
‘All That Glitters Is Not Gold’: High-Resolution Crystal Structures of Ligand-Protein Complexes Need Not Always Represent Confident Binding Poses


Abstract
1. Introduction
2. Results
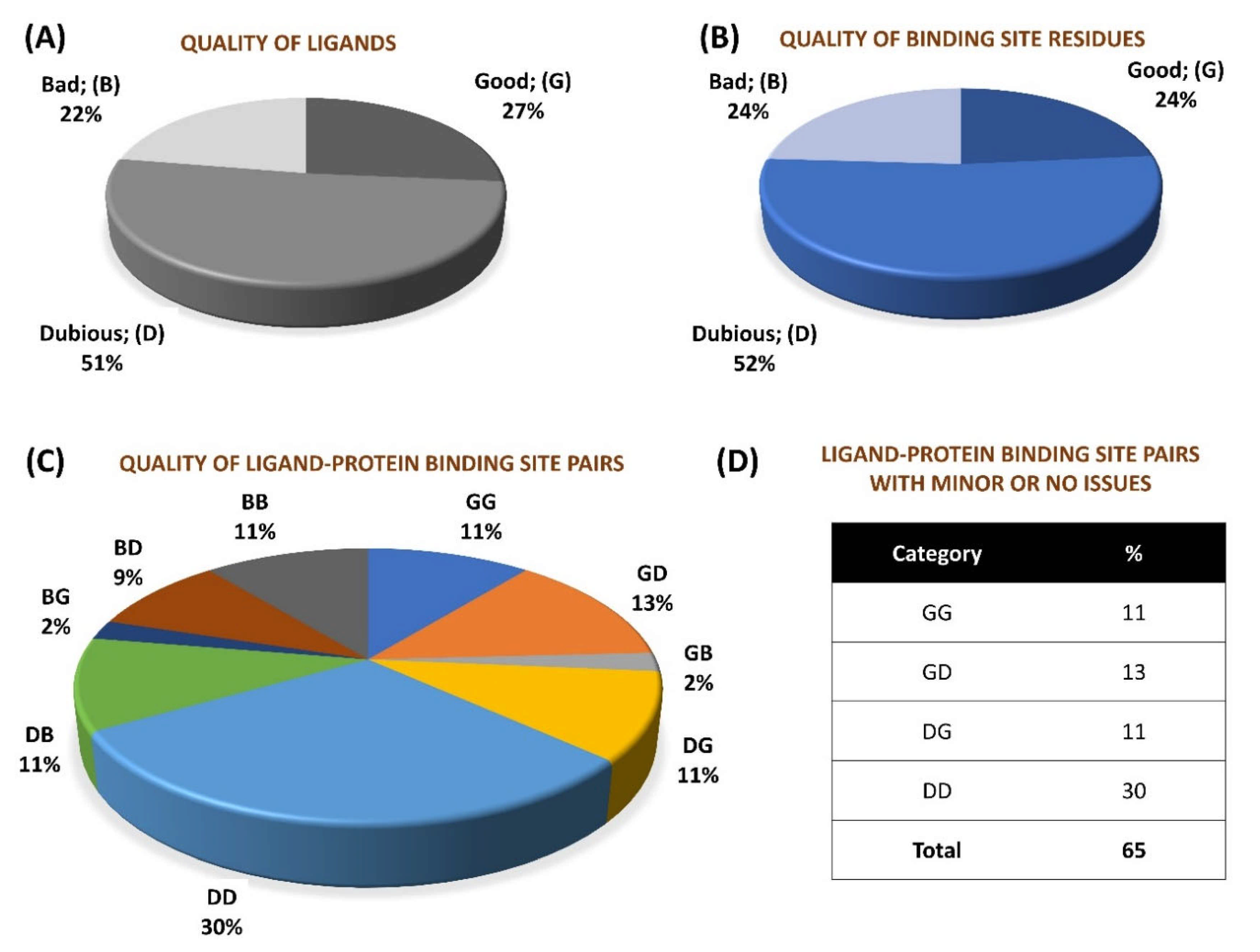
2.1. General Analyses
2.2. Local Quality and Resolution
2.3. Case Studies
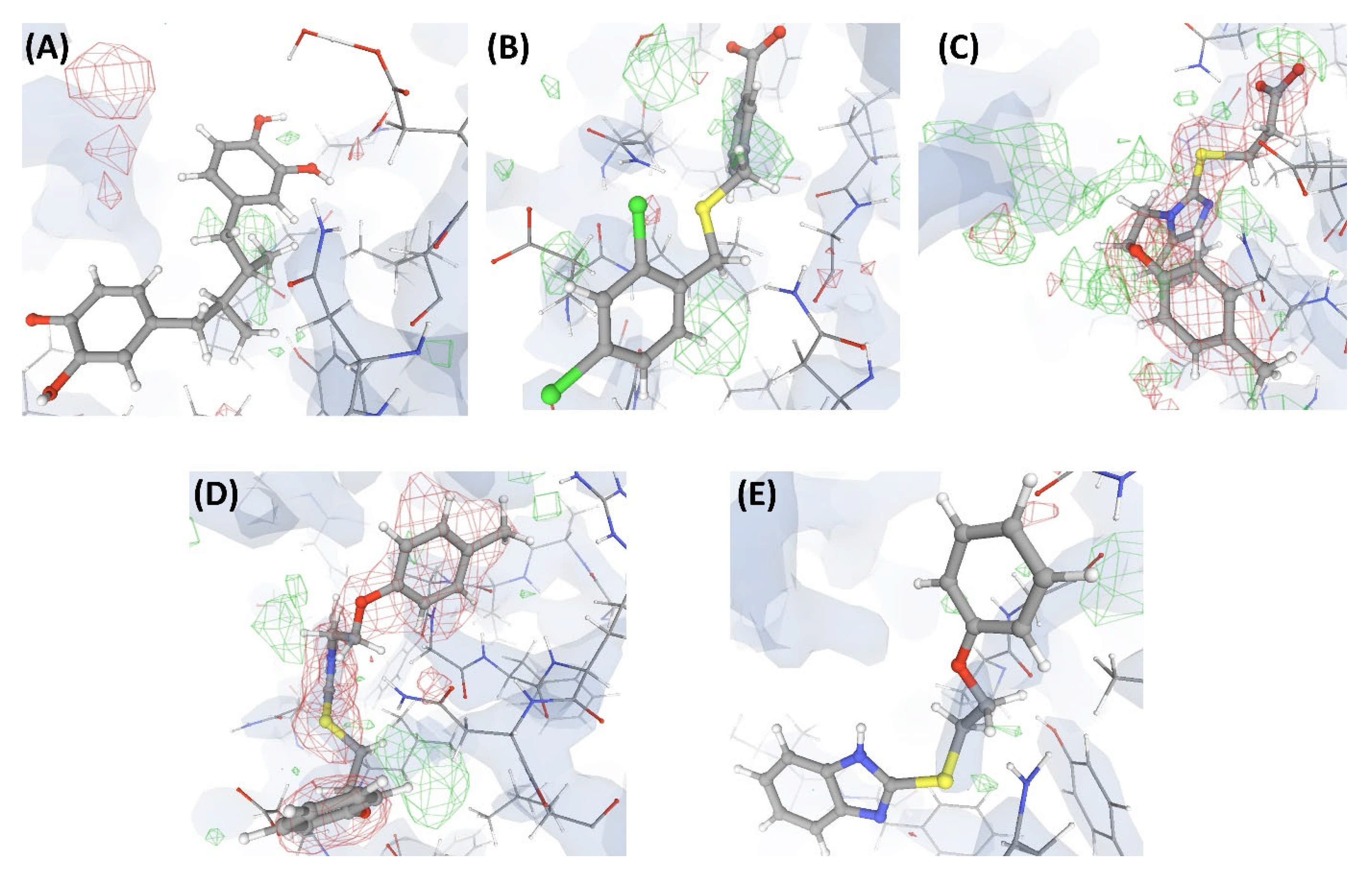
2.3.1. Case Study-1
VHELIBS and Visual Inspection
EDIA
PDB-REDO
Polder Maps
Ligand Building (with ARP/wARP)
Docking Simulations
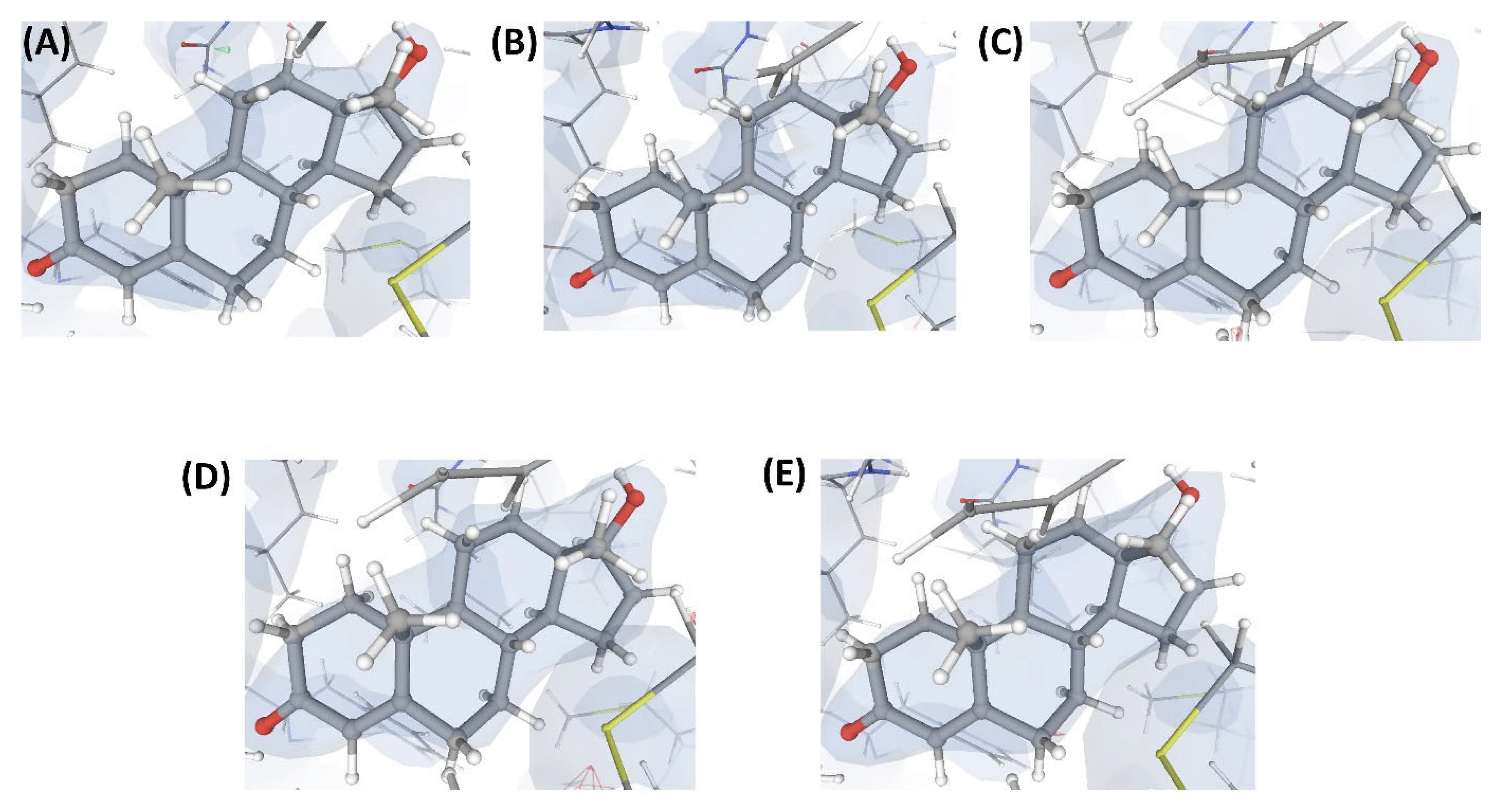
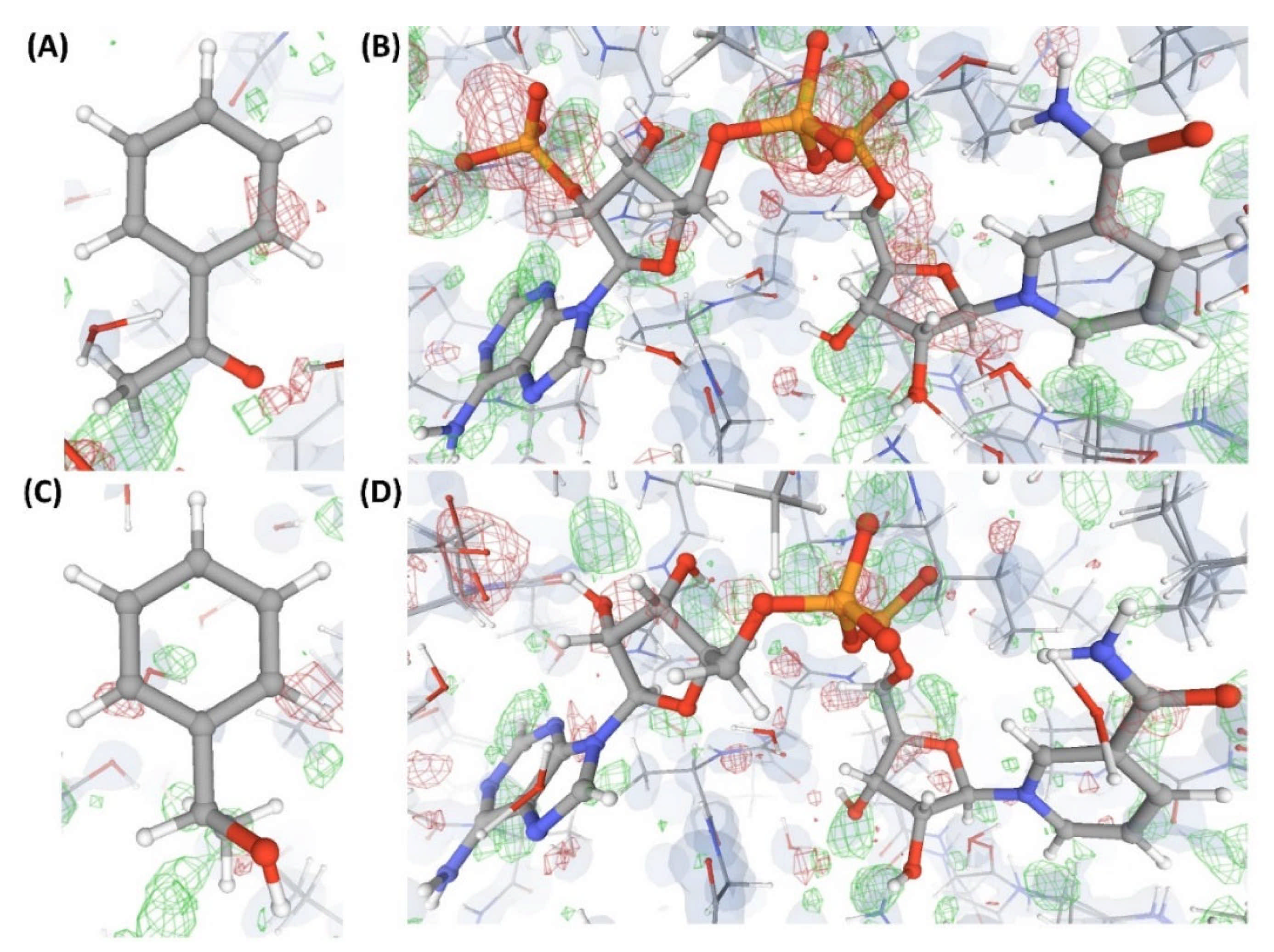
2.3.2. Case Study-2
2.3.3. Case Study-3
2.4. In Silico Assessment of Stereochemical Features of P–L Complexes
3. Discussion
3.1. Sensitizing Users
3.2. Re-Visiting the Structures of Concern by Crystallographers
3.3. Proposals to the Journals
3.4. Proposals to the Repository Community
4. Materials and Methods
4.1. Dataset
4.2. Quality Assessment of Protein–Ligand Binding Sites
- (a)
- Maximum Real Space R-factor (RSR): 0.4;
- (b)
- Maximum ‘Good’ RSR: 0.24;
- (c)
- Minimum ‘Good’ real space correlation coefficient (RSCC): 0.9;
- (d)
- Average occupancy: 1.0;
- (e)
- Occupancy weighted average B-factor (OWAB): 50 Å2;
- (f)
- Maximum Rfree: 0.3;
- (g)
- Maximum [(Rfree) − (Rwork)] value: 0.05.
4.3. Polder Map Assessment
4.4. Ligand Building
4.5. Docking Simulation
4.6. Visualization and Graph Plotting
4.7. In Silico Approach to Assess Stereochemical Quality of P–L Binding Sites
4.8. Software Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. NMR—this other method for protein and nucleic acid structure determination. Acta Crystallogr. Sect. D 1995, 51, 249–270. [Google Scholar] [CrossRef]
- Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- PDB Data Distribution by Experimental Method and Molecular Type. Available online: https://www.rcsb.org/stats/summary (accessed on 17 March 2021).
- COVID-19/SARS-CoV-2 Resources. Available online: https://www.rcsb.org/news?year=2020&article=5e74d55d2d410731e9944f52&feature=true (accessed on 17 March 2021).
