Abstract
DNA repair ensures genomic stability to achieve healthy ageing, including cognitive maintenance. Mutations on genes encoding key DNA repair proteins can lead to diseases with accelerated ageing phenotypes. Some of these diseases are xeroderma pigmentosum group A (XPA, caused by mutation of XPA), Cockayne syndrome group A and group B (CSA, CSB, and are caused by mutations of CSA and CSB, respectively), ataxia-telangiectasia (A-T, caused by mutation of ATM), and Werner syndrome (WS, with most cases caused by mutations in WRN). Except for WS, a common trait of the aforementioned progerias is neurodegeneration. Evidence from studies using animal models and patient tissues suggests that the associated DNA repair deficiencies lead to depletion of cellular nicotinamide adenine dinucleotide (NAD+), resulting in impaired mitophagy, accumulation of damaged mitochondria, metabolic derailment, energy deprivation, and finally leading to neuronal dysfunction and loss. Intriguingly, these features are also observed in Alzheimer’s disease (AD), the most common type of dementia affecting more than 50 million individuals worldwide. Further studies on the mechanisms of the DNA repair deficient premature ageing diseases will help to unveil the mystery of ageing and may provide novel therapeutic strategies for AD.
1. An Overview of DNA Damage and DNA Damage Response (DDR)
1.1. DNA Damage and DDR
DNA carries genetic instructions for the development, functioning, growth, and reproduction of cells. DNA is inherently unstable due to both spontaneous chemical instability and modifications caused by either exogenous or endogenous agents causing DNA damage [1,2]. It has been estimated that each individual cell is subjected to up to one million DNA changes per day [3,4,5,6]. DNA damage is well known to affect both DNA replication, transcription, and a broad spectrum of signaling pathways including the nucleus to the mitochondria signaling pathway [1,7]. In this review, we update the progress of mechanistic studies on the key DNA repair pathways, including base excision repair (BER), nucleotide excision repair (NER), and double-strand break repair (DSBR). Furthermore, we focus on the role of DNA damage in ageing-related neurodegenerative diseases, with particular attention to its role in both rare premature ageing diseases, and the age-predisposed condition Alzheimer’s disease (AD).
Unlike other macromolecules of the cell, DNA cannot be replaced, but must be repaired to remain intact and functional. To avert deleterious consequences of DNA damage, cells have evolved several mechanisms, collectively termed the DNA damage response (DDR), to detect DNA damage, signal its presence and promote its repair. A variety of DDR pathways have been identified in organisms ranging from bacteria to humans, and are essential to life [8]. Three main repair pathways in mammalian neurons, including base excision repair (BER), nucleotide excision repair (NER), and double strand break repair (DSBR), are discussed below.
1.2. Major DNA Repair Pathways and Their Roles in Neurons and Microglia
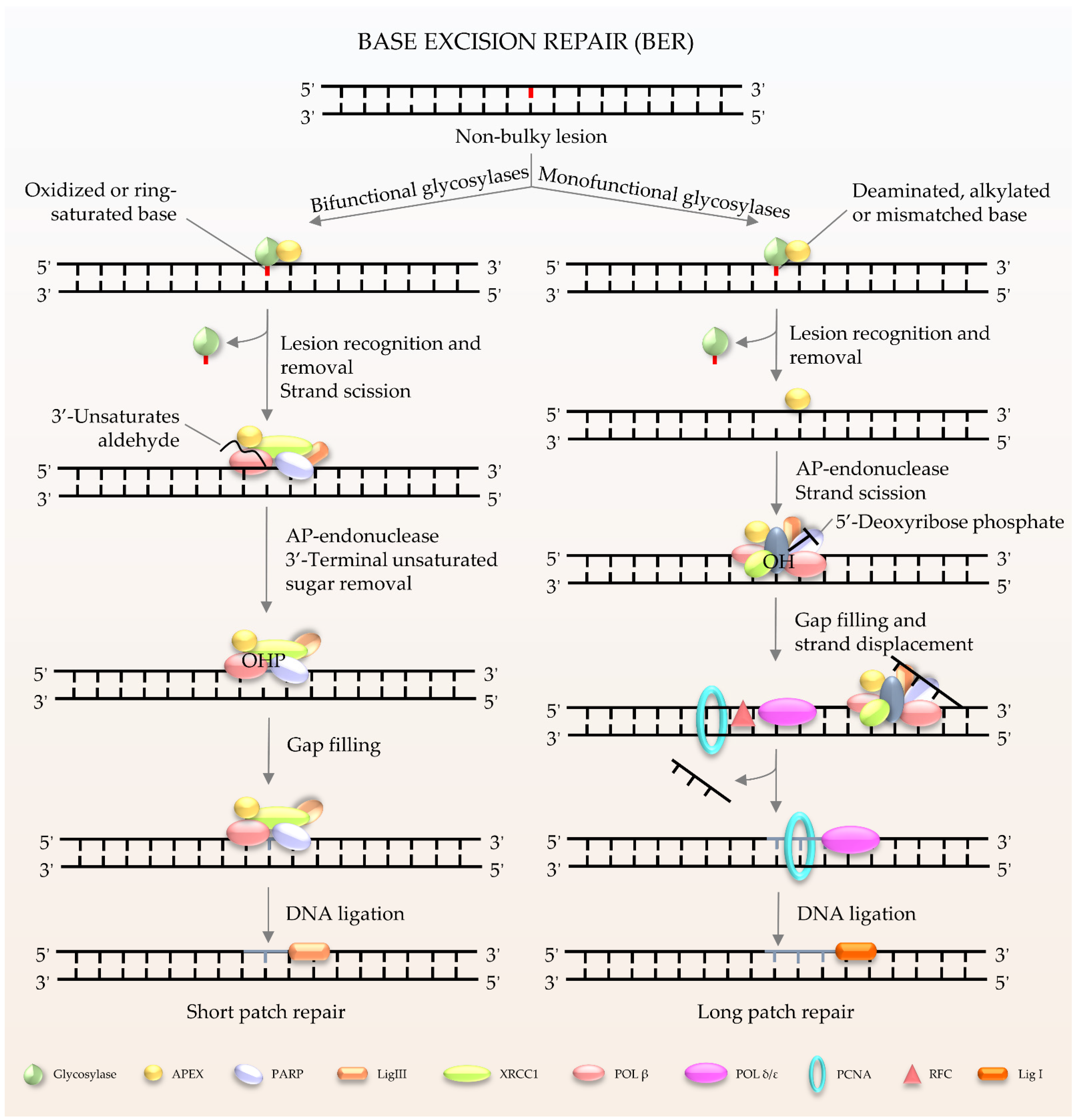
BER (Figure 1) is the major pathway involved in the repair of oxidative lesions and is responsible for the repair of non-bulky DNA oxidation, deamination, and alkylation [9,10,11]. The first step in the BER pathway uses a lesion-specific DNA glycosylase to recognize and eliminate damaged base pairs, which initiates the pathway [12]. Either a bifunctional or monofunctional DNA glycosylase catalyzes the cleavage of the N-glycosidic bond by flipping the damaged base out of the double helix to release a free base and create an abasic site (AP site). The repair is further processed by AP endonuclease 1 (APE1) to cleave the DNA backbone 5′ to the AP site, hereby producing 3′-hydroxyl and 5′-2-deoxyribose-5′-phosphate (5′-dRP). The gap is then filled by DNA polymerases using 3′-hydroxyl through template-directed synthesis via either short-patch or long-patch repair, depending on the number of inserted nucleotides. In the case of short-patch BER, DNA polymerase β (Polβ) and the inherent dRP-lyase activity of Polβ detaches the 5′-dRP to replace a single nucleotide. In general, long-patch repair requires the assistance of Polδ/ε and flap endonuclease 1 (FEN1) to substitute the displaced 5′-end structures with 2–13 nucleotides [10]. The importance of BER has been exemplified by the lethality of Ape1−/− or Polβ−/− mouse models. Deletion of mouse APE1 (also known as Apex1) leads to embryonic lethality, and deficiency in cells can promote cellular senescence and premature ageing features [13]. It has been demonstrated that Polβ defects can hamper amyloid β (Aβ)-induced neurogenesis in mice [14]. In particular, knockdown of Polβ inhibited the 42 amino acids form of Aβ peptide (Aβ1–42)-promoted differentiation of nestin+ progenitor cells into nestin+/Distal-less homeobox 2 (Dlx-2+) neuroblasts [14]. Moreover, pharmacological blockage of Polβ prevented Aβ1–42-induced differentiation of progenitors into MAP-2+ neurons [14]. It is worth noting that many proteins involved in BER not only exist in the nucleus, but also in the mitochondria [8]. Mitochondrial DNA (mtDNA) is more susceptible to the adverse effects of reactive oxygen species (ROS) produced during oxidative phosphorylation than nuclear DNA [15]. Thus, BER is considered to be the main repair pathway for DNA damage in mitochondria [16,17].
