; Sanguinetti, M.; et al. Influence of hepatitis C virus eradication with direct-acting antivirals on the gut microbiota in patients with cirrhosis. Aliment. Pharmacol. Ther. 2018, 48, 1301–1311. [Google Scholar] [CrossRef]
- Pérez-Matute, P.; Íñiguez, M.; Villanueva-Millán, M.J.; Recio-Fernández, E.; Vázquez, A.M.; Sánchez, S.C.; Morano, L.E.; Oteo, J.A. Short-term effects of direct-acting antiviral agents on inflammation and gut microbiota in hepatitis C-infected patients. Eur. J. Intern. Med. 2019, 67, 47–58. [Google Scholar] [CrossRef]
- Dore, G.J.; Ward, J.; Thursz, M. Hepatitis C disease burden and strategies to manage the burden (Guest Editors Mark Thursz, Gregory Dore and John Ward). J. Viral Hepat. 2014, 21 (Suppl. 1), 1–4. [Google Scholar] [CrossRef]
- Doskali, M.; Tanaka, Y.; Ohira, M.; Ishiyama, K.; Tashiro, H.; Chayama, K.; Ohdan, H. Possibility of adoptive immunotherapy with pe-ripheral blood derived CD3−CD56+ and CD3+CD56+ cells for inducing anti-hepatocellular carcinoma and anti-hepatitis C virus activity. J. Immunother. 2011, 34, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Preveden, T.; Scarpellini, E.; Milić, N.; Luzza, F.; Abenavoli, L. Gut microbiota changes and chronic hepatitis C virus infection. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Takahashi, A.; Fujita, M.; Imaizumi, H.; Hayashi, M.; Okai, K.; Ohira, H. Dysbiosis of oral microbiota and its association with salivary immunological biomarkers in autoimmune liver disease. PLoS ONE 2018, 13, e0198757. [Google Scholar] [CrossRef]
- Lv, L.-X.; Fang, D.-Q.; Shi, D.; Chen, D.-Y.; Yan, R.; Zhu, Y.-X.; Chen, Y.-F.; Shao, L.; Guo, F.-F.; Wu, W.-R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Furukawa, M.; Moriya, K.; Nakayama, J.; Inoue, T.; Momoda, R.; Kawaratani, H.; Namisaki, T.; Sato, S.; Douhara, A.; Kaji, K.; et al. Gut dysbiosis associated with clinical prognosis of patients with primary biliary cholangitis. Hepatol. Res. 2020, 50, 840–852. [Google Scholar] [CrossRef]
- Lin, R.; Zhou, L.; Zhang, J.; Wang, B. Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int. J. Clin. Exp. Pathol. 2015, 8, 5153–5160. [Google Scholar]
