
From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
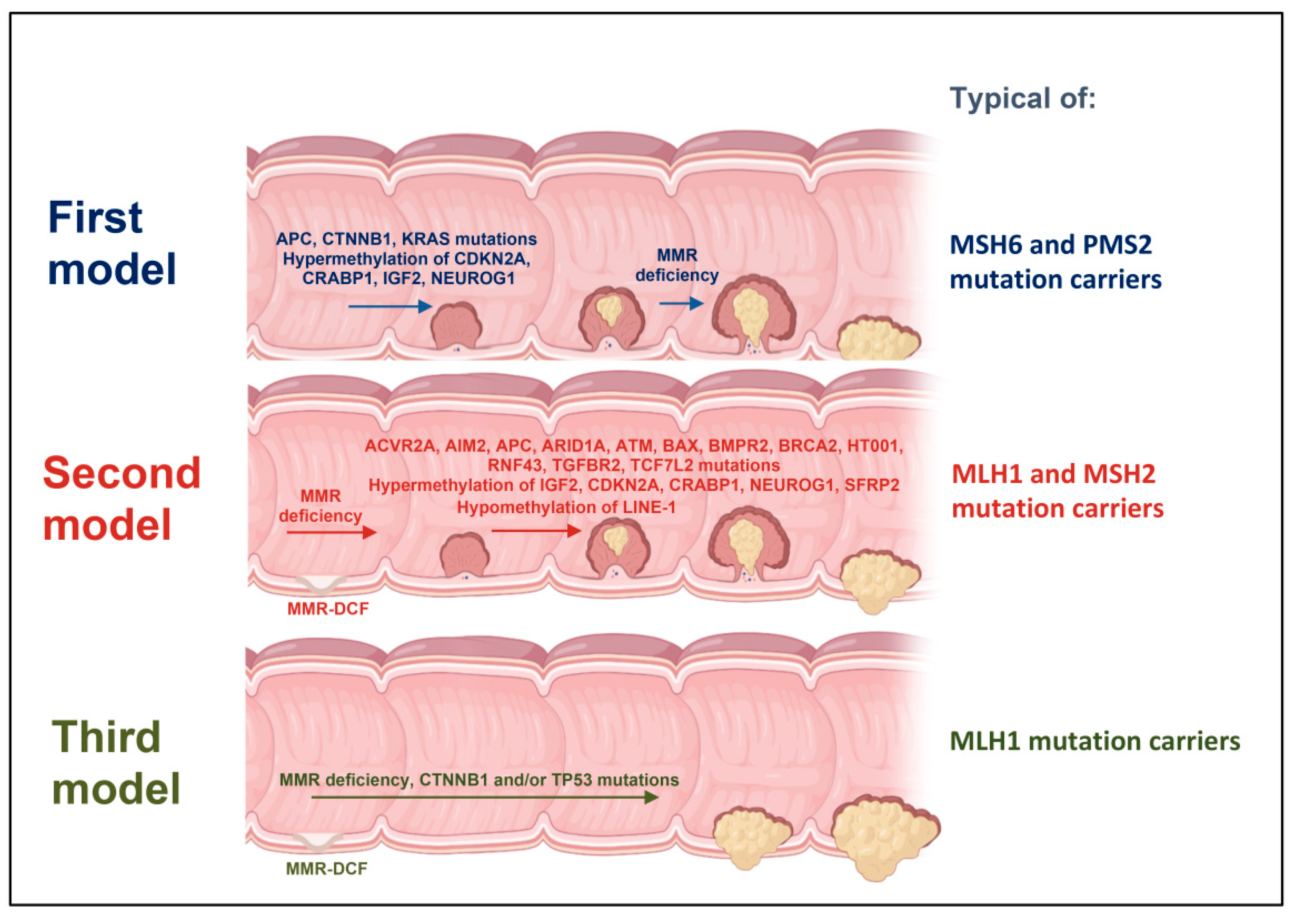
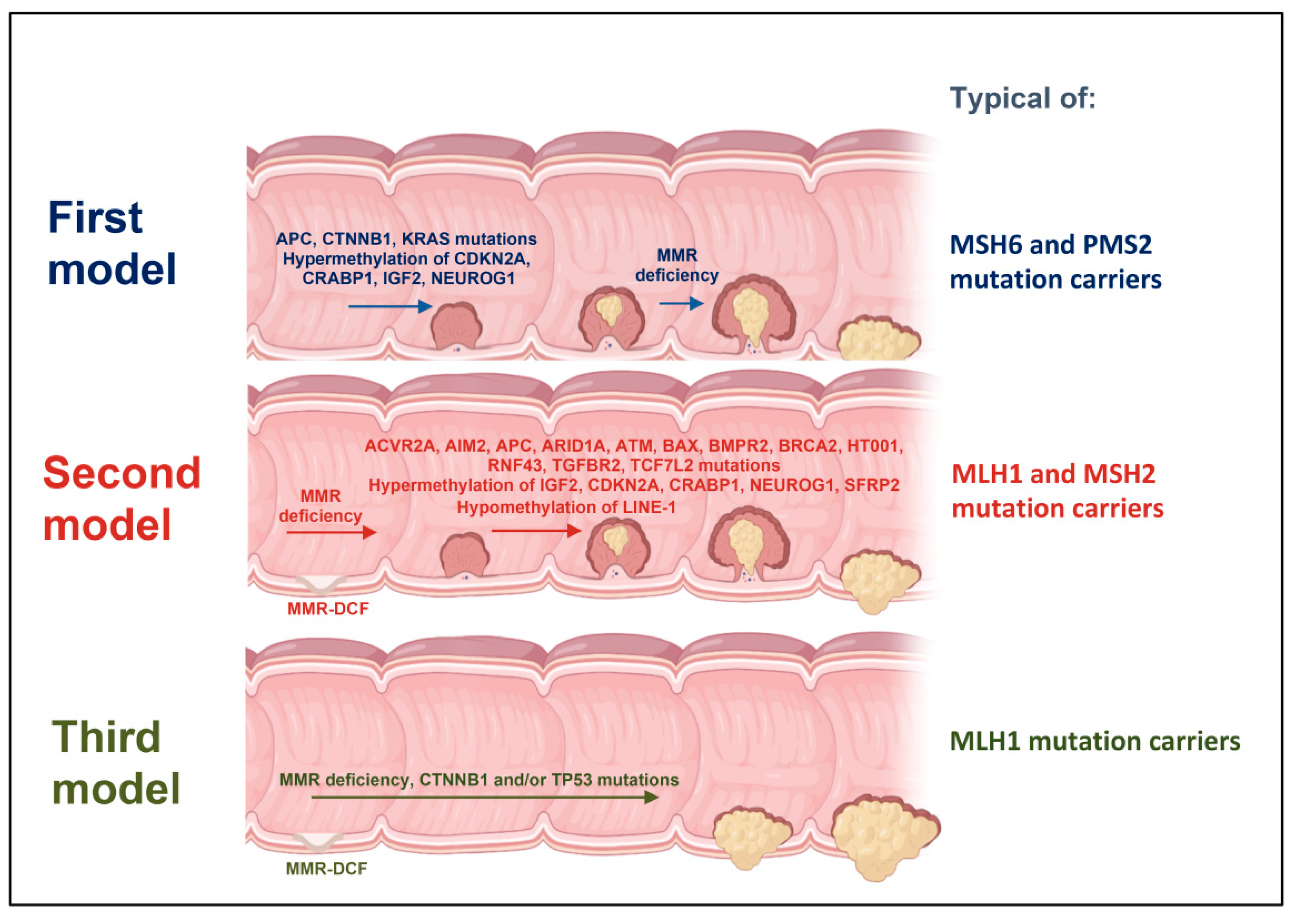
2. First Model of LS Colorectal Carcinogenesis: Adenoma Growth in an MMR-Proficient Background
3. Second Model of LS Colorectal Carcinogenesis: MMR-DCF Leading to Adenoma Formation and Transition to Carcinoma
4. Third Model of LS Colorectal Carcinogenesis: MMR-DCF Showing Direct Transition to Carcinoma
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bansidhar, B.J. Extracolonic Manifestations of Lynch Syndrome. Clin. Colon Rectal Surg. 2012, 25, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, E.M.; Turgeon, D.K.; Stockwell, D.H.; Zhao, L.; Normolle, D.P.; Tuck, M.K.; Bresalier, R.S.; Marcon, N.E.; Baron, J.A.; Ruffin, M.T.; et al. Missed Adenomas during Colonoscopic Surveillance in Individuals with Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). Cancer Prev. Res. 2008, 1, 470–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, Diagnosis and Treatment of Lynch Syndrome: Old Lessons and Current Challenges. Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morak, M.; Käsbauer, S.; Kerscher, M.; Laner, A.; Nissen, A.M.; Benet-Pagès, A.; Schackert, H.K.; Keller, G.; Massdorf, T.; Holinski-Feder, E. Loss of MSH2 and MSH6 Due to Heterozygous Germline Defects in MSH3 and MSH6. Fam. Cancer 2017, 16, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yan, H.; Kuismanen, S.; Percesepe, A.; Bisgaard, M.L.; Pedroni, M.; Benatti, P.; Kinzler, K.W.; Vogelstein, B.; Ponz de Leon, M.; et al. The Role of HPMS1 and HPMS2 in Predisposing to Colorectal Cancer. Cancer Res. 2001, 61, 7798–7802. [Google Scholar] [PubMed]
- Wu, Y.; Berends, M.J.; Sijmons, R.H.; Mensink, R.G.; Verlind, E.; Kooi, K.A.; van der Sluis, T.; Kempinga, C.; van der Zee, A.G.; Hollema, H.; et al. A Role for MLH3 in Hereditary Nonpolyposis Colorectal Cancer. Nat. Genet. 2001, 29, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Rasmussen, M.; Westers, H.; Andersen, S.D.; Jager, P.O.; Kooi, K.A.; Niessen, R.C.; Eggen, B.J.L.; Nielsen, F.C.; Kleibeuker, J.H.; et al. Biochemical Characterization of MLH3 Missense Mutations Does Not Reveal an Apparent Role of MLH3 in Lynch Syndrome. Genes. Chromosomes Cancer 2009, 48, 340–350. [Google Scholar] [CrossRef] [Green Version]
- Mäki-Nevala, S.; Valo, S.; Ristimäki, A.; Sarhadi, V.; Knuutila, S.; Nyström, M.; Renkonen-Sinisalo, L.; Lepistö, A.; Mecklin, J.-P.; Peltomäki, P. DNA Methylation Changes and Somatic Mutations as Tumorigenic Events in Lynch Syndrome-Associated Adenomas Retaining Mismatch Repair Protein Expression. EBioMedicine 2019, 39, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Satorres, C.; García-Campos, M.; Bustamante-Balén, M. Molecular Features of the Serrated Pathway to Colorectal Cancer: Current Knowledge and Future Directions. Gut Liver 2021, 15, 31–43. [Google Scholar] [CrossRef]
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New Clinical Criteria for Hereditary Nonpolyposis Colorectal Cancer (HNPCC, Lynch Syndrome) Proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Cohen, S.A. Current Lynch Syndrome Tumor Screening Practices: A Survey of Genetic Counselors. J. Genet. Couns. 2014, 23, 38–47. [Google Scholar] [CrossRef]