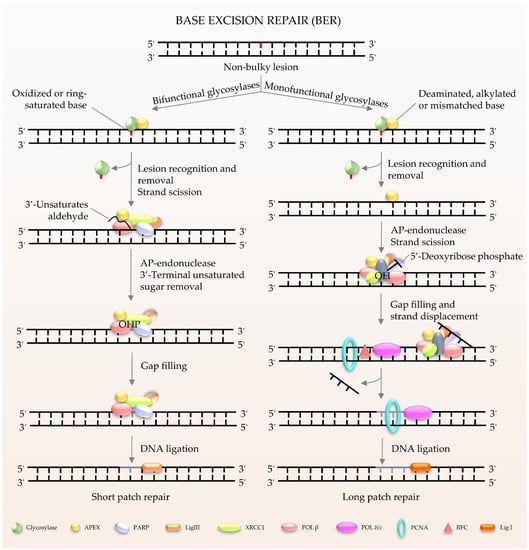
Figure 1.
Schematic of BER in mammalian cells. BER is the key pathway to remove and repair damaged bases. It starts with a DNA glycosylase to recognize and eliminate the damaged base, creating an abasic site. The gap is finally filled by DNA polymerases. The short patch is repaired with a single nucleotide, and the long patch is synthesized with 2–13 nucleotides.
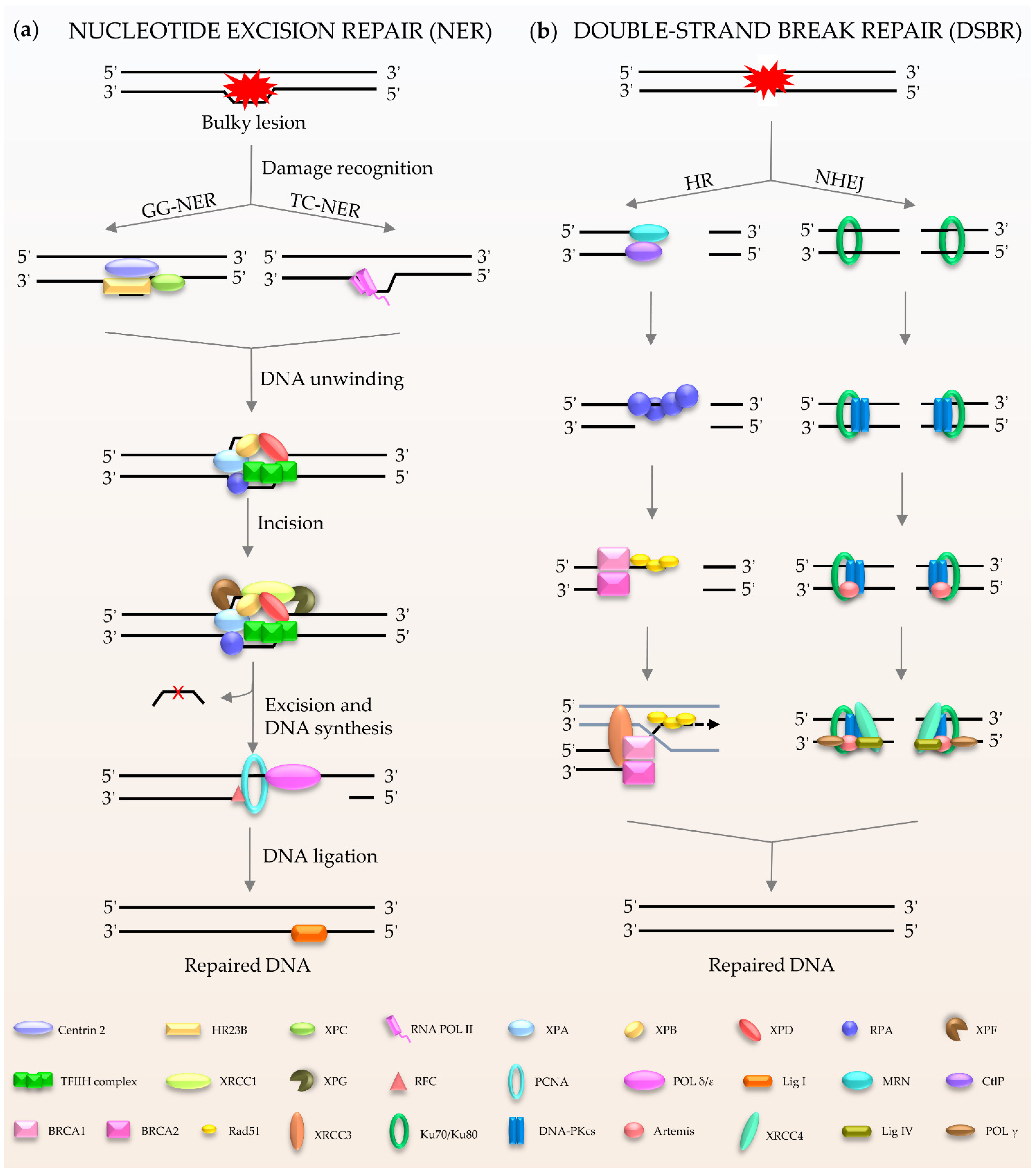
NER (Figure 2a) is a central DNA repair pathway and a highly dynamic process responsible for removing bulky lesions from the genome [18,19,20]. In humans, NER is composed of two branches, global genome NER (GG-NER) [21] and transcription-coupled NER (TC-NER) [22,23], which are distinguished by their different recognition methods. XPC, as the main damage recognition factor, scans the double helix to identify the lesion, and forms a complex with centrins, HR23B, etc., to induce GC-NER activity [24]. In the case of TC-NER, it is primed by transcribing RNA polymerase II [25]. After recognition, the two factors share the same pathway for repairing the damage. The repair factor XPA and the large multi-subunit transcription factor IIH (TFIIH) act as translocase and helicase, respectively, to unwind the DNA [19,26]. The DNA lesion is then excised through double DNA nicks with two endonucleases, XPF and XPG. Finally, the gap is filled by Polδ/ε and ligase I [21,22].
