
Treatment with the Bacterial Toxin CNF1 Selectively Rescues Cognitive and Brain Mitochondrial Deficits in a Female Mouse Model of Rett Syndrome Carrying a MeCP2-Null Mutation

 ,
,  ,
,  , ,
, ,  and
and 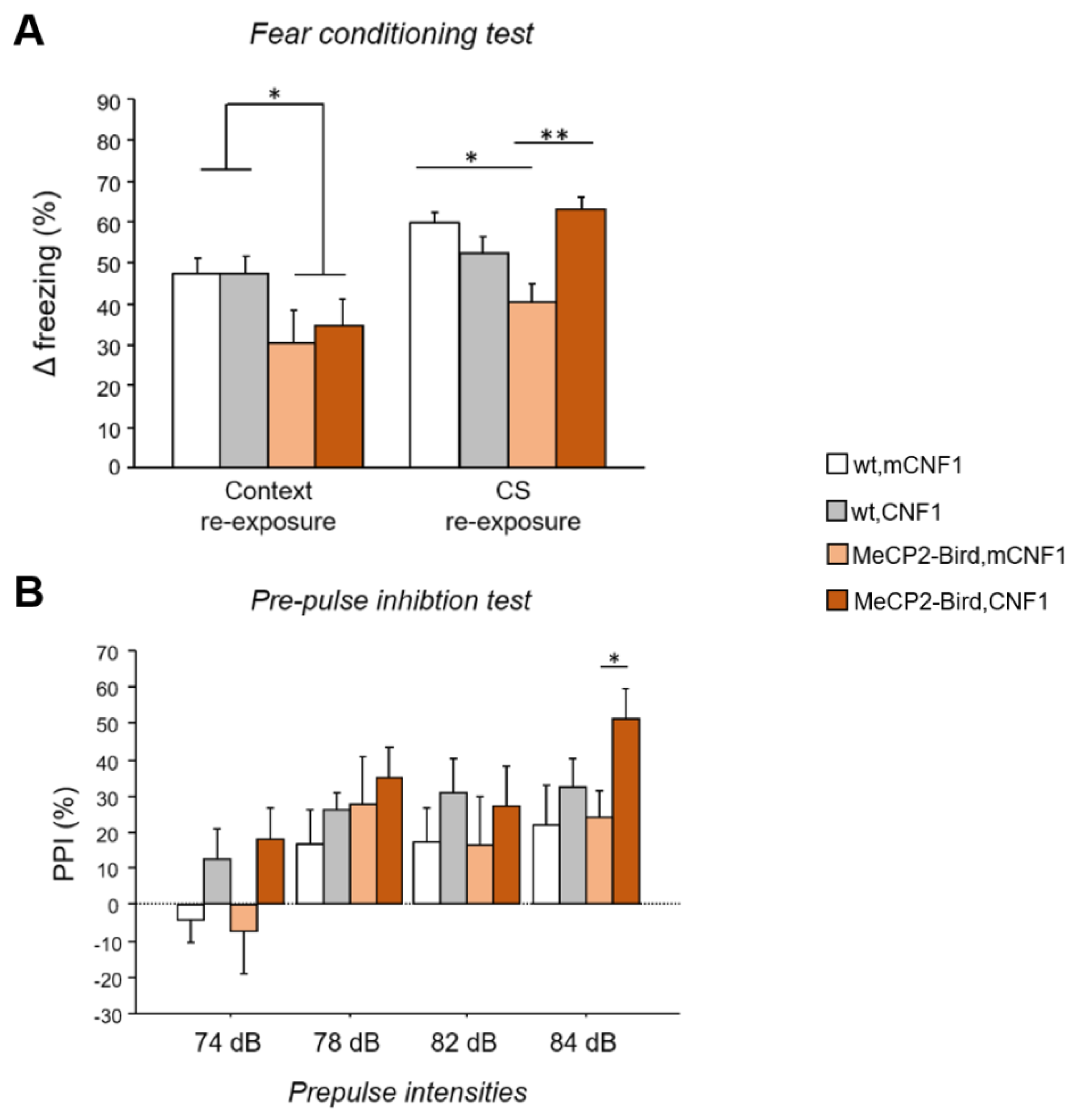{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
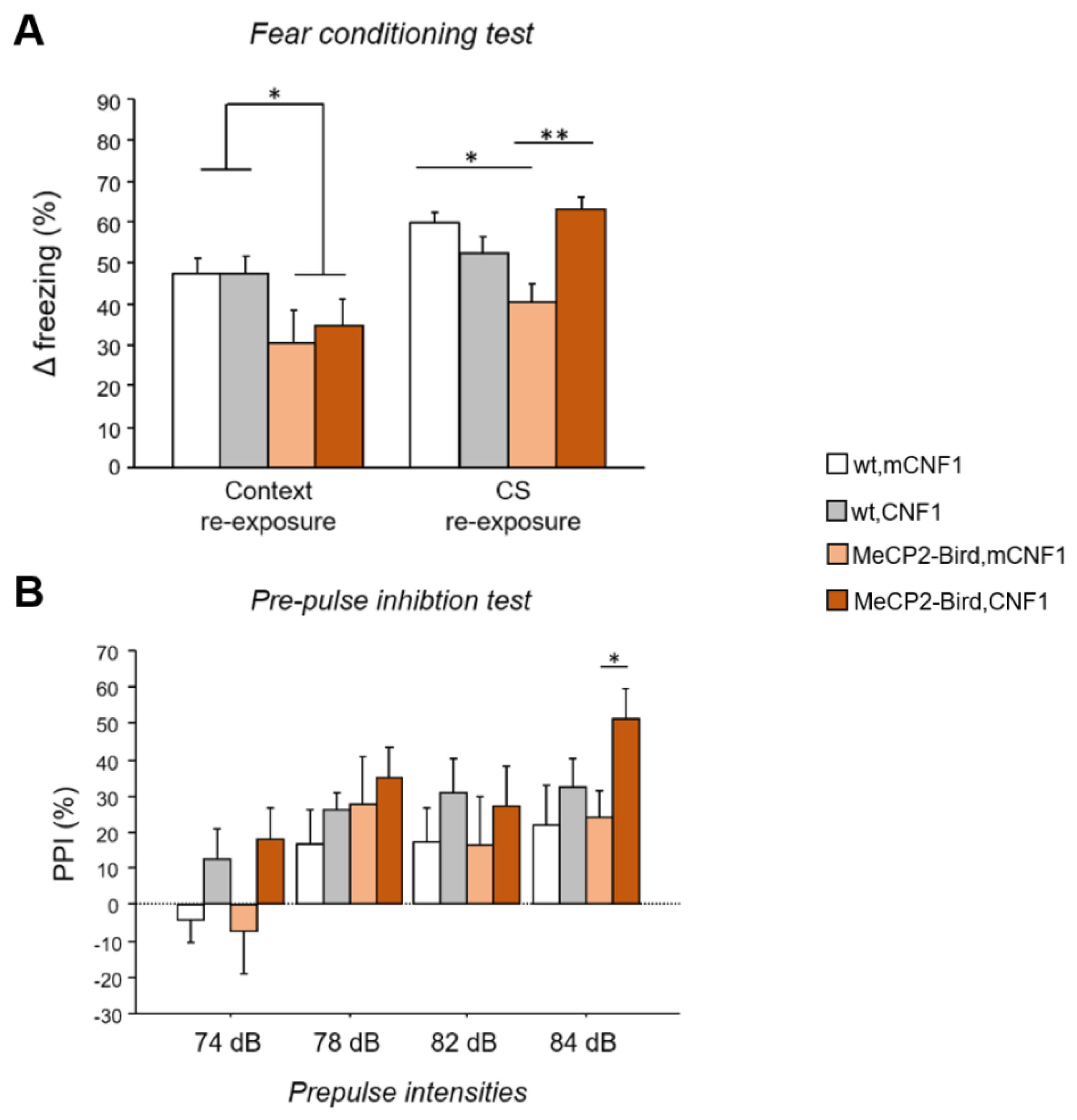
2.1. CNF1 Treatment Improves the Fear Memory Deficits in MeCP2-Bird Mice
2.2. CNF1 Treatment Increases Pre-Pulse Inhibition in MeCP2-Bird Mice
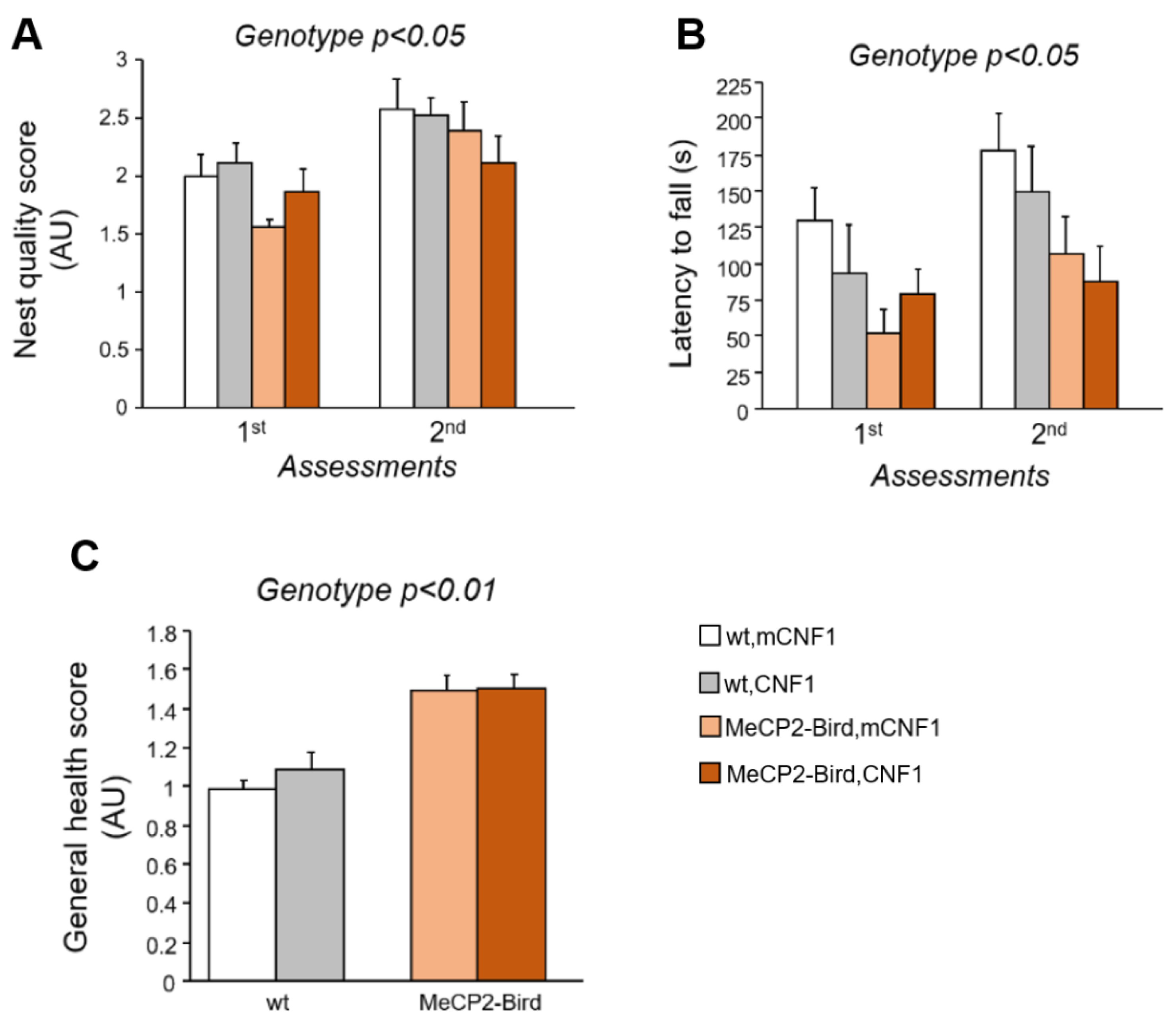
2.3. CNF1 Treatment Does Not Rescue the Motor Deficits and the General Health Status of MeCP2-Bird Mice
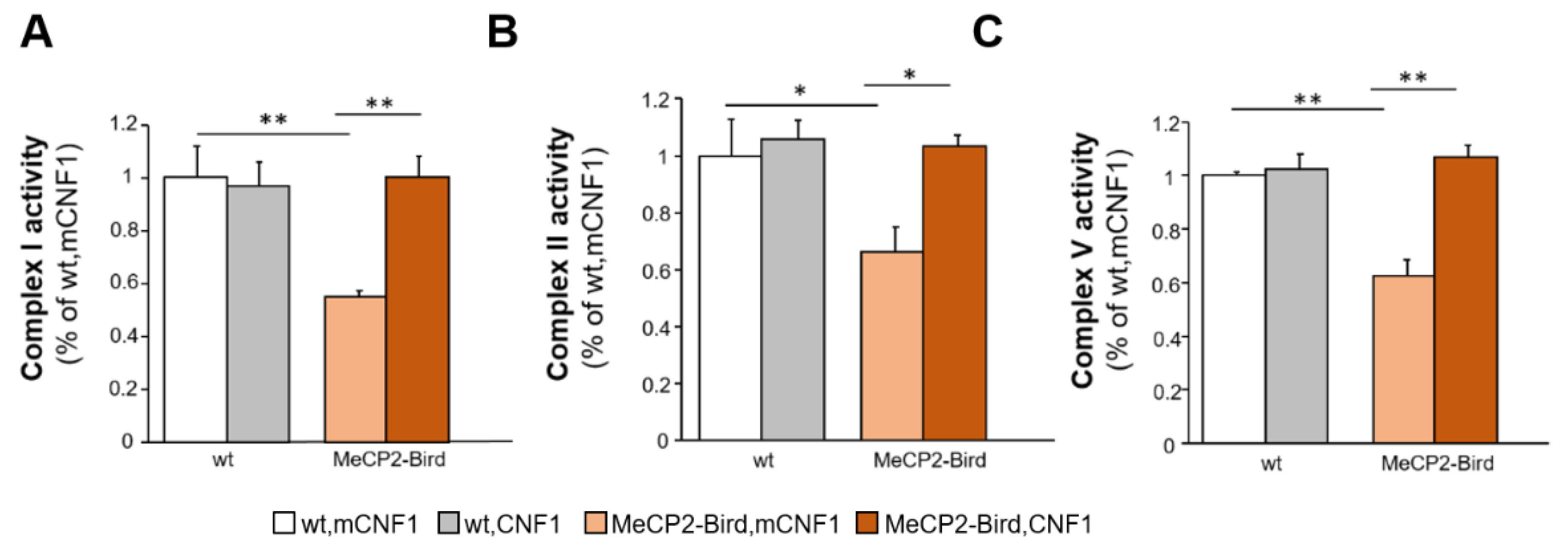
2.4. CNF1 Treatment Rescues the Defective Activity of Mitochondrial Respiratory Chain Complexes I, II and V in MeCP2-Bird Mice
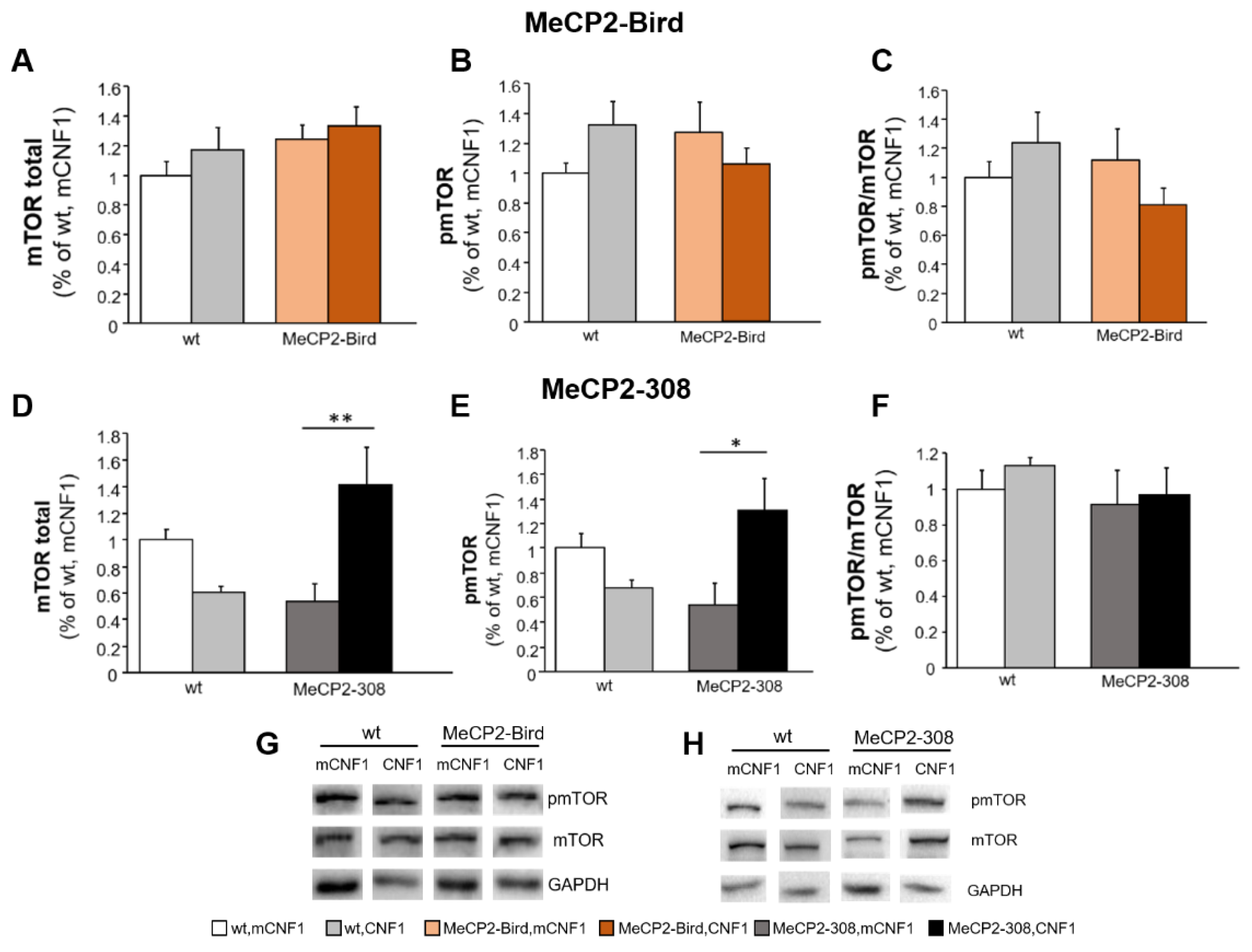
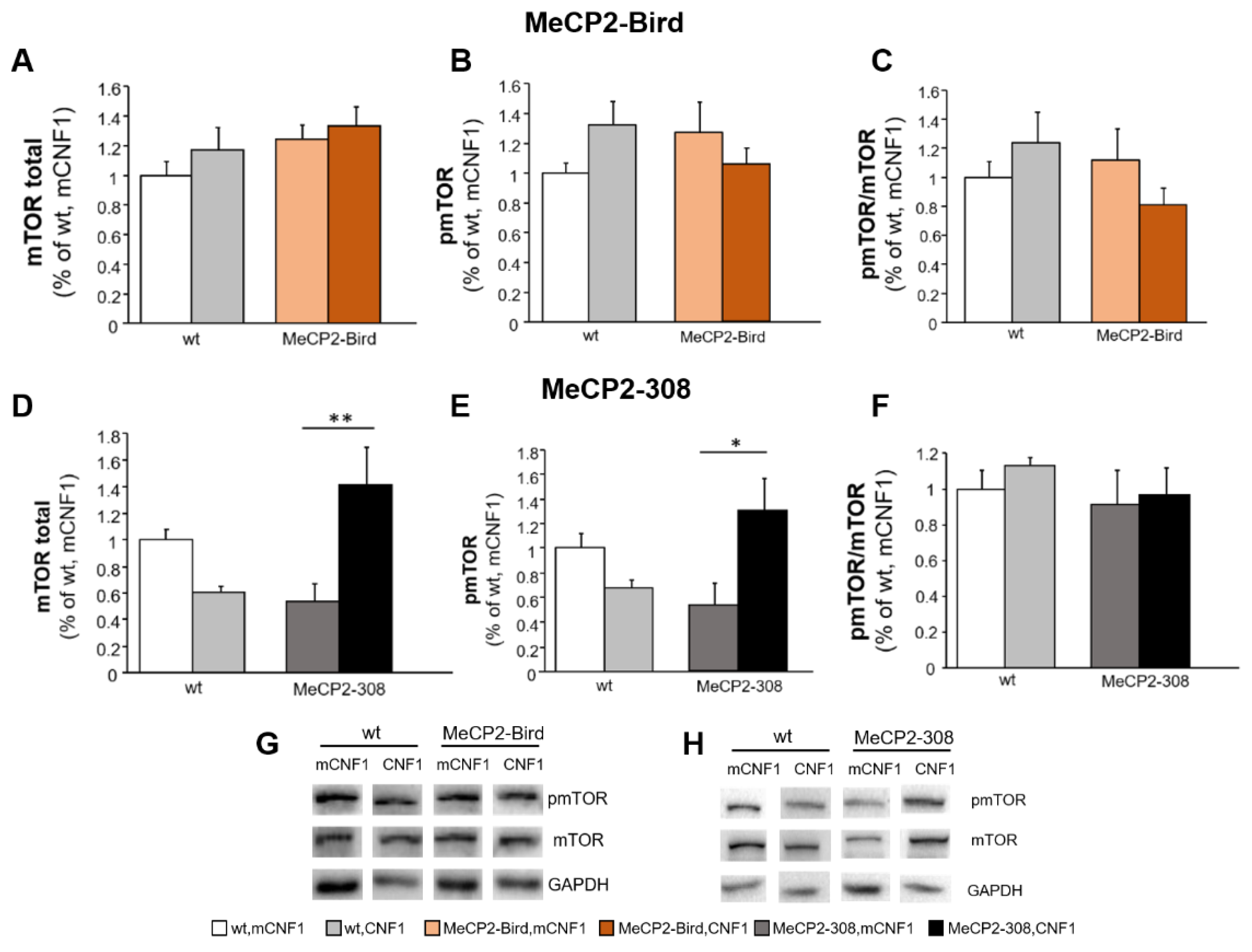
2.5. CNF1 Treatment Provides a Significant Boost of mTOR Activation in MeCP2-308 Mouse Hippocampus, Which Is Not Recapitulated in the Bird Model
2.6. CNF1 Treatment Does Not Affect Expression Levels of Mitochondrial and Synaptic Markers in RTT Mouse Brain
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. CNF1 Preparation and Intracerebroventricular Injection
4.3. Behavioural Testing
4.4. Nest Building Test
4.5. Accelerating Rotarod Test
4.6. General Health Assessment
4.7. Pre-Pulse Inhibition Test
4.8. Fear Conditioning Test
4.9. Mitochondrial Analyses
4.10. Western Blotting
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial Dysfunction in the Pathogenesis of Rett Syndrome: Implications for Mitochondria-Targeted Therapies. Front. Cell. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Ricceri, L.; De Filippis, B.; Laviola, G. Rett syndrome treatment in mouse models: Searching for effective targets and strategies. Neuropharmacology 2013, 68, 106–115. [Google Scholar] [CrossRef]