- Förster, A.; Schulze-Briese, C. A shared vision for macromolecular crystallography over the next five years. Struct. Dyn. 2019, 6, 64302. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray Crystallography and Drug Discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef]
- Feng, Z.; Verdiguel, N.; Di Costanzo, L.; Goodsell, D.S.; Westbrook, J.D.; Burley, S.K.; Zardecki, C. Impact of the Protein Data Bank Across Scientific Disciplines. Data Sci. J. 2020, 19, 25. [Google Scholar] [CrossRef]
- Pozharski, E.; Deller, M.C.; Rupp, B. Validation of Protein-Ligand Crystal Structure Models: Small Molecule and Peptide Ligands. Methods Mol. Biol. 2017, 1607, 611–625. [Google Scholar] [CrossRef]
- Deller, M.C.; Rupp, B. Models of protein-ligand crystal structures: Trust, but verify. J. Comput. Aided. Mol. Des. 2015, 29, 817–836. [Google Scholar] [CrossRef]
- Pozharski, E.; Weichenberger, C.X.; Rupp, B. Techniques, tools and best practices for ligand electron-density analysis and results from their application to deposited crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 150–167. [Google Scholar] [CrossRef]
- Wlodawer, A.; Dauter, Z.; Shabalin, I.G.; Gilski, M.; Brzezinski, D.; Kowiel, M.; Minor, W.; Rupp, B.; Jaskolski, M. Ligand-centered assessment of SARS-CoV-2 drug target models in the Protein Data Bank. FEBS J. 2020, 287, 3703–3718. [Google Scholar] [CrossRef] [PubMed]
- Smart, O.S.; Horský, V.; Gore, S.; Svobodová Va\vreková, R.; Bendová, V.; Kleywegt, G.J.; Velankar, S. Validation of ligands in macromolecular structures determined by X-ray crystallography. Acta Crystallogr. Sect. D 2018, 74, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Yang, H.; Westbrook, J.D.; Young, J.Y.; Zardecki, C.; Burley, S.K. Multivariate Analyses of Quality Metrics for Crystal Structures in the PDB Archive. Structure 2017, 25, 458–468. [Google Scholar] [CrossRef]
- Lamb, A.L.; Kappock, T.J.; Silvaggi, N.R. You are lost without a map: Navigating the sea of protein structures. Biochim. Biophys. Acta 2015, 1854, 258–268. [Google Scholar] [CrossRef]
- Tickle, I.J. Statistical quality indicators for electron-density maps. Acta Crystallogr. Sect. D 2012, 68, 454–467. [Google Scholar] [CrossRef]