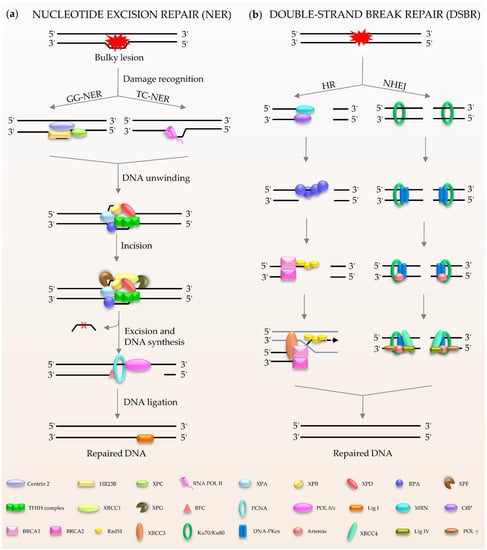
Figure 2.
Schematic of NER and DSBR in mammalian cells. (a) NER is composed of GG-NER and TC-NER. XPC scans the double helix to identify the lesion, and forms a complex with centrins, HR23B among others, to induce NER activity. Subsequently XPA and TFIIH act as translocase and helicase to unwind the DNA. The DNA lesion is excised by XPF and XPG. Finally, the gap is filled by DNA polymerases and DNA ligase I. (b) DSBR consists HR and NHEJ. HR is initiated by MRN. Rad51 participates in the search for homologous copies, and its homologues are involved in DNA strand invasion and subsequent homologous recombination to repair. NHEJ is activated by Ku80/Ku70. The damaged ends are trimmed by artemis; after that, the gap is filled by the action of DNA polymerase. The final repair is mediated by DNA ligase IV and cofactor XRCC4.
Double-stranded breaks (DSBs) are the most harmful type of DNA damage in terms of genomic integrity [27]. Mammalian cells use two main DSBR pathways: homologous recombination (HR) and non-homologous DNA end joining (NHEJ) (Figure 2b) [28,29,30,31,32]. HR is the main method for DSBR utilized during embryogenesis and embryonic development [33]. The damaged ends of DNA are recognized by the Mre11-Rad50-Nbs1 (MRN) complex [34]. This complex performs extensive DNA processing, which together with CtIP generates 3′ single-stranded DNA (ssDNA). Rad51 participates in the search for homologous copies, and its homologues are involved in DNA strand invasion and subsequent HR to achieve highly accurate damage repair [35]. The cyclic heterodimer Ku70/Ku80 triggers NHEJ and then recruits DNA-PKcs [36,37,38,39]. Afterwards, nucleases (such as Artemis) deal with the damaged ends, and the gap is filled by DNA polymerases, such as Pol μ/λ/γ [40,41]. The final step of the repair pathway is mediated by DNA ligase IV and X-ray repair cross-complementing protein (XRCC4) [39,41]. It is well known that the process of DDR requires ATP, especially for DNA ligation. In humans, in response to DNA damage, cells mobilize more than ten thousand ATP molecules to repair just one DSB [42]. Interestingly, DNA ligase IV, the key enzyme of DSBR, uses NAD+ as a substrate for double-strand connection to mediate the final repair [43].
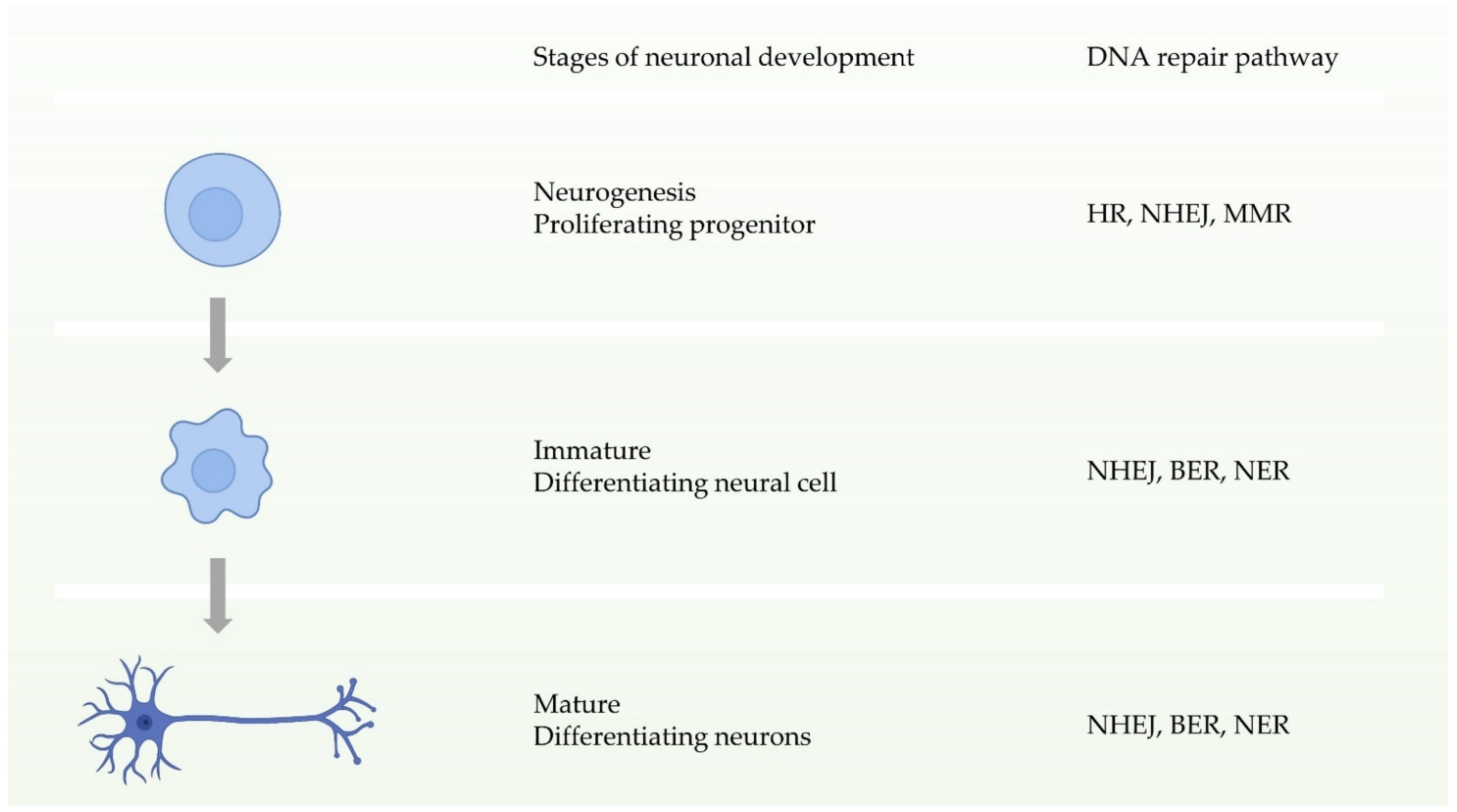
Neuronal homeostasis is a prerequisite for the development and function of the nervous system, and requires high fidelity and stable inheritance [44]. In normal cellular activities or DNA replication, the high precision and integrity of the genome must be maintained after DNA damage. Multiple DDR pathways in cells drive the biological functions ensuring this procedure. During the early stages of neuronal development, that is, neural progenitor proliferation, the nervous system has entire DDR pathways. In other words, it can repair double-strand breaks through two pathways of HR and NHEJ before neuronal maturation (Figure 3) [11,44]. In contrast, during neuronal maturation, NHEJ becomes the only way to repair double-strand break damage [45]. Defects in these DDRs can cause neurological disorders. Studies have shown that defects in NHEJ, NER, or BER increase risks to neurodegenerative disorders or neurodevelopmental defects [46,47,48,49,50,51].