- Wei, Y.; Li, Y.; Yan, L.; Sun, C.; Miao, Q.; Wang, Q.; Xiao, X.; Lian, M.; Li, B.; Chen, Y.; et al. Alterations of gut microbiome in autoimmune hepatitis. Gut 2020, 69, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Goeser, F.; Münch, P.; Lesker, T.R.; Lutz, P.L.; Krämer, B.; Kaczmarek, D.J.; Finnemann, C.; Nischalke, H.D.; Geffers, R.; Parcina, M.; et al. Neither black nor white: Do altered intestinal microbiota reflect chronic liver disease severity? Gut 2020. [Google Scholar] [CrossRef]
- Wei, Y.; Zeng, B.; Chen, J.; Cui, G.; Lu, C.; Wu, W.; Yang, J.; Wei, H.; Xue, R.; Bai, L.; et al. Enterogenous bacterial glycolipids are required for the generation of natural killer T cells mediated liver injury. Sci. Rep. 2016, 6, 36365. [Google Scholar] [CrossRef] [Green Version]
- Pinzani, M.; Rosselli, M.; Zuckermann, M. Liver cirrhosis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 281–290. [Google Scholar] [CrossRef]
- Tandon, P.; Garcia-Tsao, G. Bacterial Infections, Sepsis, and Multiorgan Failure in Cirrhosis. Semin. Liver Dis. 2008, 28, 026–042. [Google Scholar] [CrossRef] [Green Version]
- Prytz, H.; Holst-Christensen, J.; Korner, B.; Liehr, H. Portal venous and systemic endotoxemia in patients without liver disease and systemic endotoxemia in patients with cirrhosis. Scand. J. Gastroenterol. 1976, 11, 857–863. [Google Scholar] [CrossRef]
- Fukui, H.; Tsujita, S.; Matsumoto, M.; Kitano, H.; Hoppou, K.; Morimura, M.; Takaya, A.; Okamoto, S.; Tsujii, T.; Bode, C. Endotoxemia in chronic hepatitis and cirrhosis: Epiphenomenon or of pathological relevence? In Gut and the Liver; Blum, H., Bode, C., Bode, J.C., Sartor, R.B., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1998; pp. 251–262. [Google Scholar]
- Lin, R.S.; Lee, F.Y.; Lee, S.D.; Tsai, Y.T.; Lin, H.C.; Lu, R.H.; Hsu, W.C.; Huang, C.C.; Wang, S.S.; Lo, K.J. Endotoxemia in patients with chronic liver diseases: Relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J. Hepatol. 1995, 22, 165–172. [Google Scholar] [CrossRef]
- Fukui, H. Endotoxin and Other Microbial Translocation Markers in the Blood: A Clue to Understand Leaky Gut Syndrome. Cell. Mol. Med. Open Access 2016, 2, 3. [Google Scholar] [CrossRef]