- Ricceri, L.; De Filippis, B.; Laviola, G. Mouse models of Rett syndrome: From behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav. Pharmacol. 2008, 19, 501–517. [Google Scholar] [CrossRef] [Green Version]
- Vashi, N.; Justice, M.J. Treating Rett syndrome: From mouse models to human therapies. Mamm. Genome 2019, 30, 90–110. [Google Scholar] [CrossRef] [Green Version]
- Gold, W.A.; Williamson, S.L.; Kaur, S.; Hargreaves, I.P.; Land, J.M.; Pelka, G.J.; Tam, P.P.L.; Christodoulou, J. Mitochondrial dysfunction in the skeletal muscle of a mouse model of Rett syndrome (RTT): Implications for the disease phenotype. Mitochondrion 2014, 15, 10–17. [Google Scholar] [CrossRef]
- Cobb, S.; Guy, J.; Bird, A. Reversibility of functional deficits in experimental models of Rett syndrome. Biochem. Soc. Trans. 2010, 38, 498–506. [Google Scholar] [CrossRef]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of Rett syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; de Bari, L.; De Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free. Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef]
- De Filippis, B.; Romano, E.; Laviola, G. Aberrant Rho GTPases signaling and cognitive dysfunction: In vivo evidence for a compelling molecular relationship. Neurosci. Biobehav. Rev. 2014, 46, 285–301. [Google Scholar] [CrossRef] [PubMed]
- De Viti, S.; Martino, A.; Musilli, M.; Fiorentini, C.; Diana, G. The Rho GTPase activating CNF1 improves associative working memory for object-in-place. Behav. Brain Res. 2010, 212, 78–83. [Google Scholar] [CrossRef]