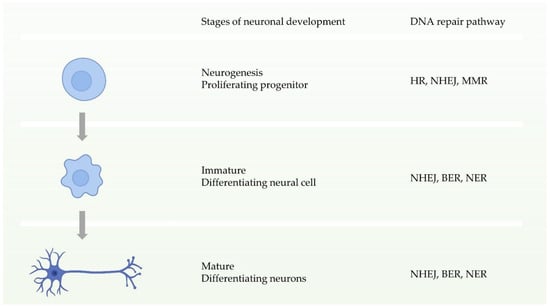
Figure 3.
Schematic of the availability of different DNA repair pathways during different stages of neuronal development.
Microglia are glial cells widely distributed in the brain and spinal cord. They are the main form of active immune defense in the central nervous system, and are key cells involved in the neuroprotective functions that maintain normal brain function; however, microglia can be hostile to neurons in disease conditions [52,53]. Studies have shown that DDR-related proteins have an impact on the activity, function, and survival status of microglia. In DDR, especially in DSBR, the deficiency of one crucial protein, ataxia-telangiectasia mutated (ATM) [54], results in abnormally active microglia, and stimulates excessive production of pro-inflammatory factors, which result in neurotoxicity [55]. More specifically, ATM dysfunction causes damage to DNA repair, and leads to the further accumulation of impaired cytoplasmic DNA. In microglia, cytoplasmic DNA can subsequently activate an antiviral defense system via the DNA sensor stimulator of interferon genes (STING). Cytoplasmic DNA can also trigger absent in melanoma 2 (AIM2) inflammasomes and, in parallel, induce elevated levels of cytokine precursors, such as pro-IL-1, through proteolytic processing. These processes create an extreme environment of neurotoxic inflammation [55]. In addition, the DNA excision repair protein, ERCC1, is very important for NER and DSBR [56]. Loss of ERCC1 results in microglial death and a compensatory increase in proliferation [57]. Of note, it is speculated that, in a mouse model of Cx3cr1-Ercc1ko/loxP and Cx3cr1-Ercc1wt/loxP, Ercc1-deficient microglia might have a link with ageing-related phenotypes [57].
2. Crosstalk between Nucleus and Mitochondria in DNA Damage
DNA damage not only accumulates in chromosomal DNA, but also in mtDNA, leading to mitochondrial dysfunction [58]. Dysfunctional mitochondria are targeted for lysosomal destruction through mitophagy and are recycled for cell utilization, as well as being degraded by the ubiquitin-proteasome system (UPS) [59]. Mitochondrial dysfunction and mitophagy defects are likely key features of age-related neurodegenerative disorders [60]. The accumulation of mtDNA damage and the reduction of mitophagy are also hallmarks of premature ageing diseases, such as XP and A-T [61,62,63]. Although organisms have a large number of responses to DNA damage, not only will DNA lesions increase during ageing, but the efficiency of DDR will also decrease [7,64,65,66,67]. Many accelerated ageing diseases, such as XP, A-T, CS, and WS, and neurodegenerative diseases such as AD, are closely related to the mutation of DDR proteins and DNA damage in both the nucleus and mitochondria [68,69,70,71,72].