- Remo, A.; Fassan, M.; Lanza, G. Immunohistochemical Evaluation of Mismatch Repair Proteins in Colorectal Carcinoma: The AIFEG/GIPAD Proposal. Pathologica 2016, 108, 104–109. [Google Scholar]
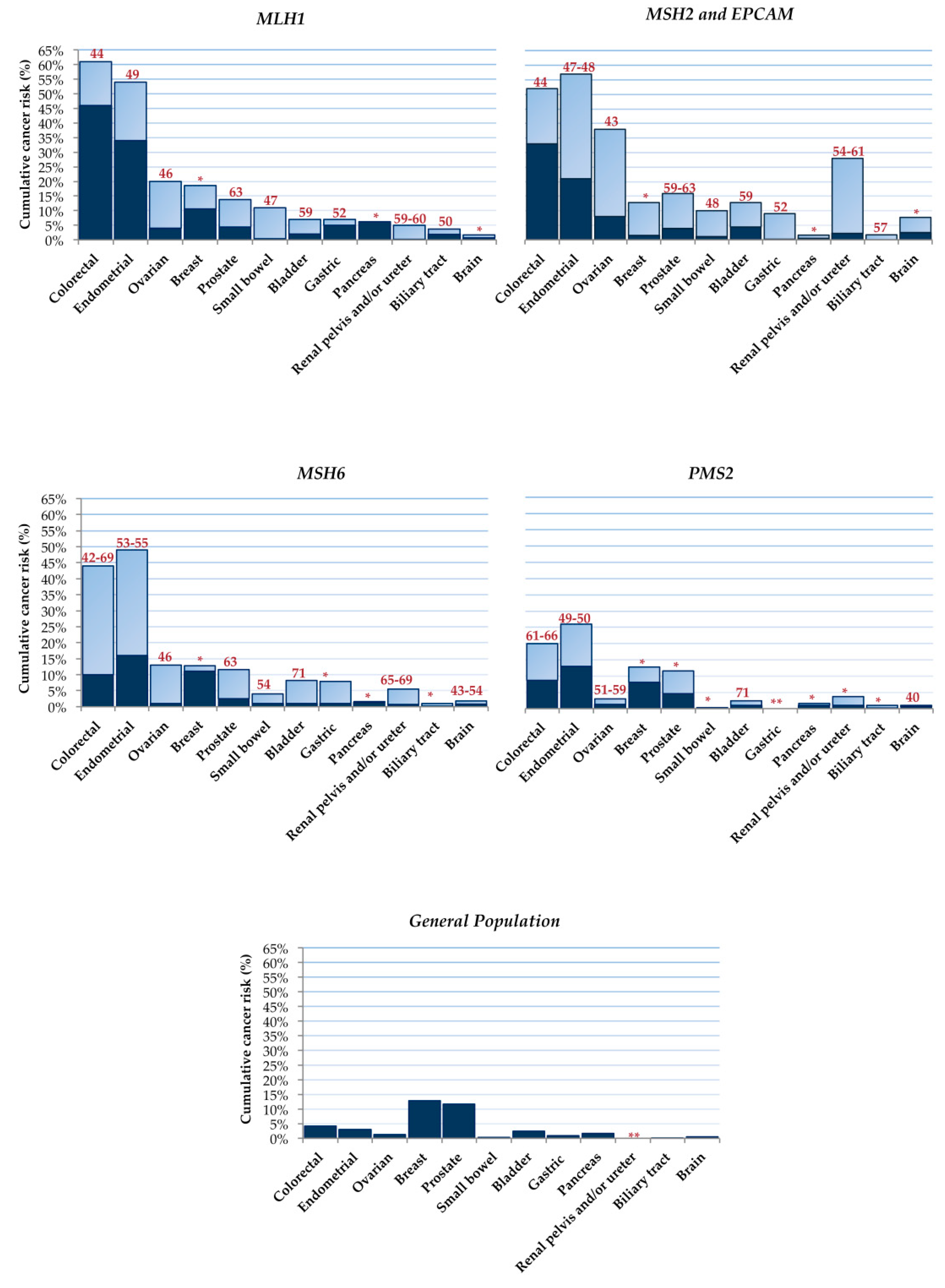
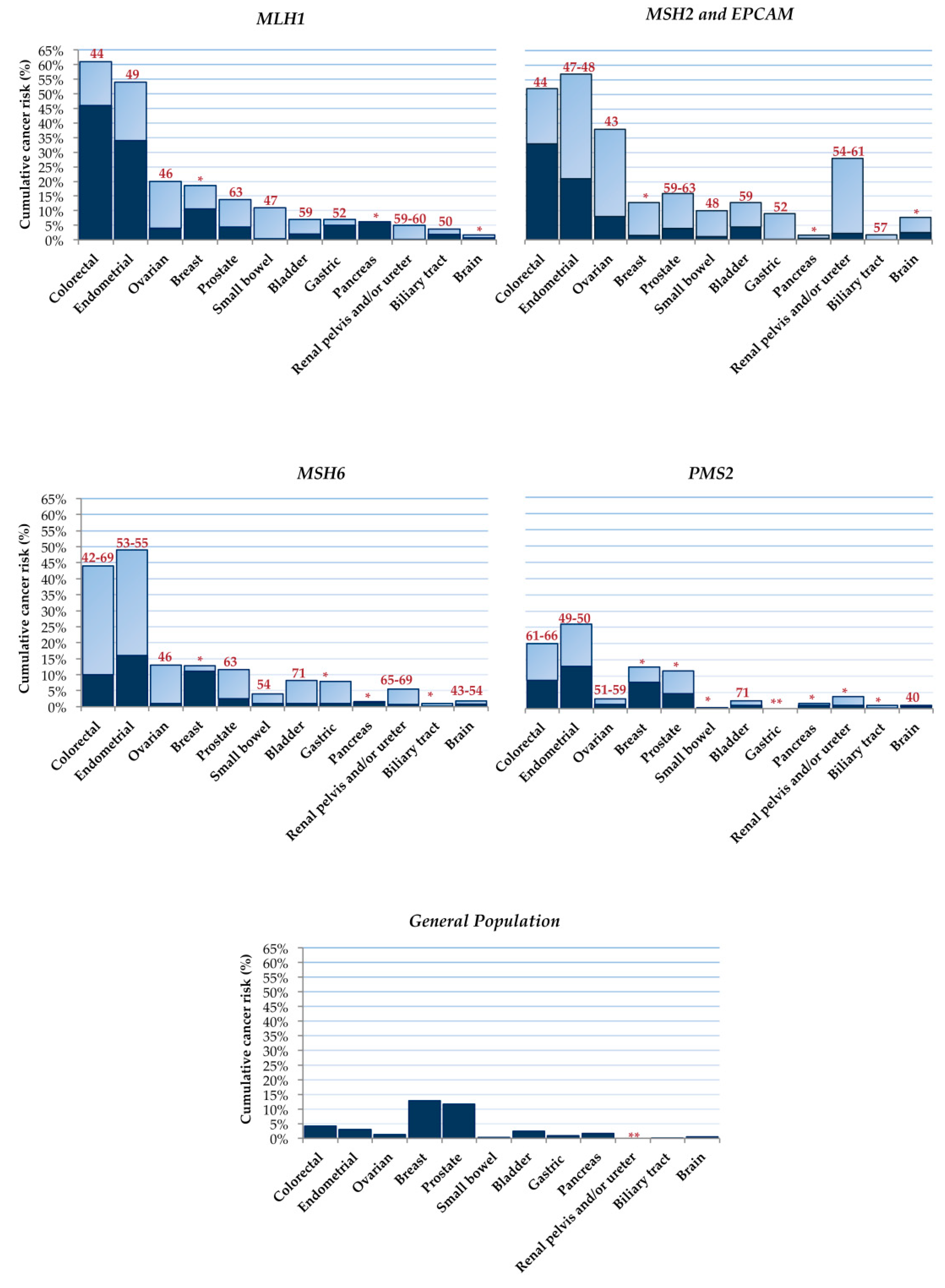
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer Risks Associated with Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer Risk and Survival in Path_MMR Carriers by Gene and Gender up to 75 Years of Age: A Report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, N.A.J.; Morris, J.; Green, K.; Lalloo, F.; Woodward, E.R.; Hill, J.; Crosbie, E.J.; Evans, D.G. Association of Mismatch Repair Mutation With Age at Cancer Onset in Lynch Syndrome: Implications for Stratified Surveillance Strategies. JAMA Oncol. 2017, 3, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Barrow, E.; Robinson, L.; Alduaij, W.; Shenton, A.; Clancy, T.; Lalloo, F.; Hill, J.; Evans, D.G. Cumulative Lifetime Incidence of Extracolonic Cancers in Lynch Syndrome: A Report of 121 Families with Proven Mutations. Clin. Genet. 2009, 75, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer Incidence and Survival in Lynch Syndrome Patients Receiving Colonoscopic and Gynaecological Surveillance: First Report from the Prospective Lynch Syndrome Database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Baglietto, L.; Lindor, N.M.; Dowty, J.G.; White, D.M.; Wagner, A.; Gomez Garcia, E.B.; Vriends, A.H.J.T.; Dutch Lynch Syndrome Study Group; Cartwright, N.R.; Barnetson, R.A.; et al. Risks of Lynch Syndrome Cancers for MSH6 Mutation Carriers. J. Natl. Cancer Inst. 2010, 102, 193–201. [Google Scholar] [CrossRef]
- Suerink, M.; Rodríguez-Girondo, M.; van der Klift, H.M.; Colas, C.; Brugieres, L.; Lavoine, N.; Jongmans, M.; Munar, G.C.; Evans, D.G.; Farrell, M.P.; et al. An Alternative Approach to Establishing Unbiased Colorectal Cancer Risk Estimation in Lynch Syndrome. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 2706–2712. [Google Scholar] [CrossRef]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The Clinical Phenotype of Lynch Syndrome Due to Germ-Line PMS2 Mutations. Gastroenterology 2008, 135, 419–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Broeke, S.W.; van der Klift, H.M.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; de la Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, C.; Loeffler, M.; Steinke, V.; Rahner, N.; Holinski-Feder, E.; Dietmaier, W.; Schackert, H.K.; Goergens, H.; von Knebel Doeberitz, M.; Goecke, T.O.; et al. Risks of Less Common Cancers in Proven Mutation Carriers with Lynch Syndrome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Joost, P.; Therkildsen, C.; Jonsson, M.; Rambech, E.; Nilbert, M. Frequent Mismatch-Repair Defects Link Prostate Cancer to Lynch Syndrome. BMC Urol. 2016, 16, 15. [Google Scholar] [CrossRef] [Green Version]