- Diana, G.; Valentini, G.; Travaglione, S.; Falzano, L.; Pieri, M.; Zona, C.; Meschini, S.; Fabbri, A.; Fiorentini, C. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 636–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loizzo, S.; Rimondini, R.; Travaglione, S.; Fabbri, A.; Guidotti, M.; Ferri, A.; Campana, G.; Fiorentini, C. CNF1 increases brain energy level, counteracts neuroinflammatory markers and rescues cognitive deficits in a murine model of Alzheimer’s disease. PLoS ONE 2013, 8, e65898. [Google Scholar] [CrossRef]
- Cerri, C.; Fabbri, A.; Vannini, E.; Spolidoro, M.; Costa, M.; Maffei, L.; Fiorentini, C.; Caleo, M. Activation of rho GTPases triggers structural remodeling and functional plasticity in the adult rat visual cortex. J. Neurosci. 2011, 31, 15163–15172. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Katz, D.M.; Berger-Sweeney, J.E.; Eubanks, J.H.; Justice, M.J.; Neul, J.L.; Pozzo-Miller, L.; Blue, M.E.; Christian, D.; Crawley, J.N.; Giustetto, M.; et al. Preclinical research in Rett syndrome: Setting the foundation for translational success. Dis. Models Mech. 2012, 5, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardi, S.; Boggio, E.M.; Grosso, S.; Lonetti, G.; Forlani, G.; Stefanelli, G.; Calcagno, E.; Morello, N.; Landsberger, N.; Biffo, S.; et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 2011, 20, 1182–1196. [Google Scholar] [CrossRef] [Green Version]
- Troca-Marin, J.A.; Alves-Sampaio, A.; Montesinos, M.L. Deregulated mTOR-mediated translation in intellectual disability. Prog. Neurobiol. 2012, 96, 268–282. [Google Scholar] [CrossRef]
- Yuan, Z.-F.; Mao, S.-S.; Shen, J.; Jiang, L.-H.; Xu, L.; Xu, J.-L.; Gao, F. Insulin-Like Growth Factor-1 Down-Regulates the Phosphorylation of FXYD1 and Rescues Behavioral Deficits in a Mouse Model of Rett Syndrome. Front. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef]