- Kalaitzakis, E. Gastrointestinal dysfunction in liver cirrhosis. World J. Gastroenterol. 2014, 20, 14686–14695. [Google Scholar] [CrossRef]
- Campillo, B.; Pernet, P.; Bories, P.; Richardet, J.; Devanlay, M.; Aussel, C. Intestinal permeability in liver cirrhosis: Relationship with severe septic complications. Eur. J. Gastroenterol. Hepatol. 1999, 11, 755–759. [Google Scholar] [CrossRef]
- Pascual, S.; Such, J.; Esteban, A.; Zapater, P.; Casellas, J.A.; Aparicio, J.R.; Girona, E.; Gutierrez, A.; Carnices, F.; Palazon, J.M. Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology 2003, 50, 1482–1486. [Google Scholar]
- Scarpellini, E.; Valenza, V.; Gabrielli, M.; Lauritano, E.C.; Perotti, G.; Merra, G.; Lago, A.D.; Ojetti, V.; Ainora, M.E.; Santoro, M.; et al. Intestinal Permeability in Cirrhotic Patients with and Without Spontaneous Bacterial Peritonitis: Is the Ring Closed? Am. J. Gastroenterol. 2010, 105, 323–327. [Google Scholar] [CrossRef]
- Bajaj, S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. The cirrhosis dysbiosis ratio defines changes in the gut microbiome associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Campillo, B.; Richardet, J.-P.; Kheo, T.; Dupeyron, C. Nosocomial Spontaneous Bacterial Peritonitis and Bacteremia in Cirrhotic Patients: Impact of Isolate Type on Prognosis and Characteristics of Infection. Clin. Infect. Dis. 2002, 35, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-S.; Chen, G.-H.; Lien, H.-C.; Yeh, H.-Z. Small intestine dysmotility and bacterial overgrowth in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology 1998, 28, 1187–1190. [Google Scholar] [CrossRef]
- Francés, R.; González-Navajas, J.M.; Zapater, P.; Muñoz, C.; Cano, R.; Pascual, S.; Santana, F.; Márquez, D.; Pérez-Mateo, M.; Such, J. Translocation of bacterial DNA from Gram-positive microorganisms is associated with a species-specific inflammatory response in serum and ascitic fluid of patients with cirrhosis. Clin. Exp. Immunol. 2007, 150, 230–237. [Google Scholar] [CrossRef]