- Joost, P.; Therkildsen, C.; Dominguez-Valentin, M.; Jönsson, M.; Nilbert, M. Urinary Tract Cancer in Lynch Syndrome; Increased Risk in Carriers of MSH2 Mutations. Urology 2015, 86, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Vasen, H.F.A.; Mecklin, J.-P.; Bernstein, I.; Aarnio, M.; Järvinen, H.J.; Myrhøj, T.; Sunde, L.; Wijnen, J.T.; Lynch, H.T. The Risk of Extra-Colonic, Extra-Endometrial Cancer in the Lynch Syndrome. Int. J. Cancer 2008, 123, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Capelle, L.G.; Van Grieken, N.C.T.; Lingsma, H.F.; Steyerberg, E.W.; Klokman, W.J.; Bruno, M.J.; Vasen, H.F.A.; Kuipers, E.J. Risk and Epidemiological Time Trends of Gastric Cancer in Lynch Syndrome Carriers in the Netherlands. Gastroenterology 2010, 138, 487–492. [Google Scholar] [CrossRef]
- Harkness, E.F.; Barrow, E.; Newton, K.; Green, K.; Clancy, T.; Lalloo, F.; Hill, J.; Evans, D.G. Lynch Syndrome Caused by MLH1 Mutations Is Associated with an Increased Risk of Breast Cancer: A Cohort Study. J. Med. Genet. 2015, 52, 553–556. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Rafnar, T.; Frankel, W.L.; Einarsdottir, S.; Sigurdsson, A.; Hampel, H.; Snaebjornsson, P.; Masson, G.; Weng, D.; Arngrimsson, R.; et al. Comprehensive Population-Wide Analysis of Lynch Syndrome in Iceland Reveals Founder Mutations in MSH6 and PMS2. Nat. Commun. 2017, 8, 14755. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. American College of Gastroenterology ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am. J. Gastroenterol. 2015, 110, 223–262, quiz 263. [Google Scholar] [CrossRef] [Green Version]
- Edelstein, D.L.; Axilbund, J.; Baxter, M.; Hylind, L.M.; Romans, K.; Griffin, C.A.; Cruz-Correa, M.; Giardiello, F.M. Rapid Development of Colorectal Neoplasia in Patients with Lynch Syndrome. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2011, 9, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, S.; Tamura, S.; Nakajima, T.; Yamano, H.; Kusaka, H.; Watanabe, H. Diagnosis of Colorectal Tumorous Lesions by Magnifying Endoscopy. Gastrointest. Endosc. 1996, 44, 8–14. [Google Scholar] [CrossRef]
- Lambert, R. The Paris Endoscopic Classification of Superficial Neoplastic Lesions: Esophagus, Stomach, and Colon. Gastrointest. Endosc. 2003, 58, S3–S43. [Google Scholar] [CrossRef]
- Ahadova, A.; Seppälä, T.T.; Engel, C.; Gallon, R.; Burn, J.; Holinski-Feder, E.; Steinke-Lange, V.; Möslein, G.; Nielsen, M.; ten Broeke, S.W.; et al. The “Unnatural” History of Colorectal Cancer in Lynch Syndrome: Lessons from Colonoscopy Surveillance. Int. J. Cancer 2021, 148, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, S.; Pan, P.; Xia, T.; Chang, X.; Yang, X.; Guo, L.; Meng, Q.; Yang, F.; Qian, W.; et al. Magnitude, Risk Factors, and Factors Associated With Adenoma Miss Rate of Tandem Colonoscopy: A Systematic Review and Meta-Analysis. Gastroenterology 2019, 156, 1661–1674.e11. [Google Scholar] [CrossRef] [PubMed]