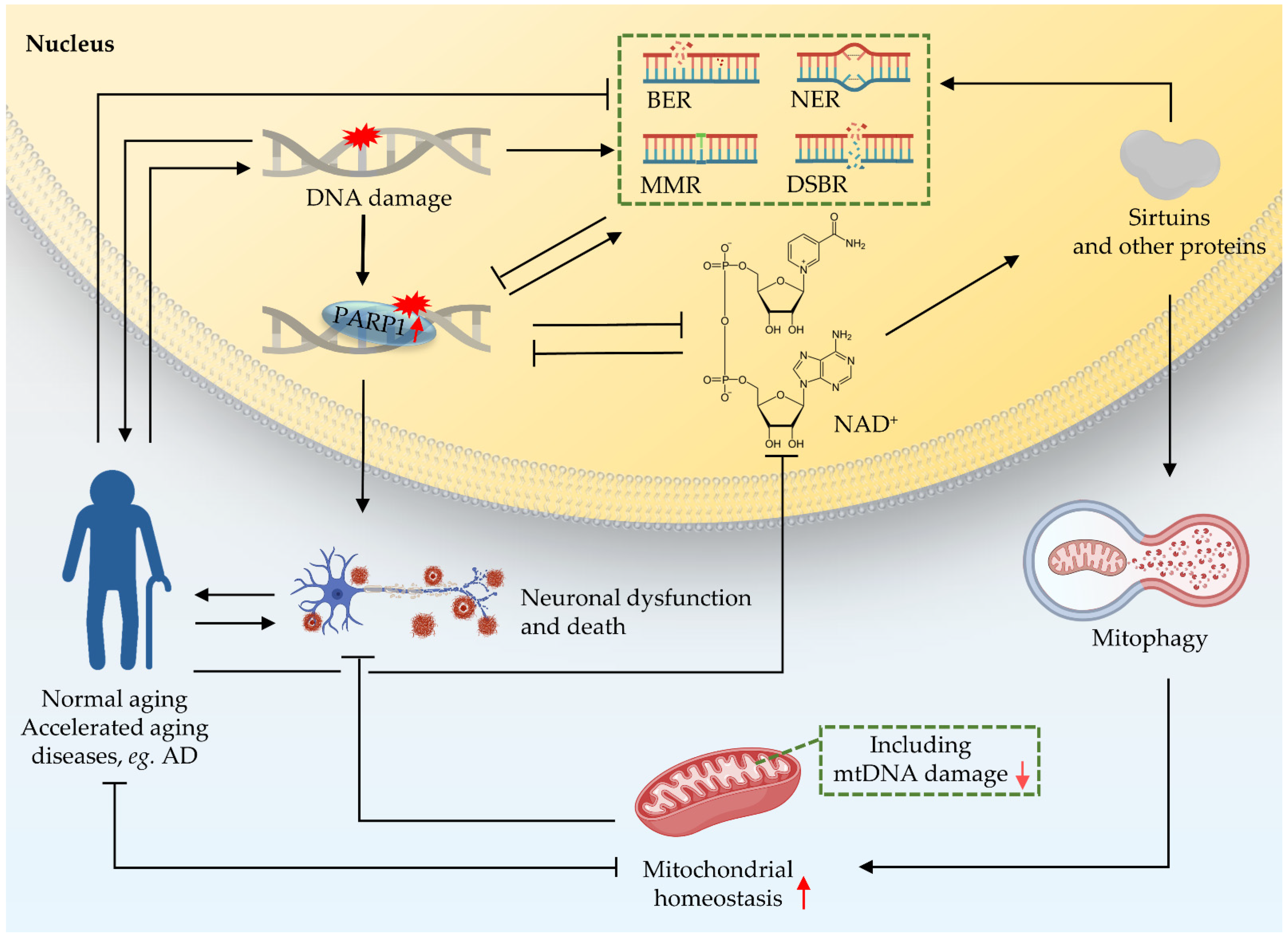
Crosstalk between the nucleus and mitochondria is essential for cellular function [7,63]; this crosstalk is a response to different ‘stimulators’ such as oxidative stress, DNA damage, and mitochondrial dysfunction [73,74]. There are accurate and rigorous regulatory mechanisms between the two organelles to control the stability of mitochondria [74]. One of them is that the nucleus regulates mitochondrial function through the poly-ADP-ribose polymerase 1 (PARP1)–NAD+–sirtuin 1 (SIRT1) signaling pathway (Figure 4). NAD+ is an important substrate for enzymes like PARPs and the NAD+-dependent deacetylases (sirtuins or SIRTs). NAD+ plays a key role in DDR. PARP1 monitors DNA lesions and subsequently recruits DNA repair proteins through PARylation, while consuming NAD+ [7,75]. PARP1 is continuously activated due to the accumulation of nuclear DNA damage. Hyperactivity of PARP1, as shown in multiple DNA repair deficient models (CS, XP), can lead to NAD+ depletion, thereby reducing the activity of sirtuins, and finally leading to mitochondrial dysfunction via impaired mitochondrial biogenesis and depleted mitophagy [76]. The sirtuin family shuttles between the nucleus, mitochondria, and cytoplasm in response to cell stimulation [74,77,78,79,80]. In addition, studies have shown that mutation of the C. elegans pme-1, the homologue of mammalian PARP1, increased NAD+ levels and Sir2.1 (the homologue of SIRT1 in C. elegans) activity, as well as increasing both healthspan and lifespan; mechanistically, this effect is at least partially contributed to by increased mitochondrial homeostasis through UPRmt activation [63,68,81]. PARP1 interacts with SIRT1 to achieve signal transduction from the nucleus to the mitochondria [7]. Further studies on the role of PARP1–NAD+–SIRT1 signaling in nucleus-mitochondria crosstalk are necessary.
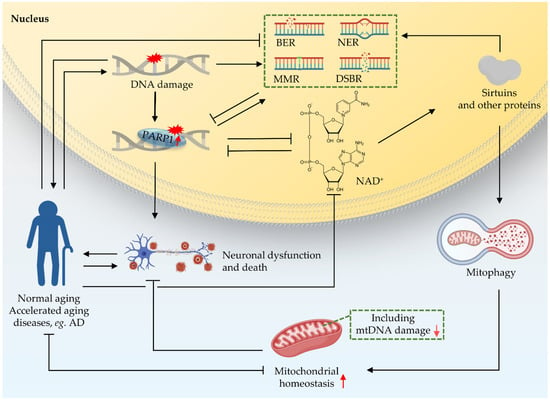
Figure 4.
Crosstalk between nucleus and mitochondria in premature ageing and AD. Nuclear DNA damage can lead to mitochondrial dysfunction. One of the pathways through which the nucleus regulates mitochondrial function is the PARP1–NAD+–SIRT1 signaling pathway. NAD+ plays an important role as a reaction substrate in various pathways of DDR. Together, these contribute to ageing and the neurodegeneration in accelerated ageing. Black arrows indicate promotion, and inverted T bars indicate repression. Red arrows indicate up- or downregulation.
4. Conclusions and Future Perspectives
DNA repair plays a fundamental role in life and health, while dysfunctional DNA repair drives or increases risks of premature ageing diseases, and a broad spectrum of other diseases, such as AD. In this review, we have updated recent progress in description of the major DNA repair pathways, including BER, NER, and DSBR. Mutations of genes involved in these pathways cause a group of premature ageing diseases, such as XPA, CS, A-T, and WS. Recent studies suggest DNA damage increases the risks of AD. Emerging questions and perspectives include (a) how cells orchestrate different DNA repair pathways to maintain genomic stability and health; (b) why is there little to no neurodegeneration in WS, while DSB is detrimental to neurons; and (c) research to find clinical evidence supporting promising drug candidates should be undertaken. Further studies on the mechanisms of DNA repair and their roles in healthy ageing and brain maintenance will shed light on the development of novel interventional strategies and treatments to support a healthier lifespan.
Funding
This review was supported by HELSE SØR-ØST (# 2017056, # 2020001, # 2021021), the Research Council of Norway (# 262175 and # 277813), the National Natural Science Foundation of China (# 81971327), an Akershus University Hospital Strategic grant (# 269901), the KAPPA programme by Technology Agency of the Czech Republic and the Research Council of Norway joint grant (T001000215), and a 3-year Ph.D. fellowship for A.G. from the Civitan Norges Forskningsfond for Alzheimers sykdom. H.W. was also sponsored by the China Scholarship Council [http:www.csc.edu.cn/], start date was 19 January 2020.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the valuable work of the many investigators whose published articles they were unable to cite owing to space limitations. We thank the Fang lab member Dawn Patrick-Brown in reading of the paper. Some of the figures were generated using the subscribed software BioRender, ©biorender.com, Toronto, ON, Canada.