- Such, J.; Francés, R.; Muñoz, C.; Zapater, P.; Casellas, J.A.; Cifuentes, A.; Rodriguez-Valera, F.; Pascual, S.; Sola-Vera, J.; Carnicer, F.; et al. Detection and identification of bacterial DNA in patients with cirrhosis and culture-negative, nonneutrocytic ascites. Hepatology 2002, 36, 135–141. [Google Scholar] [CrossRef]
- Bellot, P.; García-Pagán, J.C.; Francés, R.; Gonzalez-Abraldes, J.; Navasa, M.; Pérez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.A.; Deshpande, A.; Lopez, R.; Hall, G.S.; van Duin, D.; Carey, W.D. Increased Rate of Spontaneous Bacterial Peritonitis Among Cirrhotic Patients Receiving Pharmacologic Acid Suppression. Clin. Gastroenterol. Hepatol. 2012, 10, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Aldersley, M.A.; Howdle, P.D. Intestinal permeability and liver disease. Eur. J. Gastroenterol. Hepatol. 1999, 11, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Thalheimer, U.; Triantos, C.K.; Samonakis, D.N.; Patch, D.; Burroughs, A.K. Infection, coagulation, and variceal bleeding in cirrhosis. Gut 2005, 54, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.B.; Henderson, J.M.; Kutner, M.H. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology 1988, 8, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chiva, M.; Guarner, C.; Peralta, C.; Llovet, T.; Gómez, G.; Soriano, G.; Balanzó, J. Intestinal mucosal oxidative damage and bacterial translocation in cirrhotic rats. Eur. J. Gastroenterol. Hepatol. 2003, 15, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Clements, W.D.B.; Erwin, P.; McCaigue, M.D.; Halliday, I.; Barclay, G.R.; Rowlands, B.J. Conclusive evidence of endotoxaemia in biliary obstruction. Gut 1998, 42, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Pannen, B.H.; Bauer, M.; Zhang, J.X.; Robotham, J.L.; Clemens, M.G. Endotoxin pretreatment enhances portal venous contractile response to endothelin-1. Am. J. Physiol. Circ. Physiol. 1996, 270, H7–H15. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cao, H.; Liu, H.; Wu, Z.-Y. Role of nitric oxide synthase and cyclooxygenase in hyperdynamic splanchnic circulation of portal hypertension. Hepatobiliary Pancreat. Dis. Int. 2008, 7, 503–508. [Google Scholar]
- Dong, T.; Aronsohn, A.; Reddy, K.G.; Te, H.S. Rifaximin Decreases the Incidence and Severity of Acute Kidney Injury and Hepatorenal Syndrome in Cirrhosis. Dig. Dis. Sci. 2016, 61, 3621–3626. [Google Scholar] [CrossRef]
