
Targeting the Angiotensin II Type 1 Receptor in Cerebrovascular Diseases: Biased Signaling Raises New Hopes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
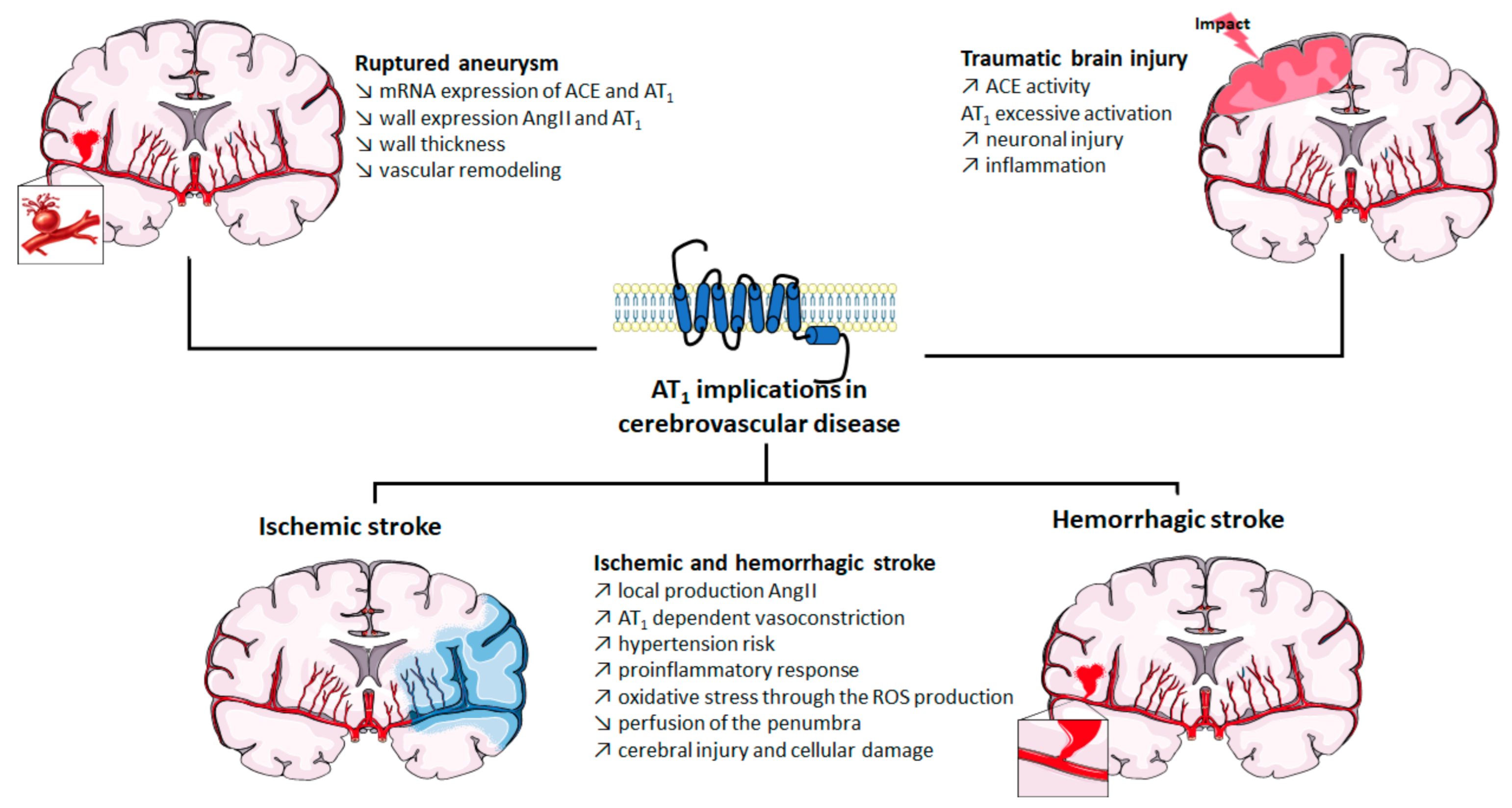
2. Implication of AT1 in Cerebrovascular Disease
2.1. Role of AT1 Activation in Strokes
2.2. Role of AT1 Activation in Cerebral Aneurysm
2.3. Role of AT1 Activation in Traumatic Brain Injury
2.4. Role of AT1 Activation in Neurodegenerative Diseases
2.5. Limitations
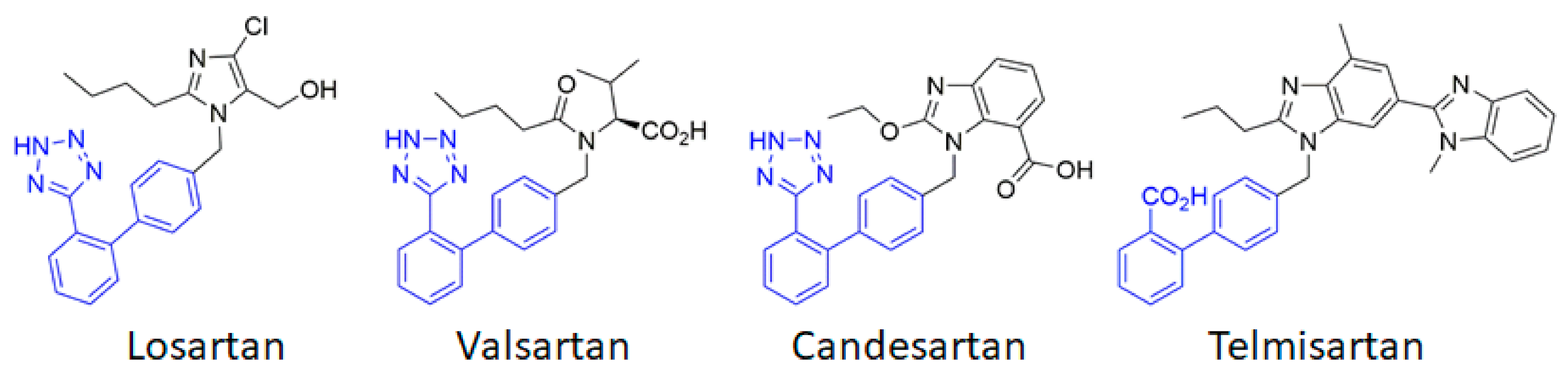
3. Blocking AT1 by ARBs in Cerebrovascular Disease: Success and Limitations