- Meyder, A.; Nittinger, E.; Lange, G.; Klein, R.; Rarey, M. Estimating Electron Density Support for Individual Atoms and Molecular Fragments in X-ray Structures. J. Chem. Inf. Model. 2017, 57, 2437–2447. [Google Scholar] [CrossRef]
- Domagalski, M.J.; Zheng, H.; Zimmerman, M.D.; Dauter, Z.; Wlodawer, A.; Minor, W. The quality and validation of structures from structural genomics. Methods Mol. Biol. 2014, 1091, 297–314. [Google Scholar] [CrossRef]
- Weichenberger, C.X.; Pozharski, E.; Rupp, B. Visualizing ligand molecules in twilight electron density. Acta Crystallogr. Sect. F 2013, 69, 195–200. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Joosten, R.P.; Valls, C.; Mulero, M.; Salvado, M.J.; Arola-Arnal, A.; Arola, L.; Garcia-Vallvé, S.; Pujadas, G. The good, the bad and the dubious: VHELIBS, a validation helper for ligands and binding sites. J. Cheminform. 2013, 5, 36. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 148–157. [Google Scholar] [CrossRef]
- Masmaliyeva, R.C.; Babai, K.H.; Murshudov, G.N. Local and global analysis of macromolecular atomic displacement parameters. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Velankar, S.; Alhroub, Y.; Alili, A.; Best, C.; Boutselakis, H.C.; Caboche, S.; Conroy, M.J.; Dana, J.M.; van Ginkel, G.; Golovin, A.; et al. PDBe: Protein Data Bank in Europe. Nucleic Acids Res. 2011, 39, D402–D410. [Google Scholar] [CrossRef]
- PDBe Brings Electron-Density Viewing to the Masses. Available online: https://www.ebi.ac.uk/pdbe/about/news/pdbe-brings-electron-density-viewing-masses (accessed on 16 March 2021).
- Liebeschuetz, J.; Hennemann, J.; Olsson, T.; Groom, C.R. The good, the bad and the twisted: A survey of ligand geometry in protein crystal structures. J. Comput. Aided. Mol. Des. 2012, 26, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Hou, J.; Zimmerman, M.D.; Wlodawer, A.; Minor, W. The future of crystallography in drug discovery. Expert Opin. Drug Discov. 2014, 9, 125–137. [Google Scholar] [CrossRef]
- Cooper, D.R.; Porebski, P.J.; Chruszcz, M.; Minor, W. X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery. Expert Opin. Drug Discov. 2011, 6, 771–782. [Google Scholar] [CrossRef]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 2013, 280, 5705–5736. [Google Scholar] [CrossRef]