- Wiest, R.; Das, S.; Cadelina, G.; Garcia-Tsao, G.; Milstien, S.; Groszmann, R.J. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J. Clin. Investig. 1999, 104, 1223–1233. [Google Scholar] [CrossRef] [Green Version]
- Tazi, K.A.; Moreau, R.; Hervé, P.; Dauvergne, A.; Cazals-Hatem, D.; Bert, F.; Poirel, O.; Rabiller, A.; Lebrec, D. Norfloxacin Reduces Aortic NO Synthases and Proinflammatory Cytokine Up-Regulation in Cirrhotic Rats: Role of Akt Signaling. Gastroenterology 2005, 129, 303–314. [Google Scholar] [CrossRef]
- Kim, B.I.; Kim, H.J.; Park, J.H.; Park, D.I.; Cho, Y.K.; Sohn, C.I.; Jeon, W.K.; Kim, D.J.; Kim, H.S. Increased intestinal permeability as a predictor of bacterial infections in patients with decompensated liver cirrhosis and hemorrhage. J. Gastroenterol. Hepatol. 2010, 26, 550–557. [Google Scholar] [CrossRef]
- Fukui, H.; Matsumoto, M.; Tsujita, S.; Takaya, A.; Kojima, H.; Matsumura, M.; Tsujii, T. Plasma endotoxin concentration and endo-toxin binding capacity of plasma acute phase proteins in cirrhotics with variceal bleeding: An analysis by new methods. J. Gastroenterol. Hepatol. 1994, 9, 582–586. [Google Scholar] [CrossRef]
- Aguirre Valadez, J.M.; Rivera-Espinosa, L.; Méndez-Guerrero, O.; Chávez-Pacheco, J.L.; García Juárez, I.; Torre, A. Intestinal per-meability in a patient with liver cirrhosis. Ther. Clin. Risk Manag. 2016, 12, 1729–1748. [Google Scholar] [CrossRef] [Green Version]
- Hou, M.-C.; Lin, H.-C.; Liu, T.-T.; Kuo, B.I.-T.; Lee, F.-Y.; Chang, F.-Y.; Lee, S.-D. Antibiotic prophylaxis after endoscopic therapy prevents rebleeding in acute variceal hemorrhage: A randomized trial. Hepatology 2004, 39, 746–753. [Google Scholar] [CrossRef]
- Bernard, B.; Grangé, J.D.; Khac, E.N.; Amiot, X.; Opolon, P.; Poynard, T. Antibiotic prophylaxis for the prevention of bacterial infec-tions in cirrhotic patients with gastrointestinal bleeding: A meta-analysis. Hepatology 1999, 29, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Rau, P.; Liu, D.Q.; Holzwanger, E.; Mathew, J.P.; Guilarte-Walker, Y.; Szabo, G. Proton pump inhibitors increase the se-verity of hepatic encephalopathy in cirrhotic patients. World J. Hepatol. 