- Rijcken, F.E.M.; Hollema, H.; Kleibeuker, J.H. Proximal Adenomas in Hereditary Non-Polyposis Colorectal Cancer Are Prone to Rapid Malignant Transformation. Gut 2002, 50, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.D.; Buchanan, D.D.; Pearson, S.-A.; Clendenning, M.; Jenkins, M.A.; Win, A.K.; Walters, R.J.; Spring, K.J.; Nagler, B.; Pavluk, E.; et al. Immunohistochemical Testing of Conventional Adenomas for Loss of Expression of Mismatch Repair Proteins in Lynch Syndrome Mutation Carriers: A Case Series from the Australasian Site of the Colon Cancer Family Registry. Mod. Pathol. 2012, 25, 722–730. [Google Scholar] [CrossRef]
- Sánchez, A.; Roos, V.H.; Navarro, M.; Pineda, M.; Caballol, B.; Moreno, L.; Carballal, S.; Rodríguez-Alonso, L.; Ramon Y Cajal, T.; Llort, G.; et al. Quality of Colonoscopy Is Associated With Adenoma Detection and Postcolonoscopy Colorectal Cancer Prevention in Lynch Syndrome. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020. [Google Scholar] [CrossRef]
- Ballester, V.; Taylor, W.R.; Slettedahl, S.W.; Mahoney, D.W.; Yab, T.C.; Sinicrope, F.A.; Boland, C.R.; Lidgard, G.P.; Cruz-Correa, M.R.; Smyrk, T.C.; et al. Novel Methylated DNA Markers Accurately Discriminate Lynch Syndrome Associated Colorectal Neoplasia. Epigenomics 2020, 12, 2173–2187. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three Molecular Pathways Model Colorectal Carcinogenesis in Lynch Syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Engel, C.; Ahadova, A.; Seppälä, T.T.; Aretz, S.; Bigirwamungu-Bargeman, M.; Bläker, H.; Bucksch, K.; Büttner, R.; de Vos Tot Nederveen Cappel, W.T.; Endris, V.; et al. Associations of Pathogenic Variants in MLH1, MSH2, and MSH6 With Risk of Colorectal Adenomas and Tumors and With Somatic Mutations in Patients With Lynch Syndrome. Gastroenterology 2020, 158, 1326–1333. [Google Scholar] [CrossRef]
- Møller, P. The Prospective Lynch Syndrome Database Reports Enable Evidence-Based Personal Precision Health Care. Hered. Cancer Clin. Pract. 2020, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L. Cancer Epigenetics. Oncogene 2003, 22, 6479–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG Island Methylator Phenotype in Colorectal Cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valo, S.; Kaur, S.; Ristimäki, A.; Renkonen-Sinisalo, L.; Järvinen, H.; Mecklin, J.-P.; Nyström, M.; Peltomäki, P. DNA Hypermethylation Appears Early and Shows Increased Frequency with Dysplasia in Lynch Syndrome-Associated Colorectal Adenomas and Carcinomas. Clin. Epigenet. 2015, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Flatin, B.T.B.; Vedeld, H.M.; Pinto, R.; Langerud, J.; Lind, G.E.; Lothe, R.A.; Sveen, A.; Jeanmougin, M. Multiregional Assessment of CIMP in Primary Colorectal Cancers: Phenotype Concordance but Marker Variability. Int. J. Cancer 2021, 148, 1652–1657. [Google Scholar] [CrossRef]
- Otero-Estévez, O.; Gallardo-Gomez, M.; de la Cadena, M.P.; Rodríguez-Berrocal, F.J.; Cubiella, J.; Ramirez, V.H.; García-Nimo, L.; Chiara, L.D. Value of Serum NEUROG1 Methylation for the Detection of Advanced Adenomas and Colorectal Cancer. Diagnostics 2020, 10, 437. [Google Scholar] [CrossRef]
- Herbst, A.; Rahmig, K.; Stieber, P.; Philipp, A.; Jung, A.; Ofner, A.; Crispin, A.; Neumann, J.; Lamerz, R.; Kolligs, F.T. Methylation of NEUROG1 in Serum Is a Sensitive Marker for the Detection of Early Colorectal Cancer. Am. J. Gastroenterol. 2011, 106, 1110–1118. [Google Scholar] [CrossRef]