- Kellenberger, E.; Muller, P.; Schalon, C.; Bret, G.; Foata, N.; Rognan, D. sc-PDB: An annotated database of druggable binding sites from the Protein Data Bank. J. Chem. Inf. Model. 2006, 46, 717–727. [Google Scholar] [CrossRef]
- Singh, S.; Srivastava, H.K.; Kishor, G.; Singh, H.; Agrawal, P.; Raghava, G.P.S. Evaluation of protein-ligand docking methods on peptide-ligand complexes for docking small ligands to peptides. bioRxiv 2017. [Google Scholar] [CrossRef]
- Bhojwan, H.R.; Joshi, U.J. Selecting protein structure/s for docking-based virtual screening: A case study on type II inhibitors of VEGFR-2 kinase. Int. J. Pharm. Sci. Res. 2019, 10, 2998–3011. [Google Scholar] [CrossRef]
- Kirchmair, J.; Wolber, G.; Laggner, C.; Langer, T. Comparative Performance Assessment of the Conformational Model Generators Omega and Catalyst: A Large-Scale Survey on the Retrieval of Protein-Bound Ligand Conformations. J. Chem. Inf. Model. 2006, 46, 1848–1861. [Google Scholar] [CrossRef]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, High-Quality Test Set for the Validation of Protein−Ligand Docking Performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Beshnova, D.A.; Pereira, J.; Lamzin, V.S. Estimation of the protein-ligand interaction energy for model building and validation. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Boittier, E.D.; Tang, Y.Y.; Buckley, M.E.; Schuurs, Z.P.; Richard, D.J.; Gandhi, N.S. Assessing Molecular Docking Tools to Guide Targeted Drug Discovery of CD38 Inhibitors. Int. J. Mol. Sci. 2020, 21, 5183. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Spyrakis, F.; Cozzini, P.; Kellogg, G.E. Docking and Scoring in Drug Discovery. Burger’s Med. Chem. Drug Discov. 2010, 601–684. [Google Scholar] [CrossRef]
- Elokely, K.M.; Doerksen, R.J. Docking Challenge: Protein Sampling and Molecular Docking Performance. J. Chem. Inf. Model. 2013, 53, 1934–1945. [Google Scholar] [CrossRef]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008, 275, 1–21. [Google Scholar] [CrossRef]