Conflicts of Interest
E.F.F. has CRADA arrangement with ChromaDex, and is consultant to Aladdin Healthcare Technologies, Vancouver Dementia Prevention Centre, and Intellectual Labs.
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, V.; Wilson, D.M., 3rd. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nat. Cell Biol. 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T.; Barnes, D.E. Repair of Endogenous DNA Damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, T.A. Celebrating DNA’s Repair Crew. Cell 2015, 163, 1301–1303. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nat. Cell Biol. 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Lee, T.-H.; Kang, T.-H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar] [CrossRef] [Green Version]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA Damage and Its Links to Neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, E.A.; Rodriguez, A.A.; Bradley, N.P.; Eichman, B.F. Emerging Roles of DNA Glycosylases and the Base Excision Repair Pathway. Trends Biochem. Sci. 2019, 44, 765–781. [Google Scholar] [CrossRef]
- Li, M.; Yang, X.; Lu, X.; Dai, N.; Zhang, S.; Cheng, Y.; Zhang, L.; Yang, Y.; Liu, Y.; Yang, Z.; et al. APE1 deficiency promotes cellular senescence and premature aging features. Nucleic Acids Res. 2018, 46, 5664–5677. [Google Scholar] [CrossRef]
- Calafiore, M.; Copani, A.; Deng, W. DNA polymerase-beta mediates the neurogenic effect of beta-amyloid protein in cultured subventricular zone neurospheres. J. Neurosci. Res. 2012, 90, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Doublie, S. Base Excision Repair in the Mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boguszewska, K.; Szewczuk, M.; Kaźmierczak-Barańska, J.; Karwowski, B.T. The Similarities between Human Mitochondria and Bacteria in the Context of Structure, Genome, and Base Excision Repair System. Molecules 2020, 25, 2857. [Google Scholar] [CrossRef]
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Topolska-Woś, A.M.; Sugitani, N.; Cordoba, J.J.; Le Meur, K.V.; Le Meur, R.A.A.; Kim, H.S.; Yeo, J.-E.; Rosenberg, D.; Hammel, M.; Schärer, O.D.; et al. A key interaction with RPA orients XPA in NER complexes. Nucleic Acids Res. 2020, 48, 2173–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, M.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 2008, 18, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohr, V.A.; Smith, C.A.; Okumoto, D.S.; Hanawalt, P.C. DNA repair in an active gene: Removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 1985, 40, 359–369. [Google Scholar] [CrossRef]
- Riedl, T.; Hanaoka, F.; Egly, J.M. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003, 22, 5293–5303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petruseva, I.O.; Evdokimov, A.N.; Lavrik, O.I. Molecular Mechanism of Global Genome Nucleotide Excision Repair. Acta Naturae 2014, 6, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Kokic, G.; Chernev, A.; Tegunov, D.; Dienemann, C.; Urlaub, H.; Cramer, P. Structural basis of TFIIH activation for nucleotide excision repair. Nat. Commun. 2019, 10, 2885. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef]
- Kaniecki, K.; De Tullio, L.; Greene, E.C. A change of view: Homologous recombination at single-molecule resolution. Nat. Rev. Genet. 2018, 19, 191–207. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Piazza, A.; Heyer, W.-D. Homologous Recombination and the Formation of Complex Genomic Rearrangements. Trends Cell Biol. 2019, 29, 135–149. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nat. Cell Biol. 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Wyman, C.; Ristic, D.; Kanaar, R. Homologous recombination-mediated double-strand break repair. DNA Repair 2004, 3, 827–833. [Google Scholar] [CrossRef]
- Doherty, A.J.; Jackson, S.P. DNA repair: How Ku makes ends meet. Curr. Biol. 2001, 11, R920–R924. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Ci, S.; Li, M.; Wang, M.; Dianov, G.L.; Ma, Z.; Li, L.; Hua, K.; Alagamuthu, K.K.; Qing, L.; et al. Two-way crosstalk between BER and c-NHEJ repair pathway is mediated by Pol-beta and Ku70. FASEB J. 2019, 33, 11668–11681. [Google Scholar] [CrossRef] [Green Version]
- Brochier, C.; Langley, B. Chromatin Modifications Associated with DNA Double-strand Breaks Repair as Potential Targets for Neurological Diseases. Neurotherapeutics 2013, 10, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Gu, X.; Zhang, X.; Kong, J.; Ding, N.; Qi, Y.; Zhang, Y.; Wang, J.; Huang, D. Cadmium delays non-homologous end joining (NHEJ) repair via inhibition of DNA-PKcs phosphorylation and downregulation of XRCC4 and Ligase IV. Mutat. Res. Mol. Mech. Mutagen. 2015, 779, 112–123. [Google Scholar] [CrossRef]
- Liang, L.; Feng, J.; Zuo, P.; Yang, J.; Lu, Y.; Yin, Y. Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat. Commun. 2020, 11, 1972. [Google Scholar] [CrossRef]
- Gerodimos, C.A.; Chang, H.H.Y.; Watanabe, G.; Lieber, M.R. Effects of DNA end configuration on XRCC4-DNA ligase IV and its stimulation of Artemis activity. J. Biol. Chem. 2017, 292, 13914–13924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeijmakers, J.H. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Yu, X. Human DNA ligase IV is able to use NAD+ as an alternative adenylation donor for DNA ends ligation. Nucleic Acids Res. 2018, 47, 1321–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, P.J. Maintaining genome stability in the nervous system. Nat. Neurosci. 2013, 16, 1523–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, H.-M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Cerosaletti, K.M.; Girard, P.-M.; Dai, Y.; Stumm, M.; Kysela, B.; Hirsch, B.; Gennery, A.; Palmer, S.E.; Seidel, J.; et al. DNA Ligase IV Mutations Identified in Patients Exhibiting Developmental Delay and Immunodeficiency. Mol. Cell 2001, 8, 1175–1185. [Google Scholar] [CrossRef]
- Woodbine, L.; Neal, J.A.; Sasi, N.K.; Shimada, M.; Deem, K.; Coleman, H.; Dobyns, W.B.; Ogi, T.; Meek, K.; Davies, E.G.; et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J. Clin. Investig. 2013, 123, 2969–2980. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; van den Heuvel, D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M.; et al. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180, 1228–1244. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, Z.; Liu, L.; Liu, J.; Wang, Y. Rat Model of Cockayne Syndrome Neurological Disease. Cell Rep. 2019, 29, 800–809. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.; Houlden, H.; Tabrizi, S.J. DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 2017, 16, 88–96. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2018, 216, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Song, X.; Ma, F.; Herrup, K. Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J. Neurosci. 2019, 39, 6378–6394. [Google Scholar] [CrossRef] [Green Version]