- Powell, S.B.; Weber, M.; Geyer, M.A. Genetic models of sensorimotor gating: Relevance to neuropsychiatric disorders. Curr. Top. Behav. Neurosci. 2012, 12, 251–318. [Google Scholar] [PubMed] [Green Version]
- De Filippis, B.; Musto, M.; Altabella, L.; Romano, E.; Canese, R.; Laviola, G. Deficient Purposeful Use of Forepaws in Female Mice Modelling Rett Syndrome. Neural Plast. 2015, 326184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; de Bari, L.; Vigli, D.; Lacivita, E.; Leopoldo, M.; Laviola, G.; Vacca, R.A.; De Filippis, B. Stimulation of the brain serotonin receptor 7 rescues mitochondrial dysfunction in female mice from two models of Rett syndrome. Neuropharmacology 2017, 121, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Maroccia, Z.; Guidotti, M.; Pierri, C.L.; Primiano, G.; Servidei, S.; Loizzo, S.; Fiorentini, C. The Bacterial Protein CNF1 as a Potential Therapeutic Strategy against Mitochondrial Diseases: A Pilot Study. Int. J. Mol. Sci. 2018, 19, 1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travaglione, S.; Ballan, G.; Fortuna, A.; Ferri, A.; Guidotti, M.; Campana, G.; Fiorentini, C.; Loizzo, S. CNF1 Enhances Brain Energy Content and Counteracts Spontaneous Epileptiform Phenomena in Aged DBA/2J Mice. PLoS ONE 2015, 10, e0140495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.E.; Jensen, R.E. Barreling through the membrane. Nat. Struct. Mol. Biol. 2004, 11, 113–114. [Google Scholar] [CrossRef]
- Feng, J.; Yan, Z.; Ferreira, A.; Tomizawa, K.; Liauw, J.A.; Zhuo, M.; Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. USA 2000, 97, 9287–9292. [Google Scholar] [CrossRef] [Green Version]