- Yao, S.; Moseley, H.N.B. A chemical interpretation of protein electron density maps in the worldwide protein data bank. PLoS ONE 2020, 15, e0236894. [Google Scholar] [CrossRef]
- Joosten, R.P.; Joosten, K.; Murshudov, G.N.; Perrakis, A. PDB_REDO: Constructive validation, more than just looking for errors. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 484–496. [Google Scholar] [CrossRef]
- Merz, K.M., Jr. Using quantum mechanical approaches to study biological systems. Acc. Chem. Res. 2014, 47, 2804–2811. [Google Scholar] [CrossRef]
- Fu, Z.; Li, X.; Miao, Y.; Merz, K.M., Jr. Conformational analysis and parallel QM/MM X-ray refinement of protein bound anti-Alzheimer drug donepezil. J. Chem. Theory Comput. 2013, 9, 1686–1693. [Google Scholar] [CrossRef]
- Li, X.; Fu, Z.; Merz, K.M., Jr. QM/MM refinement and analysis of protein bound retinoic acid. J. Comput. Chem. 2012, 33, 301–310. [Google Scholar] [CrossRef]
- Li, X.; Hayik, S.A.; Merz, K.M., Jr. QM/MM X-ray refinement of zinc metalloenzymes. J. Inorg. Biochem. 2010, 104, 512–522. [Google Scholar] [CrossRef][Green Version]
- Xu, Z.; Zhang, Q.; Shi, J.; Zhu, W. Underestimated Noncovalent Interactions in Protein Data Bank. J. Chem. Inf. Model. 2019, 59, 3389–3399. [Google Scholar] [CrossRef]
- Peters, M.B.; Raha, K.; Merz, K.M.J. Quantum mechanics in structure-based drug design. Curr. Opin. Drug Discov. Devel. 2006, 9, 370–379. [Google Scholar]
- Brown, E.N.; Ramaswamy, S. Quality of protein crystal structures. Acta Crystallogr. Sect. D 2007, 63, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Chakravarthi, P.; Srinivasan, N. Chapter 2-A ligand-centric approach to identify potential drugs for repurposing: Case study with aurora kinase inhibitors. In Drug Repurposing in Cancer Therapy; To, K., Cho, W., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 15–54. ISBN 978-0-12-819668-7. [Google Scholar]