- Tada, T.; Watanabe, T.; Kazama, S.; Kanazawa, T.; Hata, K.; Komuro, Y.; Nagawa, H. Reduced P16 Expression Correlates with Lymphatic Invasion in Colorectal Cancers. Hepatogastroenterology 2003, 50, 1756–1760. [Google Scholar]
- Samowitz, W.S.; Albertsen, H.; Herrick, J.; Levin, T.R.; Sweeney, C.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Evaluation of a Large, Population-Based Sample Supports a CpG Island Methylator Phenotype in Colon Cancer. Gastroenterology 2005, 129, 837–845. [Google Scholar] [CrossRef]
- Xing, X.; Cai, W.; Shi, H.; Wang, Y.; Li, M.; Jiao, J.; Chen, M. The Prognostic Value of CDKN2A Hypermethylation in Colorectal Cancer: A Meta-Analysis. Br. J. Cancer 2013, 108, 2542–2548. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A.; Adamek, A. Insulin-Like Growth Factor 2 (IGF2) Signaling in Colorectal Cancer-From Basic Research to Potential Clinical Applications. Int. J. Mol. Sci. 2019, 20, 4915. [Google Scholar] [CrossRef] [Green Version]
- Jirtle, R.L. IGF2 Loss of Imprinting: A Potential Heritable Risk Factor for Colorectal Cancer. Gastroenterology 2004, 126, 1190–1193. [Google Scholar] [CrossRef]
- De Jong, A.E.; Morreau, H.; Van Puijenbroek, M.; Eilers, P.H.; Wijnen, J.; Nagengast, F.M.; Griffioen, G.; Cats, A.; Menko, F.H.; Kleibeuker, J.H.; et al. The Role of Mismatch Repair Gene Defects in the Development of Adenomas in Patients with HNPCC. Gastroenterology 2004, 126, 42–48. [Google Scholar] [CrossRef]
- Rondagh, E.J.A.; Gulikers, S.; Gómez-García, E.B.; Vanlingen, Y.; Detisch, Y.; Winkens, B.; Vasen, H.F.A.; Masclee, A.A.M.; Sanduleanu, S. Nonpolypoid Colorectal Neoplasms: A Challenge in Endoscopic Surveillance of Patients with Lynch Syndrome. Endoscopy 2013, 45, 257–264. [Google Scholar] [CrossRef]
- Andersen, S.H.; Lykke, E.; Folker, M.B.; Bernstein, I.; Holck, S. Sessile Serrated Polyps of the Colorectum Are Rare in Patients with Lynch Syndrome and in Familial Colorectal Cancer Families. Fam. Cancer 2008, 7, 157–162. [Google Scholar] [CrossRef]
- Lanspa, S.J.; Jenkins, J.X.; Cavalieri, R.J.; Smyrk, T.C.; Watson, P.; Lynch, J.; Lynch, H.T. Surveillance in Lynch Syndrome: How Aggressive? Am. J. Gastroenterol. 1994, 89, 1978–1980. [Google Scholar]
- Ahadova, A.; von Knebel Doeberitz, M.; Bläker, H.; Kloor, M. CTNNB1-Mutant Colorectal Carcinomas with Immediate Invasive Growth: A Model of Interval Cancers in Lynch Syndrome. Fam. Cancer 2016, 15, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Goel, A.; Hornick, J.L.; Sen, A.; Turgeon, D.K.; Ruffin, M.T.; Marcon, N.E.; Baron, J.A.; Bresalier, R.S.; Syngal, S.; et al. Microsatellite Instability and DNA Mismatch Repair Protein Deficiency in Lynch Syndrome Colorectal Polyps. Cancer Prev. Res. 2012, 5, 574–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Broeke, S.W.; van Bavel, T.C.; Jansen, A.M.L.; Gómez-García, E.; Hes, F.J.; van Hest, L.P.; Letteboer, T.G.W.; Olderode-Berends, M.J.W.; Ruano, D.; Spruijt, L.; et al. Molecular Background of Colorectal Tumors From Patients With Lynch Syndrome Associated With Germline Variants in PMS2. Gastroenterology 2018, 155, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Bläker, H. Prevalence of Mismatch Repair-Deficient Crypt Foci in Lynch Syndrome: A Pathological Study. Lancet Oncol. 