2019, 11, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.J.; Betrapally, N.; Ghosh, S.A.; Sartor, R.B.; Hylemon, P.B.P.B.; Gillevet, P.M.P.M.; Sanyal, A.J.A.J.; Heuman, D.M.D.M.; Carl, D.; Zhou, H.; et al. Gut microbiota drive the development of neuroinflammatory response in cirrhosis in mice. Hepatology 2016, 64, 1232–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Guo, J.-Y.; Cao, W.-K. Inflammation: A novel target of current therapies for hepatic encephalopathy in liver cirrhosis. World J. Gastroenterol. 2015, 21, 11815–11824. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Rao, K.V.R.; Norenberg, M.D. Neuroinflammation in Hepatic Encephalopathy: Mechanistic Aspects. J. Clin. Exp. Hepatol. 2015, 5, S21–S28. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Liver Physiol. 2012, 303, G675–G685. [Google Scholar] [CrossRef]
- Gupta, A.; Dhiman, R.K.; Kumari, S.; Rana, S.; Agarwal, R.; Duseja, A.; Chawla, Y. Role of small intestinal bacterial overgrowth and delayed gastrointestinal transit time in cirrhotic patients with minimal hepatic encephalopathy. J. Hepatol. 2010, 53, 849–855. [Google Scholar] [CrossRef]
- Bajaj, J.S. Review article: Potential mechanisms of action of rifaximin in the management of hepatic encephalopathy and other complications of cirrhosis. Aliment. Pharmacol. Ther. 2015, 43, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Zamparelli, M.S.; Rocco, A.; Compare, D.; Nardone, G. Faculty Opinions recommendation of the gut microbiota: A new potential driving force in liver cirrhosis and hepatocellular carcinoma. United Eur. Gastroenterol. J. 2017, 5, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-X.; Yan, H.-X.; Liu, Q.; Yang, W.; Wu, H.-P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.-Q.; Zou, S.-S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef]
- Fukui, H. Improve gut microbiome: A new horizon of cancer therapy. Hepatobiliary Surg. Nutr. 2017, 6, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of Gut Microbiota in Hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, T.E.; Grander, C.; Moschen, A.R.; Tilg, H. Liver–Microbiome Axis in Health and Disease. Trends Immunol. 2018, 39, 712–723. [Google Scholar] [CrossRef] [PubMed]