- Chakraborti, S.; Bheemireddy, S.; Srinivasan, N. Repurposing drugs against the main protease of SARS-CoV-2: Mechanism-based insights supported by available laboratory and clinical data. Mol. Omi. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Chakraborty, M.; Bose, A.; Srinivasan, N.; Visweswariah, S.S. Identification of potential binders of Mtb Universal Stress Protein (Rv1636) through an in silico approach and insights into compound selection for experimental validation. Front. Mol. Biosci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Dauter, Z.; Porebski, P.J.; Minor, W.; Stanfield, R.; Jaskolski, M.; Pozharski, E.; Weichenberger, C.X.; Rupp, B. Detect, correct, retract: How to manage incorrect structural models. FEBS J. 2018, 285, 444–466. [Google Scholar] [CrossRef]
- Minor, W.; Dauter, Z.; Helliwell, J.R.; Jaskolski, M.; Wlodawer, A. Safeguarding Structural Data Repositories against Bad Apples. Structure 2016, 24, 216–220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adams, P.D.; Aertgeerts, K.; Bauer, C.; Bell, J.A.; Berman, H.M.; Bhat, T.N.; Blaney, J.M.; Bolton, E.; Bricogne, G.; Brown, D.; et al. Outcome of the First wwPDB/CCDC/D3R Ligand Validation Workshop. Structure 2016, 24, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Westbrook, J.D.; Sala, R.; Smart, O.S.; Bricogne, G.; Matsubara, M.; Yamada, I.; Tsuchiya, S.; Aoki-Kinoshita, K.F.; Hoch, J.C.; et al. Enhanced validation of small-molecule ligands and carbohydrates in the Protein Data Bank. Structure 2021, 29, 393–400.e1. [Google Scholar] [CrossRef] [PubMed]
- Schöning-Stierand, K.; Diedrich, K.; Fährrolfes, R.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Steinegger, R.; Rarey, M. ProteinsPlus: Interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020, 48, W48–W53. [Google Scholar] [CrossRef]
- Waskom, M.; Botvinnik, O.; O’Kane, D.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Cole, J.B.; Warmenhoven, J.; et al. mwaskom/seaborn: v0.8.1 (September 2017). Zenodo 2017. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Mod. 1999, 17, 57–61. [Google Scholar]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.; Hildebrand, P.W.; Goede, A.; Gruening, B.; Preissner, R. Voronoia: Analyzing packing in protein structures. Nucleic Acids Res. 2009, 37, D393–D395. [Google Scholar] [CrossRef] [PubMed]
- VHELIBS, version 4.3; Software for quality assessment/validation of protein-ligand binding sites; GitHub: San Francisco, CA, USA, 2012.