2012, 13, 598–606. [Google Scholar] [CrossRef]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch Repair Deficiency Commonly Precedes Adenoma Formation in Lynch Syndrome-Associated Colorectal Tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Staffa, L.; Echterdiek, F.; Nelius, N.; Benner, A.; Werft, W.; Lahrmann, B.; Grabe, N.; Schneider, M.; Tariverdian, M.; von Knebel Doeberitz, M.; et al. Mismatch Repair-Deficient Crypt Foci in Lynch Syndrome--Molecular Alterations and Association with Clinical Parameters. PLoS ONE 2015, 10, e0121980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaltonen, L.A.; Salovaara, R.; Kristo, P.; Canzian, F.; Hemminki, A.; Peltomäki, P.; Chadwick, R.B.; Kääriäinen, H.; Eskelinen, M.; Järvinen, H.; et al. Incidence of Hereditary Nonpolyposis Colorectal Cancer and the Feasibility of Molecular Screening for the Disease. N. Engl. J. Med. 1998, 338, 1481–1487. [Google Scholar] [CrossRef]
- Perucho, M. Microsatellite Instability: The Mutator That Mutates the Other Mutator. Nat. Med. 1996, 2, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Woerner, S.M.; Kloor, M.; von Knebel Doeberitz, M.; Gebert, J.F. Microsatellite Instability in the Development of DNA Mismatch Repair Deficient Tumors. Cancer Biomark. Sect. Dis. Markers 2006, 2, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFbeta Signaling in Growth Control, Cancer, and Heritable Disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J. TGF Beta Signaling in Health and Disease. Nat. Genet. 2004, 36, 790–792. [Google Scholar] [CrossRef]
- Xu, Y.; Pasche, B. TGF-Beta Signaling Alterations and Susceptibility to Colorectal Cancer. Hum. Mol. Genet. 2007, 16 Spec No 1, R14–R20. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal Cancer: Genetics of Development and Metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef]
- Pinheiro, M.; Pinto, C.; Peixoto, A.; Veiga, I.; Lopes, P.; Henrique, R.; Baldaia, H.; Carneiro, F.; Seruca, R.; Tomlinson, I.; et al. Target Gene Mutational Pattern in Lynch Syndrome Colorectal Carcinomas According to Tumour Location and Germline Mutation. Br. J. Cancer 2015, 113, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, R.; He, Y.; Yi, Y.; Wu, H.; Liang, Z. Next-Generation Sequencing Reveals Heterogeneous Genetic Alterations in Key Signaling Pathways of Mismatch Repair Deficient Colorectal Carcinomas. Mod. Pathol. 2020, 33, 2591–2601. [Google Scholar] [CrossRef]
- Fennell, L.J.; Clendenning, M.; McKeone, D.M.; Jamieson, S.H.; Balachandran, S.; Borowsky, J.; Liu, J.; Kawamata, F.; Bond, C.E.; Rosty, C.; et al. RNF43 Is Mutated Less Frequently in Lynch Syndrome Compared with Sporadic Microsatellite Unstable Colorectal Cancers. Fam. Cancer 2018, 17, 63–69. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-Spondin Fusions in Colon Cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Estécio, M.R.H.; Gharibyan, V.; Shen, L.; Ibrahim, A.E.K.; Doshi, K.; He, R.; Jelinek, J.; Yang, A.S.; Yan, P.S.; Huang, T.H.-M.; et al. LINE-1 Hypomethylation in Cancer Is Highly Variable and Inversely Correlated with Microsatellite Instability. PLoS ONE 2007, 2, e399. [Google Scholar] [CrossRef] [PubMed]