- Orelli, B.; McClendon, T.B.; Tsodikov, O.V.; Ellenberger, T.; Niedernhofer, L.J.; Schärer, O.D. The XPA-binding domain of ERCC1 Is Required for Nucleotide Excision Repair but Not Other DNA Repair Pathways. J. Biol. Chem. 2010, 285, 3705–3712. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Heng, Y.; Kooistra, S.M.; Van Weering, H.R.J.; Brummer, M.L.; Gerrits, E.; Wesseling, E.M.; Brouwer, N.; Nijboer, T.W.; Dubbelaar, M.L.; et al. Intrinsic DNA damage repair deficiency results in progressive microglia loss and replacement. Glia 2021, 69, 729–745. [Google Scholar] [CrossRef]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14, 612757. [Google Scholar] [CrossRef]
- Saito, T.; Sadoshima, J. Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186, 111207. [Google Scholar] [CrossRef] [PubMed]
- Valentin-Vega, Y.A.; MacLean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, I.; Yoshida, Y.; Suda, M.; Minamino, T. DNA Damage Response and Metabolic Disease. Cell Metab. 2014, 20, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Ou, H.-L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, T.; Suart, C.; Hung, C.L.-K.; Graham, K.; Bazan, C.A.B.; Truant, R. DNA Damage Repair in Huntington’s Disease and Other Neurodegenerative Diseases. Neurotherapeutics 2019, 16, 948–956. [Google Scholar] [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.-Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nat. Cell Biol. 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD + Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Lederman, H.M. Ataxia-telangiectasia (A-T): An emerging dimension of premature ageing. Ageing Res. Rev. 2017, 33, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Caponio, D.; Cheung, H.H.; Piccoli, C.; Stevnsner, T.; Chan, W.-Y.; Fang, E.F. Studying Werner syndrome to elucidate mechanisms and therapeutics of human aging and age-related diseases. Biogerontology 2019, 20, 255–269. [Google Scholar] [CrossRef]
- Misiak, M.; Vergara Greeno, R.; Baptiste, B.A.; Sykora, P.; Liu, D.; Cordonnier, S.; Fang, E.F.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. DNA polymerase β decrement triggers death of olfactory bulb cells and impairs olfaction in a mouse model of Alzheimer’s disease. Aging Cell 2017, 16, 162–172. [Google Scholar] [CrossRef]
- Sykora, P.; Misiak, M.; Wang, Y.; Ghosh, S.; Leandro, G.S.; Liu, D.; Tian, J.; Baptiste, B.A.; Cong, W.N.; Brenerman, B.M.; et al. DNA polymerase β deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res. 2015, 43, 943–959. [Google Scholar] [CrossRef]
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA damage and mitochondria in cancer and aging. Carcinogenesis 2020, 41, 1625–1634. [Google Scholar] [CrossRef]
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Free. Radic. Biol. Med. 2017, 107, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Iv, F.T.B.; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ. Res. 2020, 126, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Cao, Y.; Zheng, Q.; Tu, J.; Zhou, W.; He, J.; Zhong, J.; Chen, Y.; Wang, J.; Cai, R.; et al. SENP1-Sirt3 Signaling Controls Mitochondrial Protein Acetylation and Metabolism. Mol. Cell 2019, 75, 823–834.e5. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.; Seebode, C.; Martens, M.C.; Emmert, S. Xeroderma Pigmentosum—Facts and Perspectives. Anticancer Res. 2018, 38, 1159–1164. [Google Scholar]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a Light on Xeroderma Pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Bojanini, E.; Hernandez-Quiceno, S.; Cock-Rada, A.M. Xeroderma Pigmentosum with Severe Neurological Manifestations/De Sanctis–Cacchione Syndrome and a Novel XPC Mutation. Case Rep. Med. 2017, 2017, 7162737. [Google Scholar] [CrossRef]
- Abeti, R.; Zeitlberger, A.; Peelo, C.; Fassihi, H.; Sarkany, R.P.E.; Lehmann, A.R.; Giunti, P. Xeroderma pigmentosum: Overview of pharmacology and novel therapeutic strategies for neurological symptoms. Br. J. Pharmacol. 2019, 176, 4293–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA Repair and Neurodegenerative Disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef] [Green Version]
- Fassihi, H.; Sethi, M.; Fawcett, H.; Wing, J.; Chandler, N.; Mohammed, S.; Craythorne, E.; Morley, A.M.S.; Lim, R.; Turner, S.; et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc. Natl. Acad. Sci. USA 2016, 113, E1236–E1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleaver, J.E. Defective Repair Replication of DNA in Xeroderma Pigmentosum. Nat. Cell Biol. 1968, 218, 652–656. [Google Scholar] [CrossRef]
- Sugasawa, K.; Akagi, J.-I.; Nishi, R.; Iwai, S.; Hanaoka, F. Two-Step Recognition of DNA Damage for Mammalian Nucleotide Excision Repair: Directional Binding of the XPC Complex and DNA Strand Scanning. Mol. Cell 2009, 36, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Sabatella, M.; Pines, A.; Slyskova, J.; Vermeulen, W.; Lans, H. ERCC1-XPF targeting to psoralen-DNA crosslinks depends on XPA and FANCD2. Cell. Mol. Life Sci. 2020, 77, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Sugitani, N.; Sivley, R.M.; Perry, K.E.; Capra, J.A.; Chazin, W.J. XPA: A key scaffold for human nucleotide excision repair. DNA Repair 2016, 44, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Elledge, S.J.; Peterson, C.A.; Bales, E.S.; Legerski, R.J. Specific association between the human DNA repair proteins XPA and ERCC1. Proc. Natl. Acad. Sci. USA 1994, 91, 5012–5016. [Google Scholar] [CrossRef] [Green Version]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Schärer, O.D.; Wagner, G.; Ellenberger, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volker, M.; Moné, M.J.; Karmakar, P.; van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.; van Driel, R.; van Zeeland, A.A.; Mullenders, L.H. Sequential Assembly of the Nucleotide Excision Repair Factors In Vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef]
- Huang, J.; Liu, W.; Doycheva, D.M.; Gamdzyk, M.; Lu, W.; Tang, J.; Zhang, J.H. Ghrelin attenuates oxidative stress and neuronal apoptosis via GHSR-1α/AMPK/Sirt1/PGC-1α/UCP2 pathway in a rat model of neonatal HIE. Free Radic. Biol. Med. 2019, 141, 322–337. [Google Scholar] [CrossRef]
- Luo, G.; Xiao, L.; Wang, D.; Wang, N.; Luo, C.; Yang, X.; Hao, L. Resveratrol protects against ethanol-induced impairment of insulin secretion in INS-1 cells through SIRT1-UCP2 axis. Toxicol. In Vitro 2020, 65, 104808. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R. NAD+ Depletion Is Necessary and Sufficient forPoly(ADP-Ribose) Polymerase-1-Mediated Neuronal Death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- Manandhar, M.; Lowery, M.G.; Boulware, K.S.; Lin, K.H.; Lu, Y.; Wood, R.D. Transcriptional consequences of XPA disruption in human cell lines. DNA Repair 2017, 57, 76–90. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Bohr, V.A. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy 2014, 10, 1468–1469. [Google Scholar] [CrossRef] [Green Version]