- AutoDock Vina; Version 1.1.2; Software for Docking Ligands into Protein Binding Sites; Molecular Graphics Laboratory, The Scripps Research Institute: La Jolla, CA, USA, 2011.
- Phenix, version 1.19.2-4158; Software suite for macromolecular structure determination and analysis; Phenix: Berkeley, CA, USA.
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed]
- PyMol-The PyMOL Molecular Graphics System, version 2.4.0; Software for molecular visualization; Schrödinger, LLC: New York, NY, USA, 2010.
- Voronoia, version 1.0; Software for calculating packing densities in protein; CHARITÉ: Berlin, Germany.
- MGL Tools; Version 1.5.6; Software for Visualization and Analysis of Molecular Structures; Molecular Graphics Laboratory, The Scripps Research Institute: La Jolla, CA, USA, 2012.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entity (Ligand/Residue) | VHELIBS * | EDIA Analysis | PDB Validation Report | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand Score; Binding Site Score; Category | EDIAm a | OPIA b | Mean B-Factor c | Occ. < 1.0 d | RSCC e | |||||||||||||||||||||||||
| C1 | C2 | C3 | C4 $ | C17 | C1 | C2 | C3 | C4 | C17 | C1 | C2 | C3 | C4 | C17 | C1 | C2 | C3 | C4 | C17 | C1 | C2 | C3 | C4 | C17 | C1 | C2 | C3 | C4 | C17 | |
| Ligand at S1 | 7;4; BB | 6;2; BD | 4;3; BB | NA | 5;3; BB | 0.22 | 0.38 | 0.39 | 0.27 | 0.41 | 0 | 15 | 36 | 14 | 0 | 75.01 (53.41) | 64.07 (32.05) | 19.96 (35.96) | 19.97 (39.87) | 82.52 (41.12) | 22 | 20 | 0 | 0 | 19 | 0.61 | 0.23 | 0.67 | 0.69 | 0.78 |
| S1: Phe1 | NA | 0.56 | 0.90 | 0.81 | 0.69 | 0.70 | 27 | 91 | 73 | 36 | 36 | 56.53 | 29.01 | 35.90 | 45.28 | 38.92 | 0 | 0 | 0 | 0 | 0 | 0.93 | 0.93 | 0.95 | 0.94 | 0.95 | ||||
| S1: Asn55 | NA | 0.74 | 0.67 | 0.70 | 0.67 | 0.54 | 75 | 50 | 75 | 63 | 38 | 59.71 | 41.85 | 38.72 | 46.98 | 53.29 | 0 | 0 | 0 | 0 | 0 | 0.87 | 0.87 | 0.86 | 0.79 | 0.94 | ||||
| S1: Phe154 | NA | 0.79 | 0.77 | 0.73 | 0.80 | 0.68 | 73 | 55 | 55 | 45 | 45 | 57.93 | 37.09 | 43.20 | 45.71 | 55.62 | 0 | 0 | 0 | 0 | 0 | 0.90 | 0.93 | 0.93 | 0.93 | 0.94 | ||||
| S1: Glu157 | NA | 0.36 | 0.35 | 0.70 | 0.55 | 0.76 | 44 | 56 | 56 | 44 | 44 | 54.83 | 32.04 | 36.58 | 38.39 | 35.86 | 3 | 3 | 3 | 3 | 0 | 0.96 | 0.96 | 0.94 | 0.93 | 0.98 | ||||
| S1: Leu158 | NA | 0.63 | 0.90 | 0.76 | 0.82 | 0.79 | 75 | 75 | 75 | 75 | 75 | 51.19 | 29.96 | 31.86 | 35.88 | 29.28 | 0 | 0 | 0 | 0 | 0 | 0.96 | 0.98 | 0.98 | 0.96 | 0.93 | ||||
| S1: Glu165 | NA | 0.87 | 0.84 | 0.91 | 0.91 | 0.84 | 56 | 56 | 78 | 89 | 67 | 57.10 | 36.72 | 41.59 | 47.95 | 38.21 | 0 | 0 | 0 | 0 | 0 | 0.91 | 0.95 | 0.96 | 0.96 | 0.92 | ||||
| S1: Arg168 | NA | 0.38 | 0.33 | 0.33 | 0.26 | 0.59 | 45 | 36 | 36 | 36 | 45 | 60.94 | 45.93 | 45.71 | 56.23 | 62.37 | 6 | 6 | 6 | 6 | 1 | 0.96 | 0.94 | 0.94 | 0.92 | 0.93 | ||||
| Ligand at S2 | 2;3; DB | 1;1; DD | 1;1; DD | 1;2; DD | 1;1; DD | 0.88 | 0.93 | 0.87 | 0.86 | 0.85 | 71 | 90 | 71 | 71 | 67 | 39.96 (44.30) | 23.01 (25.99) | 24.32 (29.96) | 24.14 (34.64) | 25.24 (29.73) | 0 | 0 | 0 | 0 | 0 | 0.94 | 0.95 | 0.98 | 0.97 | 0.97 |
| Protein–Ligand Complex Identifier (Resolution) | VHELIBS * | EDIA Analysis | PDB Validation Report | PDB-REDO | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand Score; Binding Site Score; Category | EDIAm a | OPIA b | Mean B-Factor c | Occ. < 1.0 d | RSCC e | ΔRSCC f | ||||||||