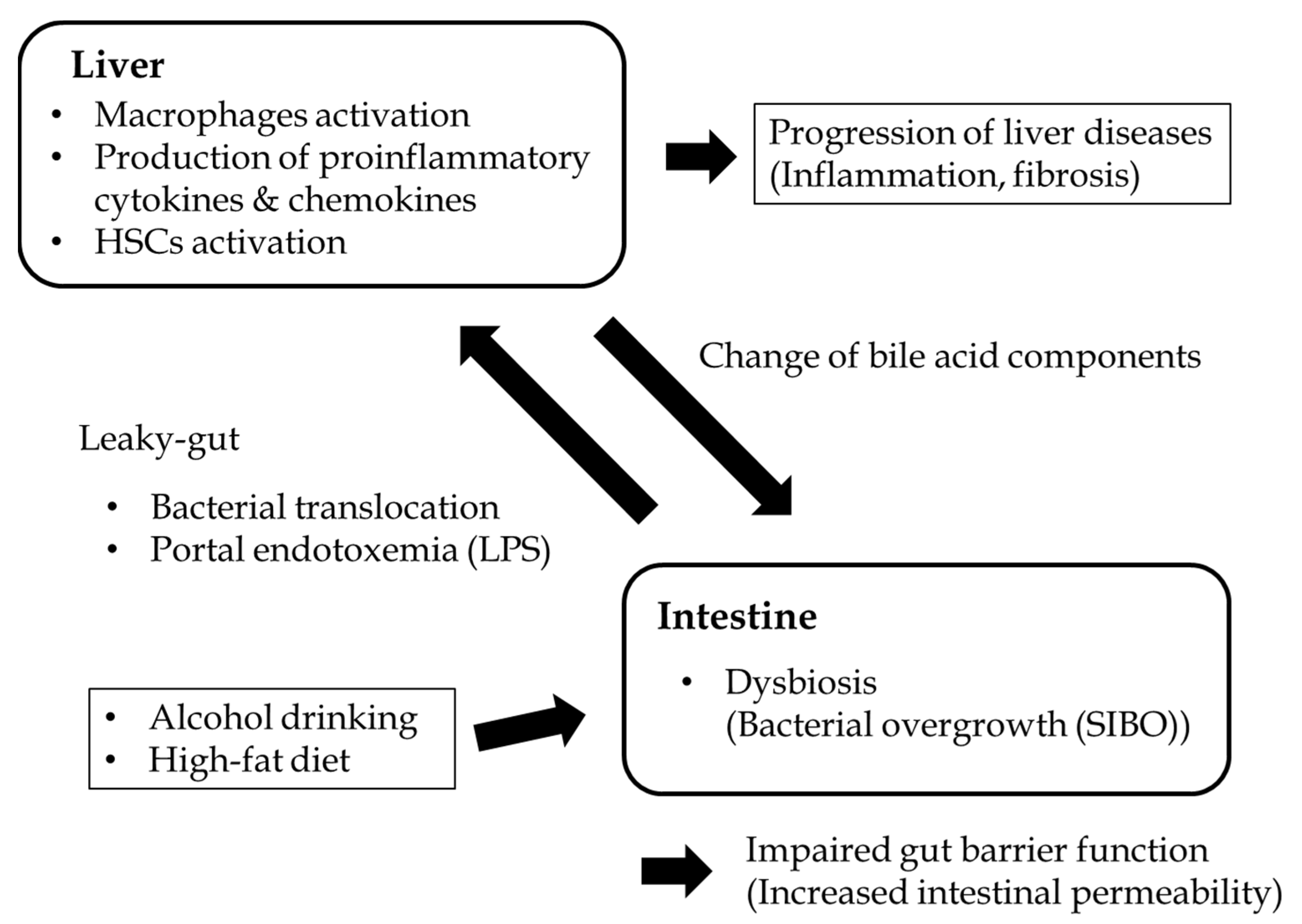


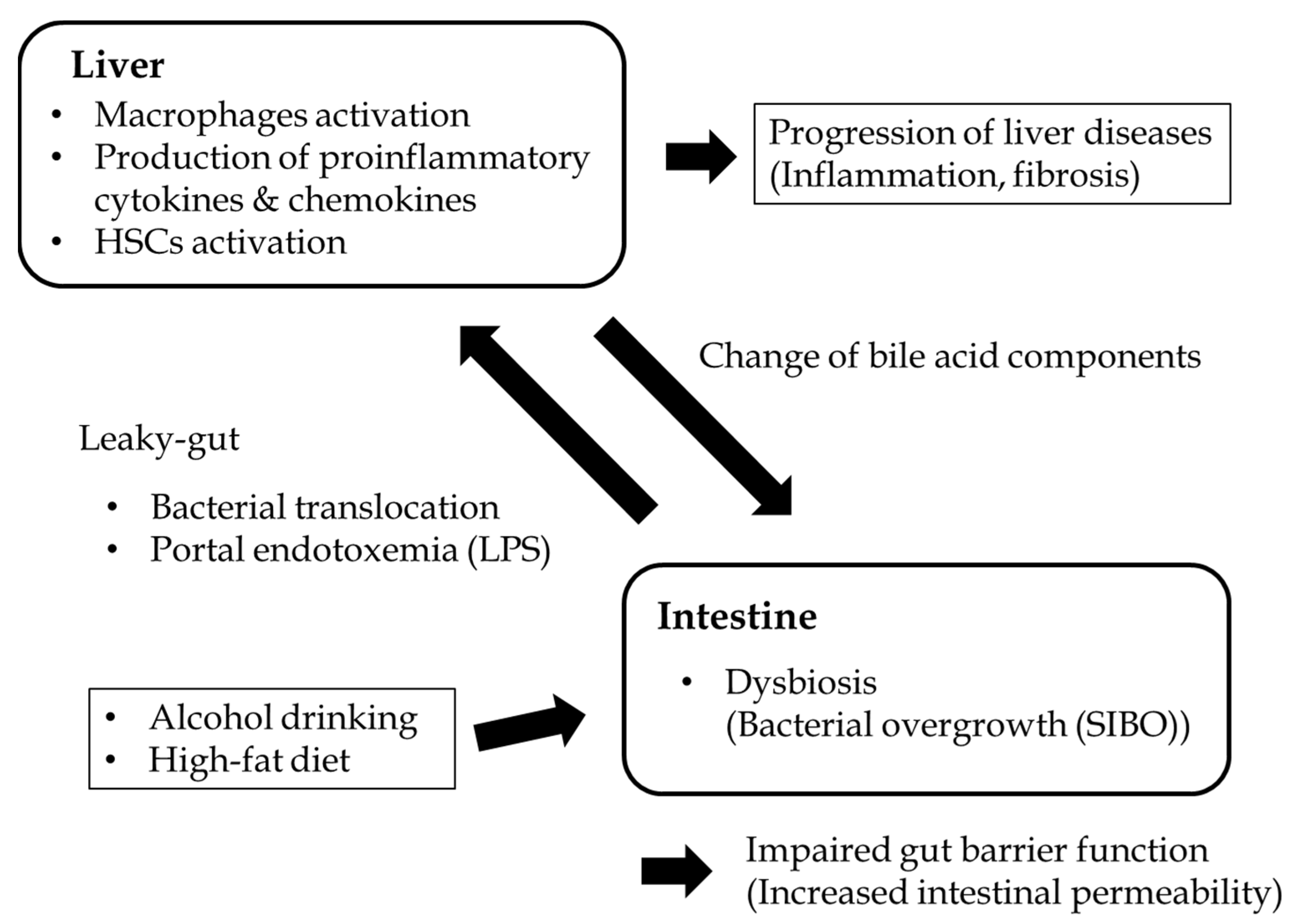{kind=link}
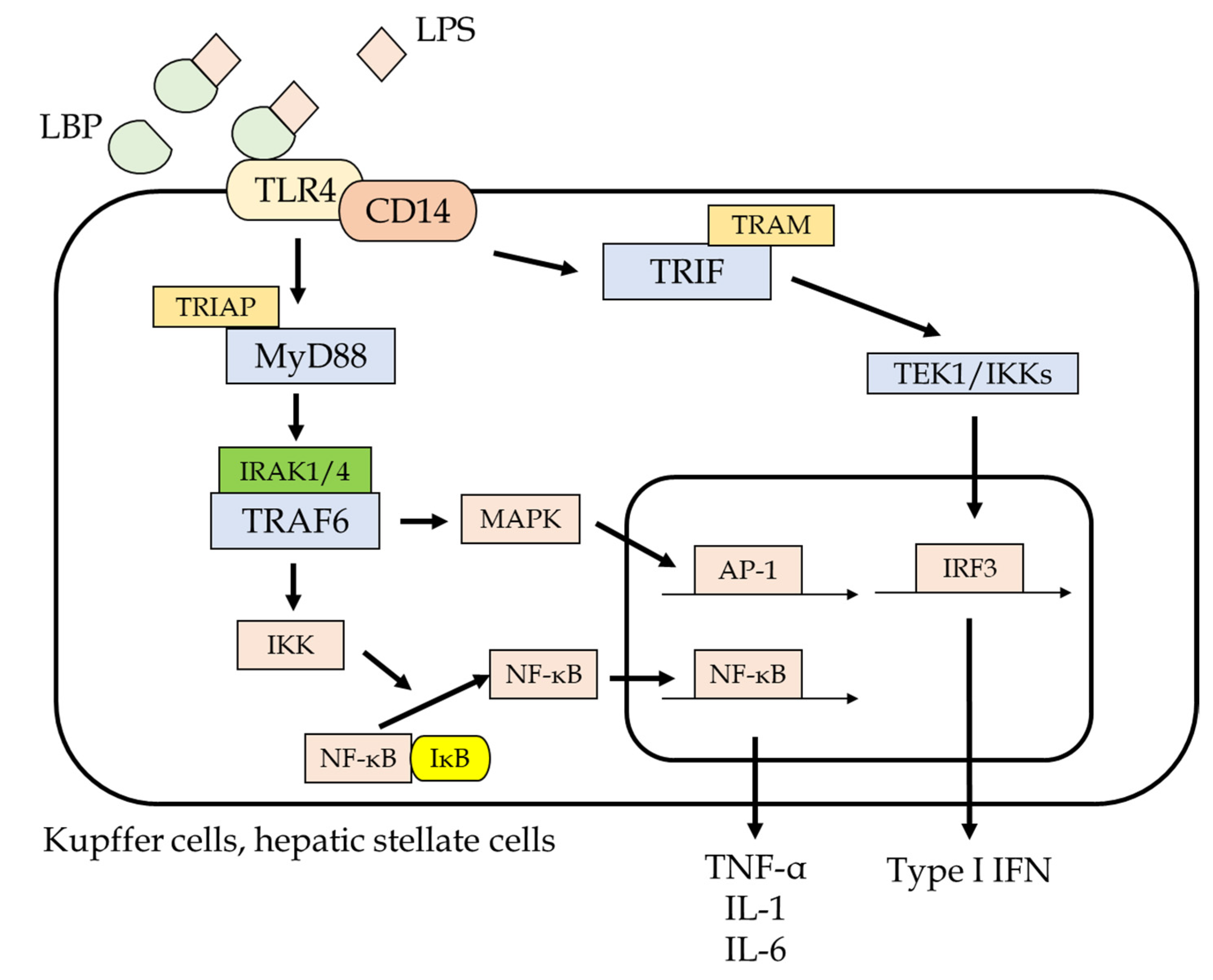{kind=link}
| Type of Diseases | Bacteria | ||||
|---|---|---|---|---|---|
| Phylum | Class | Order | Family | Genus (Species) | |
| ALD | Firmicutes Bacteroidetes ↓ Proteobacteria ↑ | Bacilli Clostridia Negativicutes Bacteroidia proteobacteria ↑ | Lactobacilales Clostridiales Selenomonadales Bacteroidales Enterobacteriales | Lactobacillaceae Clostridiaceae Ruminococcaceae ↓ Lachnospiraceae Veillonellaceae ↑ Bacteroidaceae ↓ Porphyromonadaceae ↓ Enterobacteriaceae ↑ | Lactobacillus ↓ Clostridium ↓ Faecalibacterium ↓ Dorea ↑ Blautia ↑ Klebsiella ↑ |
| NAFLD | Firmicutes Bacteroidetes ↑↓ Proteobacteria ↑ | Bacilli Clostridia ↓ Bacteroidia proteobacteria ↑ | Lactobacilales Clostridiales ↓ Bacteroidales Enterobacteriales | Lactobacillaceae ↑ Clostridiaceae Ruminococcaceae ↓ Lachnospiraceae Bacteroidaceae ↓ Porphyromonadaceae ↑↓ Enterobacteriaceae ↑ | Lactobacillus ↑ Clostridium Faecalibacterium ↓ Dorea ↑ Bacteroides ↓ Escherichia ↑ |
| HBV | Firmicutes Bacteroidetes ↓ Proteobacteria ↑ Actinobacteria | Bacilli Clostridia Bacteroidia Proteobacteria Betaproteobacteria Actinobacteriia | Lactobacilales Clostridiales Bacteroidales Enterobacteriales Neisseriales Bifidobacteriales | Lactobacillaceae ↓ Enterococcaceae ↑ Ruminococcaceae Bacteroidaceae Enterobacteriaceae ↑ Neisseriaceae ↑ Bifidobacteriaceae ↓ | Lactobacillus ↓ Enterococcus ↑ Faecalibacterium Bacteroides Bifidobacterium ↓ |
| HCV | Firmicutes ↓ Bacteroidetes ↑ Proteobacteria ↑ Actinobacteria | Bacilli Bacteroidia Proteobacteria Actinobacteriia | Lactobacilales Bacillales Enterobacteriales Bifidobacteriales | Lactobacillaceae Enterococcaceae ↑ Stphylococcaceae Enterobacteriaceae ↑ Bifidobacteriaceae | Lactobacillus Enterococcus ↑ Staphylococcus ↑ Bifidobacterium |