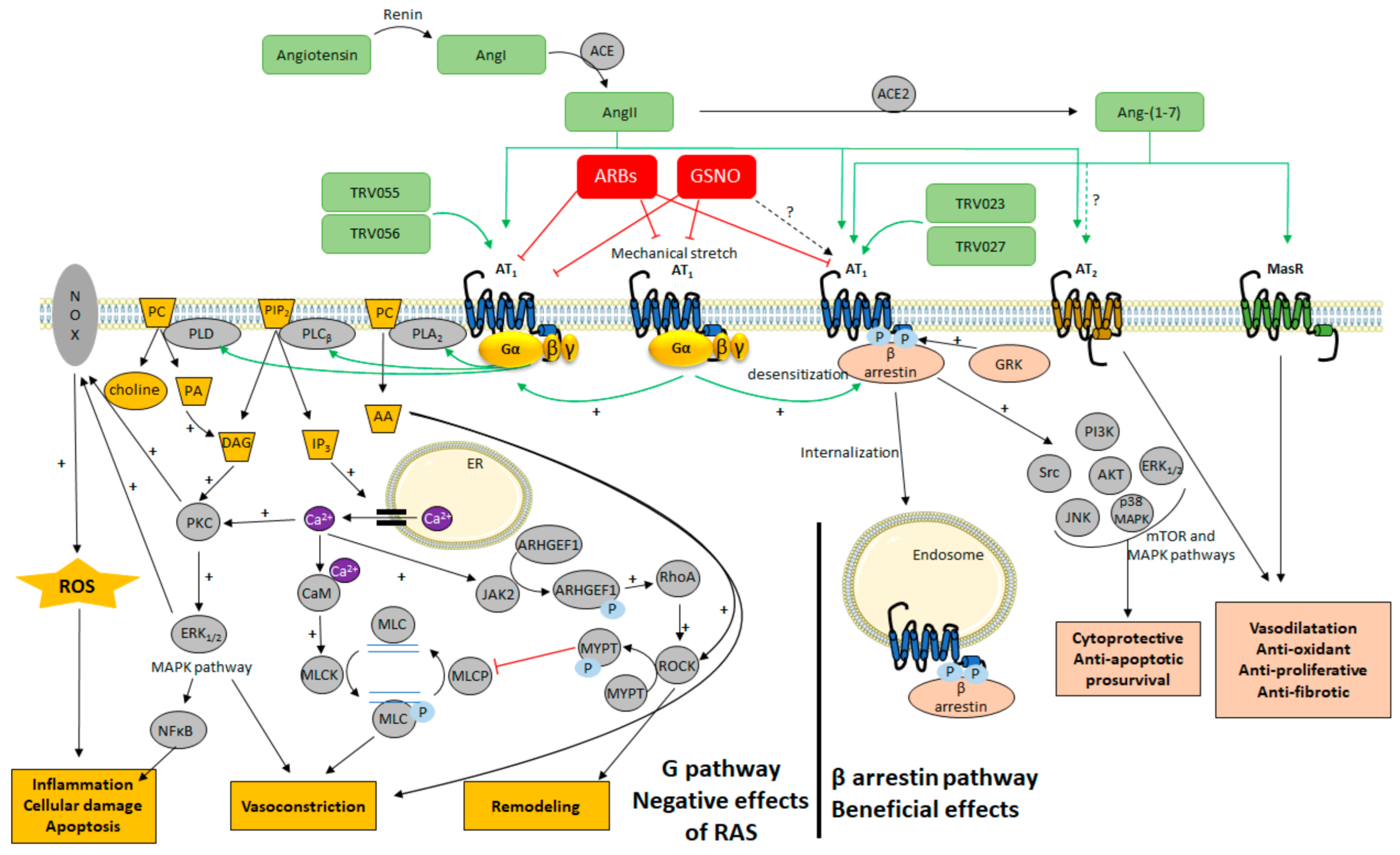
4. Alternative Ligands to Target AT1 Signaling
4.1. The Classical View of AT1 Signaling
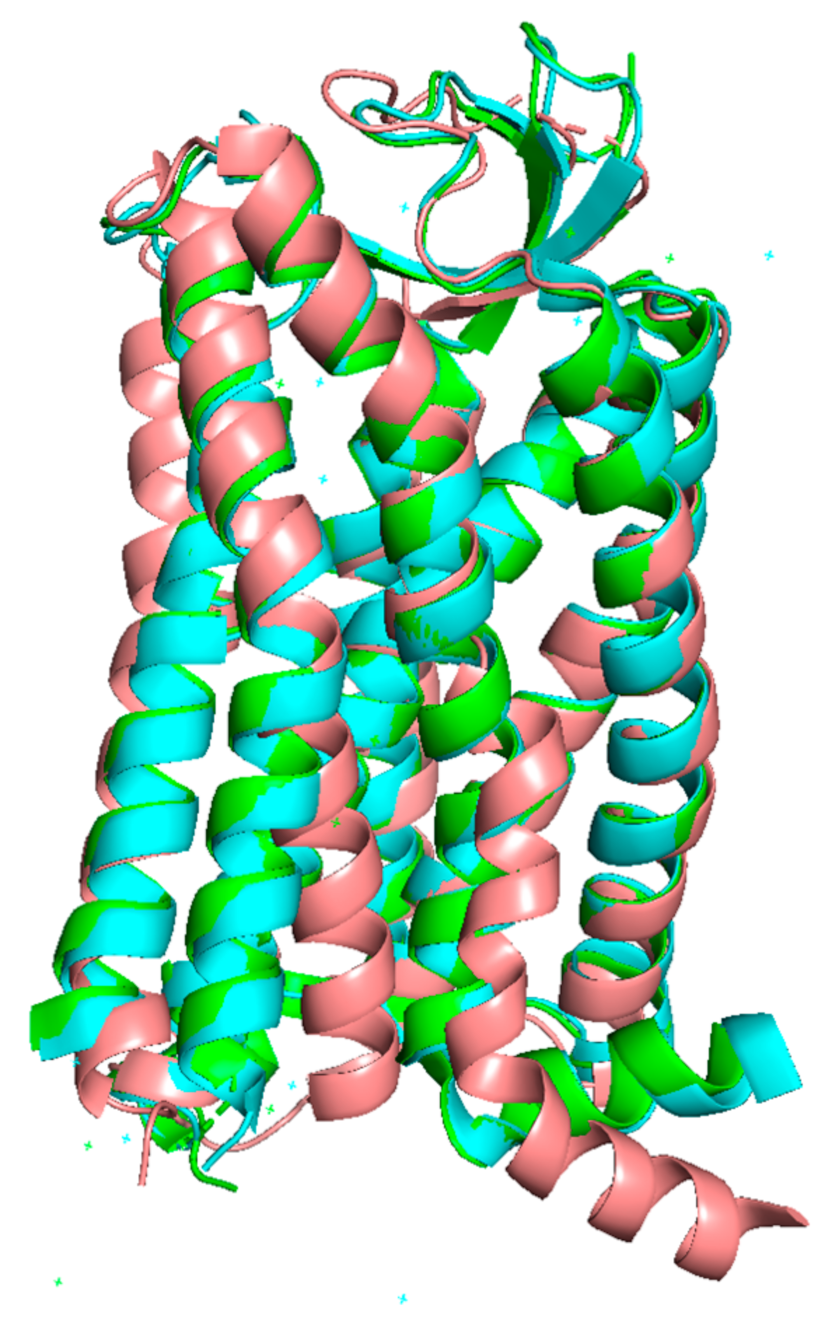
4.2. Targeting Different Active Conformations of AT1 with Biased Agonists
4.3. Targeting AT1 by Covalent S-Nitrosation
5. Promoting the β-Arrestin Pathway of AT1 to Treat Cerebrovascular Disease
5.1. Biased Agonists towards AT1/β-Arrestins in Cardiovascular Diseases
5.2. Biased Agonists towards AT1/β-Arrestins in Cerebrovascular Diseases
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| AKT | protein kinase B |