- Castro, J.; Garcia, R.I.; Kwok, S.; Banerjee, A.; Petravicz, J.; Woodson, J.; Mellios, N.; Tropea, D.; Sur, M. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 9941–9946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigli, D.; Cosentino, L.; Raggi, C.; Laviola, G.; Woolley-Roberts, M.; De Filippis, B. Chronic treatment with the phytocannabinoid Cannabidivarin (CBDV) rescues behavioural alterations and brain atrophy in a mouse model of Rett syndrome. Neuropharmacology 2018, 140, 121–129. [Google Scholar] [CrossRef]
- Zamberletti, E.; Gabaglio, M.; Piscitelli, F.; Brodie, J.S.; Woolley-Roberts, M.; Barbiero, I.; Tramarin, M.; Binelli, G.; Landsberger, N.; Kilstrup-Nielsen, C.; et al. Cannabidivarin completely rescues cognitive deficits and delays neurological and motor defects in male Mecp2 mutant mice. J. Psychopharmacol. 2019, 33, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.-M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tropea, D.; Giacometti, E.; Wilson, N.R.; Beard, C.; McCurry, C.; Fu, D.D.; Flannery, R.; Jaenisch, R.; Sur, M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2029–2034. [Google Scholar] [CrossRef] [Green Version]
- Tsujimura, K.; Irie, K.; Nakashima, H.; Egashira, Y.; Fukao, Y.; Fujiwara, M.; Itoh, M.; Uesaka, M.; Imamura, T.; Nakahata, Y.; et al. miR-199a Links MeCP2 with mTOR Signaling and Its Dysregulation Leads to Rett Syndrome Phenotypes. Cell Rep. 2015, 12, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Wu, H.; Sun, Y.E. Deciphering Rett syndrome with mouse genetics, epigenomics, and human neurons. Int. Rev. Neurobiol. 2009, 89, 147–160. [Google Scholar]
- Geary, D.C. Mitochondrial Functioning and the Relations among Health, Cognition, and Aging: Where Cell Biology Meets Cognitive Science. Int. J. Mol. Sci. 2021, 22, 3562. [Google Scholar] [CrossRef]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef]