| PBC | Firmicutes Bacteroidetes ↑↓ Proteobacteria ↑ Fusobacteria Acidobacteria ↓ | Bacilli Clostridia Negativicutes Proteobacteria Gammaproteobacteria Fusobacteriia | Lactobacilales Clostridiales Selenomonadales Enterobacteriales Pasteurellales Fusobacteriales | Lactobacillaceae Clostridiaceae Ruminococcaceae Lachnospiraceae Veillonellaceae Enterobacteriaceae ↑ Pasteurellaceae Fusobacteriaceae | Lactobacillus ↑ Eubacterium ↑ Clostridium ↓ Ruminococcus ↓ Lachnobacterium ↓ Veillonella ↑ Actinobacillus ↑ Haemophilus ↑ Fusobacterium ↑↓ |
| AIH | Firmicutes Actinobacteria Bacteroidetes Proteobacteria | Bacilli Actinobacteriia Negativicutes Proteobacteria | Lactobacilales Bifidobacteriales Selenomonadales Enterobacteriales | Lactobacillaceae Bifidobacteriaceae Veillonellaceae Streptococcaceae Enterobacteriaceae | Lactobacillus ↑↓ Bifidobacterium ↓ Veillonella ↑ Streptococcus ↑ Klebsiella ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishimura, N.; Kaji, K.; Kitagawa, K.; Sawada, Y.; Furukawa, M.; Ozutsumi, T.; Fujinaga, Y.; Tsuji, Y.; Takaya, H.; Kawaratani, H.; et al. Intestinal Permeability Is a Mechanical Rheostat in the Pathogenesis of Liver Cirrhosis. Int. J. Mol. Sci. 2021, 22, 6921. https://doi.org/10.3390/ijms22136921
Nishimura N, Kaji K, Kitagawa K, Sawada Y, Furukawa M, Ozutsumi T, Fujinaga Y, Tsuji Y, Takaya H, Kawaratani H, et al. Intestinal Permeability Is a Mechanical Rheostat in the Pathogenesis of Liver Cirrhosis. International Journal of Molecular Sciences. 2021; 22(13):6921. https://doi.org/10.3390/ijms22136921
Chicago/Turabian StyleNishimura, Norihisa, Kosuke Kaji, Koh Kitagawa, Yasuhiko Sawada, Masanori Furukawa, Takahiro Ozutsumi, Yukihisa Fujinaga, Yuki Tsuji, Hiroaki Takaya, Hideto Kawaratani, and et al. 2021. "Intestinal Permeability Is a Mechanical Rheostat in the Pathogenesis of Liver Cirrhosis" International Journal of Molecular Sciences 22, no. 13: 6921. https://doi.org/10.3390/ijms22136921
APA StyleNishimura, N., Kaji, K., Kitagawa, K., Sawada, Y., Furukawa, M., Ozutsumi, T., Fujinaga, Y., Tsuji, Y., Takaya, H., Kawaratani, H., Moriya, K., Namisaki, T., Akahane, T., Fukui, H., & Yoshiji, H. (2021). Intestinal Permeability Is a Mechanical Rheostat in the Pathogenesis of Liver Cirrhosis. International Journal of Molecular Sciences, 22(13), 6921. https://doi.org/10.3390/ijms22136921