| SL | CSL | SL | CSL | SL | CSL | SL | CSL | SL | CSL | SL | CSL | SL | CSL | |
| C5 (1.00 Å) | 4; 4; BB | 4; 4; BB | 0.10 | 0.08 | 0 | 0 | 63.29 (13.80) | 94.82 (14.32) | 0 | 0 | 0.62 | 0.32 | 0.281 (significant) | 0.226 (significant) |
| C6 (1.10 Å) | 4; 3; BB | 3; 4; BB | 0.07 | 0.11 | 0 | 0 | 55.43 (9.72) | 63.60 (10.85) | 0 | 0 | 0.10 | 0.16 | 0.188 (significant) | 0.279 (significant) |
| C7 (1.54 Å) | 4; 4; BB | 4; 4; BB | 0.24 | 0.18 | 0 | 0 | 71.94 (15.65) | 52.55 (16.45) | 0 | 0 | 0.14 | 0.34 | −0.016 (insignificant) | −0.017 (insignificant) |
| C8 (1.78 Å) | 2; 3; DB | 3; 2; BD | 0.51 | 0.18 | 33 | 0 | 49.47 (14.40) | 49.78 (14.42) | 0 | 0 | 0.62 | 0.32 | −0.034 (insignificant) | −0.024 (insignificant) |
| C9 (1.05 Å) | 2; 3; DB | 3; 2; BD | 0.04 | 0.1 | 0 | 0 | 55.04 (13.63) | 67.08 (14.27) | 0 | 0 | 0.37 | 0.12 | 0.153 (significant) | 0.240 (significant) |
| C10 (1.08 Å) | N/A | 0; 0; GG | N/A | 0.84 | N/A | 73 | N/A | 5.5 (6.25) | N/A | 0 | N/A | N/A | N/A | 0.004 (significant) |
| Protein–Ligand Complex Identifier; Ligand Code (Resolution) | VHELIBS * | EDIA Analysis | PDB Validation Report | PDB-REDO | |||
|---|---|---|---|---|---|---|---|
| Ligand Score; Binding Site Score; Category | EDIAm a | OPIA b | Mean B-factor c | Occ. < 1.0 d | RSCC e | ΔRSCC f | |
| C11; L8 (3.4 Å) | 7; 6; BB | 0.23 | 6 | 79.20 (69.83) | 31 | 0.58 | 0.080 (insignificant) |
| C12; L9 (3.4 Å) | 0; 3; GB | 0.88 | 81 | 106.33 (111.43) | 0 | 0.93 | 0.070 (insignificant) |
| C13; L8 (3.4 Å) | 0; 3; BB | 0.74 | 61 | 109.38 (98.70) | 0 | 0.95 | 0.058 (significant) |
| C14; L8 (3.4 Å) | 0; 2; GD | 0.84 | 77 | 47.55 (44.04) | 0 | 0.95 | −0.180 (significant) |
| C15; L9 (3.4 Å) | 0; 1; GD | 0.72 | 56 | 49.71 (49.17) | 0 | 0.97 | −0.055 (significant) |
| C16; L8 (2.1 Å) | 4; 1; BD | 0.31 | 19 | 61.20 (24.36) | 0 | 0.59 | 0.190 (significant) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborti, S.; Hatti, K.; Srinivasan, N. ‘All That Glitters Is Not Gold’: High-Resolution Crystal Structures of Ligand-Protein Complexes Need Not Always Represent Confident Binding Poses. Int. J. Mol. Sci. 2021, 22, 6830. https://doi.org/10.3390/ijms22136830
Chakraborti S, Hatti K, Srinivasan N. ‘All That Glitters Is Not Gold’: High-Resolution Crystal Structures of Ligand-Protein Complexes Need Not Always Represent Confident Binding Poses. International Journal of Molecular Sciences. 2021; 22(13):6830. https://doi.org/10.3390/ijms22136830
Chicago/Turabian StyleChakraborti, Sohini, Kaushik Hatti, and Narayanaswamy Srinivasan. 2021. "‘All That Glitters Is Not Gold’: High-Resolution Crystal Structures of Ligand-Protein Complexes Need Not Always Represent Confident Binding Poses" International Journal of Molecular Sciences 22, no. 13: 6830. https://doi.org/10.3390/ijms22136830
APA StyleChakraborti, S., Hatti, K., & Srinivasan, N. (2021). ‘All That Glitters Is Not Gold’: High-Resolution Crystal Structures of Ligand-Protein Complexes Need Not Always Represent Confident Binding Poses. International Journal of Molecular Sciences, 22(13), 6830. https://doi.org/10.3390/ijms22136830