- Zuliani, I.; Urbinati, C.; Valenti, D.; Quattrini, M.C.; Medici, V.; Cosentino, L.; Pietraforte, D.; Di Domenico, F.; Perluigi, M.; Vacca, R.A.; et al. The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome. J. Clin. Med. 2020, 9, 1669. [Google Scholar] [CrossRef]
- Samaco, R.C.; McGraw, C.M.; Ward, C.S.; Sun, Y.; Neul, J.L.; Zoghbi, H.Y. Female Mecp2+/− mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet. 2013, 22, 96–109. [Google Scholar] [CrossRef]
- Khacho, M.; Slack, R.S. Mitochondrial dynamics in the regulation of neurogenesis: From development to the adult brain. Dev. Dyn. 2018, 247, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, L.; Vigli, D.; Franchi, F.; Laviola, G.; De Filippis, B. Rett syndrome before regression: A time window of overlooked opportunities for diagnosis and intervention. Neurosci. Biobehav. Rev. 2019, 107, 115–135. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Young, J.I.; Yuva-Paylor, L.A.; Spencer, C.M.; Antalffy, B.A.; Noebels, J.L.; Armstrong, D.L.; Paylor, R.; Zoghbi, H.Y. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [CrossRef]
- Schmidt, G.; Selzer, J.; Lerm, M.; Aktories, K. The Rho-deamidating cytotoxic necrotizing factor 1 from Escherichia coli possesses transglutaminase activity. Cysteine 866 and histidine 881 are essential for enzyme activity. J. Biol. Chem. 1998, 273, 13669–13674. [Google Scholar] [CrossRef] [Green Version]
- Moretti, P.; Bouwknecht, J.A.; Teague, R.; Paylor, R.; Zoghbi, H.Y. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 2005, 14, 205–220. [Google Scholar] [CrossRef] [Green Version]