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med Genet. 1992, 42, 68–84. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype–phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Stevnsner, T.; Nyaga, S.; De Souza-Pinto, N.C.; Van Der Horst, G.T.J.; Gorgels, T.G.M.F.; Hogue, B.A.; Thorslund, T.; Bohr, V.A. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene 2002, 21, 8675–8682. [Google Scholar] [CrossRef] [Green Version]
- Shemen, L.J.; Mitchell, D.P.; Farkashidy, J. Cockayne syndrome—An audiologic and temporal bone analysis. Am. J. Otol. 1984, 5, 300–307. [Google Scholar]
- Wilson, B.T.; Stark, Z.; Sutton, R.E.; Danda, S.; Ekbote, A.V.; Elsayed, S.M.; Gibson, L.; Goodship, J.A.; Jackson, A.P.; Keng, W.T.; et al. The Cockayne Syndrome Natural History (CoSyNH) study: Clinical findings in 102 individuals and recommendations for care. Genet. Med. 2015, 18, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Okur, M.N.; Mao, B.; Kimura, R.; Haraczy, S.; Fitzgerald, T.; Edwards-Hollingsworth, K.; Tian, J.; Osmani, W.; Croteau, D.L.; Kelley, M.W.; et al. Short-term NAD+ supplementation prevents hearing loss in mouse models of Cockayne syndrome. NPJ Aging Mech. Dis. 2020, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; de Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M., 3rd; Stevnsner, T.; et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010, 24, 2334–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascucci, B.; Lemma, T.; Iorio, E.; Giovannini, S.; Vaz, B.; Iavarone, I.; Calcagnile, A.; Narciso, L.; Degan, P.; Podo, F.; et al. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell 2012, 11, 520–529. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Tseng, A.; Borch Jensen, M.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okur, M.N.; Fang, E.F.; Fivenson, E.M.; Tiwari, V.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD+ signaling. Aging Cell 2020, 19, e13268. [Google Scholar] [CrossRef]
- Choy, K.R.; Watters, D.J. Neurodegeneration in ataxia-telangiectasia: Multiple roles of ATM kinase in cellular homeostasis. Dev. Dyn. 2018, 247, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Amirifar, P.; Ranjouri, M.R.; Yazdani, R.; Abolhassani, H.; Aghamohammadi, A. Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatr. Allergy Immunol. 2019, 30, 277–288. [Google Scholar] [CrossRef]
- Levy, A.; Lang, A.E. Ataxia-telangiectasia: A review of movement disorders, clinical features, and genotype correlations. Mov. Disord. 2018, 33, 1238–1247. [Google Scholar] [CrossRef]
- Schon, K.; Van Os, N.J.; Oscroft, N.; Baxendale, H.; Scoffings, D.; Ray, J.; Suri, M.; Whitehouse, W.P.; Mehta, P.; Everett, N.; et al. Genotype, extrapyramidal features and severity of variant Ataxia-Telangiectasia. Ann. Neurol. 2018, 85, 170–180. [Google Scholar] [CrossRef]
- Van Os NJ, H.; Jansen AF, M.; van Deuren, M.; Haraldsson, A.; van Driel NT, M.; Etzioni, A.; van der Flier, M.; Haaxma, C.A.; Morio, T.; Rawat, A.; et al. Ataxia-telangiectasia: Immunodeficiency and survival. Clin. Immunol. 2017, 178, 45–55. [Google Scholar] [CrossRef]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504. [Google Scholar] [CrossRef]
- McKnight, K.L.; Swanson, K.V.; Austgen, K.; Richards, C.; Mitchell, J.K.; McGivern, D.R.; Fritch, E.; Johnson, J.; Remlinger, K.; Magid-Slav, M.; et al. Stimulator of interferon genes (STING) is an essential proviral host factor for human rhinovirus species A and C. Proc. Natl. Acad. Sci. USA 2020, 117, 27598–27607. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner’s syndrome gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Bohr, V.A. Rising from the RecQ-age: The role of human RecQ helicases in genome maintenance. Trends Biochem. Sci. 2008, 33, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.B.; Hickson, I.D. RecQ Helicases: Conserved Guardians of Genomic Integrity. Adv. Exp. Med. Biol. 2012, 767, 161–184. [Google Scholar] [CrossRef]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Li, B.; Gray, M.D.; Oshima, J.; Mian, I.S.; Campisi, J. The premature ageing syndrome protein, WRN, is a 3′→5′ exonuclease. Nat. Genet. 1998, 20, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; Sengupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, J.; Sidorova, J.; Monnat, R.J. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Oshima, J.; Martin, G.M.; Hisama, F.M. Werner Syndrome. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Takemoto, M.; Mori, S.; Kuzuya, M.; Yoshimoto, S.; Shimamoto, A.; Igarashi, M.; Tanaka, Y.; Miki, T.; Yokote, K. Diagnostic criteria for Werner syndrome based on Japanese nationwide epidemiological survey. Geriatr. Gerontol. Int. 2013, 13, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Sharma, N.K.; Lebedeva, M.; Thomas, T.; Kovalenko, O.A.; Stumpf, J.D.; Shadel, G.S.; Santos, J.H. Intrinsic mitochondrial DNA repair defects in Ataxia Telangiectasia. DNA Repair 2014, 13, 22–31. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fang, E.F.; Scheibye-Knudsen, M.; Cui, H.; Qiu, L.; Li, J.; He, Y.; Huang, J.; Bohr, V.A.; Ng, T.B.; et al. Di-(2-ethylhexyl) phthalate inhibits DNA replication leading to hyperPARylation, SIRT1 attenuation and mitochondrial dysfunction in the testis. Sci. Rep. 2015, 4, 6434. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, A.; Ittner, L.M. Tau-targeted treatment strategies in Alzheimer’s disease. Br. J. Pharmacol. 2012, 165, 1246–1259. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.-X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Weissman, L.; de Souza-Pinto, N.C.; Mattson, M.P.; Bohr, V.A. DNA base excision repair activities in mouse models of Alzheimer’s disease. Neurobiol. Aging 2009, 30, 2080–2081. [Google Scholar] [CrossRef] [Green Version]
- Weissman, L.; Jo, D.G.; Sørensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006, 96, 825–832. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000, 855, 116–123. [Google Scholar] [CrossRef]
- Canugovi, C.; Misiak, M.; Ferrarelli, L.K.; Croteau, D.L.; Bohr, V.A. The role of DNA repair in brain related disease pathology. DNA Repair 2013, 12, 578–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykora, P.; Wilson, D.M., 3rd; Bohr, V.A. Base excision repair in the mammalian brain: Implication for age related neurodegeneration. Mech. Ageing Dev. 2013, 134, 440–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).