- Mecklin, J.-P.; Aarnio, M.; Läärä, E.; Kairaluoma, M.V.; Pylvänäinen, K.; Peltomäki, P.; Aaltonen, L.A.; Järvinen, H.J. Development of Colorectal Tumors in Colonoscopic Surveillance in Lynch Syndrome. Gastroenterology 2007, 133, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 Mutations of CTNNB1 Drive Tumorigenesis: A Review. Oncotarget 2017, 9, 5492–5508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, C.; Vasen, H.F.; Seppälä, T.; Aretz, S.; Bigirwamungu-Bargeman, M.; de Boer, S.Y.; Bucksch, K.; Büttner, R.; Holinski-Feder, E.; Holzapfel, S.; et al. No Difference in Colorectal Cancer Incidence or Stage at Detection by Colonoscopy Among 3 Countries With Different Lynch Syndrome Surveillance Policies. Gastroenterology 2018, 155, 1400–1409.e2. [Google Scholar] [CrossRef]
- Bisschops, R.; Tejpar, S.; Willekens, H.; De Hertogh, G.; Van Cutsem, E. Virtual Chromoendoscopy (I-SCAN) Detects More Polyps in Patients with Lynch Syndrome: A Randomized Controlled Crossover Trial. Endoscopy 2017, 49, 342–350. [Google Scholar] [CrossRef]
- ASGE Technology Committee; Wong Kee Song, L.M.; Adler, D.G.; Chand, B.; Conway, J.D.; Croffie, J.M.B.; Disario, J.A.; Mishkin, D.S.; Shah, R.J.; Somogyi, L.; et al. Chromoendoscopy. Gastrointest. Endosc. 2007, 66, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Rahmi, G.; Lecomte, T.; Malka, D.; Maniere, T.; Le Rhun, M.; Guimbaud, R.; Lapalus, M.-G.; Le Sidaner, A.; Moussata, D.; Caron, O.; et al. Impact of Chromoscopy on Adenoma Detection in Patients with Lynch Syndrome: A Prospective, Multicenter, Blinded, Tandem Colonoscopy Study. Am. J. Gastroenterol. 2015, 110, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, T.; Cellier, C.; Meatchi, T.; Barbier, J.P.; Cugnenc, P.H.; Jian, R.; Laurent-Puig, P.; Landi, B. Chromoendoscopic Colonoscopy for Detecting Preneoplastic Lesions in Hereditary Nonpolyposis Colorectal Cancer Syndrome. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2005, 3, 897–902. [Google Scholar] [CrossRef]
- Hurlstone, D.P.; Karajeh, M.; Cross, S.S.; McAlindon, M.E.; Brown, S.; Hunter, M.D.; Sanders, D.S. The Role of High-Magnification-Chromoscopic Colonoscopy in Hereditary Nonpolyposis Colorectal Cancer Screening: A Prospective “Back-to-Back” Endoscopic Study. Am. J. Gastroenterol. 2005, 100, 2167–2173. [Google Scholar] [CrossRef] [PubMed]
- Hüneburg, R.; Lammert, F.; Rabe, C.; Rahner, N.; Kahl, P.; Büttner, R.; Propping, P.; Sauerbruch, T.; Lamberti, C. Chromocolonoscopy Detects More Adenomas than White Light Colonoscopy or Narrow Band Imaging Colonoscopy in Hereditary Nonpolyposis Colorectal Cancer Screening. Endoscopy 2009, 41, 316–322. [Google Scholar] [CrossRef]
- East, J.E.; Suzuki, N.; Stavrinidis, M.; Guenther, T.; Thomas, H.J.W.; Saunders, B.P. Narrow Band Imaging for Colonoscopic Surveillance in Hereditary Non-Polyposis Colorectal Cancer. Gut 2008, 57, 65–70. [Google Scholar] [CrossRef]