- Deacon, R.M.J. Assessing nest building in mice. Nat. Protoc. 2006, 1, 1117–1119. [Google Scholar] [CrossRef]
- De Filippis, B.; Ricceri, L.; Laviola, G. Early postnatal behavioral changes in the Mecp2-308 truncation mouse model of Rett syndrome. Genes brain Behav. 2010, 9, 213–223. [Google Scholar] [CrossRef]
- De Filippis, B.; Nativio, P.; Fabbri, A.; Ricceri, L.; Adriani, W.; Lacivita, E.; Leopoldo, M.; Passarelli, F.; Fuso, A.; Laviola, G. Pharmacological stimulation of the brain serotonin receptor 7 as a novel therapeutic approach for Rett syndrome. Neuropsychopharmacology 2014, 39, 2506–2518. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.R.; Geyer, M.A.; Braff, D.L. Neural circuit regulation of prepulse inhibition of startle in the rat: Current knowledge and future challenges. Psychopharmacology 2001, 156, 194–215. [Google Scholar] [CrossRef]
- Vigli, D.; Rusconi, L.; Valenti, D.; La Montanara, P.; Cosentino, L.; Lacivita, E.; Leopoldo, M.; Amendola, E.; Gross, C.; Landsberger, N.; et al. Rescue of prepulse inhibition deficit and brain mitochondrial dysfunction by pharmacological stimulation of the central serotonin receptor 7 in a mouse model of CDKL5 Deficiency Disorder. Neuropharmacology 2019, 144, 104–114. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; De Filippis, B.; Ricceri, L.; Vacca, R.A. Preservation of mitochondrial functional integrity in mitochondria isolated from small cryopreserved mouse brain areas. Anal. Biochem. 2014, 444, 25–31. [Google Scholar] [CrossRef]
- Wilcox, R.R. New Designs in Analysis of Variance. Annu. Rev. Psychol. 1987, 38, 29–60. [Google Scholar] [CrossRef]
- Grubbs, F.E. Procedures for Detecting Outlying Observations in Samples. Technometrics 1969, 11, 1–21. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbinati, C.; Cosentino, L.; Germinario, E.A.P.; Valenti, D.; Vigli, D.; Ricceri, L.; Laviola, G.; Fiorentini, C.; Vacca, R.A.; Fabbri, A.; et al. Treatment with the Bacterial Toxin CNF1 Selectively Rescues Cognitive and Brain Mitochondrial Deficits in a Female Mouse Model of Rett Syndrome Carrying a MeCP2-Null Mutation. Int. J. Mol. Sci. 2021, 22, 6739. https://doi.org/10.3390/ijms22136739
Urbinati C, Cosentino L, Germinario EAP, Valenti D, Vigli D, Ricceri L, Laviola G, Fiorentini C, Vacca RA, Fabbri A, et al. Treatment with the Bacterial Toxin CNF1 Selectively Rescues Cognitive and Brain Mitochondrial Deficits in a Female Mouse Model of Rett Syndrome Carrying a MeCP2-Null Mutation. International Journal of Molecular Sciences. 2021; 22(13):6739. https://doi.org/10.3390/ijms22136739
Chicago/Turabian StyleUrbinati, Chiara, Livia Cosentino, Elena Angela Pia Germinario, Daniela Valenti, Daniele Vigli, Laura Ricceri, Giovanni Laviola, Carla Fiorentini, Rosa Anna Vacca, Alessia Fabbri, and et al. 2021. "Treatment with the Bacterial Toxin CNF1 Selectively Rescues Cognitive and Brain Mitochondrial Deficits in a Female Mouse Model of Rett Syndrome Carrying a MeCP2-Null Mutation" International Journal of Molecular Sciences 22, no. 13: 6739. https://doi.org/10.3390/ijms22136739
APA StyleUrbinati, C., Cosentino, L., Germinario, E. A. P., Valenti, D., Vigli, D., Ricceri, L., Laviola, G., Fiorentini, C., Vacca, R. A., Fabbri, A., & De Filippis, B. (2021). Treatment with the Bacterial Toxin CNF1 Selectively Rescues Cognitive and Brain Mitochondrial Deficits in a Female Mouse Model of Rett Syndrome Carrying a MeCP2-Null Mutation. International Journal of Molecular Sciences, 22(13), 6739. https://doi.org/10.3390/ijms22136739