- Kudo, S.-E.; Mori, Y.; Misawa, M.; Takeda, K.; Kudo, T.; Itoh, H.; Oda, M.; Mori, K. Artificial Intelligence and Colonoscopy: Current Status and Future Perspectives. Dig. Endosc. Off. J. Jpn. Gastroenterol. Endosc. Soc. 2019, 31, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Jiang, A.C.; Buckingham, L.; Barbanera, W.; Korang, A.Y.; Bishesari, F.; Melson, J. LINE-1 Is Preferentially Hypomethylated within Adenomatous Polyps in the Presence of Synchronous Colorectal Cancer. Clin. Epigenet. 2017, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V. PARP Inhibitor Drugs in the Treatment of Breast, Ovarian, Prostate and Pancreatic Cancers: An Update of Clinical Trials. Curr. Drug Targets 2018, 19, 21–37. [Google Scholar] [CrossRef]
- Gupta, M.; Iyer, R.; Fountzilas, C. Poly(ADP-Ribose) Polymerase Inhibitors in Pancreatic Cancer: A New Treatment Paradigms and Future Implications. Cancers 2019, 11, 1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP Inhibitors in Pancreatic Cancer: Molecular Mechanisms and Clinical Applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef]
- Buchtel, K.M.; Vogel Postula, K.J.; Weiss, S.; Williams, C.; Pineda, M.; Weissman, S.M. FDA Approval of PARP Inhibitors and the Impact on Genetic Counseling and Genetic Testing Practices. J. Genet. Couns. 2018, 27, 131–139. [Google Scholar] [CrossRef]
- Sanese, P.; Fasano, C.; Buscemi, G.; Bottino, C.; Corbetta, S.; Fabini, E.; Silvestri, V.; Valentini, V.; Disciglio, V.; Forte, G.; et al. Targeting SMYD3 to Sensitize Homologous Recombination-Proficient Tumors to PARP-Mediated Synthetic Lethality. iScience 2020, 23, 101604. [Google Scholar] [CrossRef] [PubMed]
- Rimar, K.J.; Tran, P.T.; Matulewicz, R.S.; Hussain, M.; Meeks, J.J. The Emerging Role of Homologous Recombination Repair and PARP Inhibitors in Genitourinary Malignancies. Cancer 2017, 123, 1912–1924. [Google Scholar] [CrossRef] [PubMed]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lepore Signorile, M.; Disciglio, V.; Di Carlo, G.; Pisani, A.; Simone, C.; Ingravallo, G. From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome. Int. J. Mol. Sci. 2021, 22, 6767. https://doi.org/10.3390/ijms22136767
Lepore Signorile M, Disciglio V, Di Carlo G, Pisani A, Simone C, Ingravallo G. From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome. International Journal of Molecular Sciences. 2021; 22(13):6767. https://doi.org/10.3390/ijms22136767
Chicago/Turabian StyleLepore Signorile, Martina, Vittoria Disciglio, Gabriella Di Carlo, Antonio Pisani, Cristiano Simone, and Giuseppe Ingravallo. 2021. "From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome" International Journal of Molecular Sciences 22, no. 13: 6767. https://doi.org/10.3390/ijms22136767
APA StyleLepore Signorile, M., Disciglio, V., Di Carlo, G., Pisani, A., Simone, C., & Ingravallo, G. (2021). From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome. International Journal of Molecular Sciences, 22(13), 6767. https://doi.org/10.3390/ijms22136767