| Ang II | angiotensin II |
| ARB | AT1 receptor blocker |
| AT1 | angiotensin II type 1 receptor |
| AT2 | angiotensin II type 2 receptor; |
| GPCR | G protein-coupled receptor |
| GRKs | G protein-coupled receptor kinases |
| GSNO | S-nitrosoglutathione |
| MAPK | mitogen-activated protein kinases |
| MCAo | middle cerebral artery occlusion |
| PKC | protein kinase C |
| RAS | renin–angiotensin system |
| ROS | reactive oxygen species |
| SNP | sodium nitroprusside |
| TBI | traumatic brain injury |
| TM (1 to 7) | transmembrane domain (1 to 7); |
| VSMC | vascular smooth muscle cells |
References
- Mukoyama, M.; Nakajima, M.; Horiuchi, M.; Sasamura, H.; Pratt, R.E.; Dzau, V.J. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J. Biol. Chem. 1993, 268, 24539–24542. [Google Scholar] [CrossRef]
- Kambayashi, Y.; Bardhan, S.; Takahashi, K.; Tsuzuki, S.; Inui, H.; Hamakubo, T.; Inagami, T. Molecular cloning of a novel angiotensin II receptor isoform involved in phosphotyrosine phosphatase inhibition. J. Biol. Chem. 1993, 268, 24543–24546. [Google Scholar] [CrossRef]
- Inagami, T.; Iwai, N.; Sasaki, K.; Yamano, Y.; Bardhan, S.; Chaki, S.; Guo, D.F.; Furuta, H.; Ohyama, K.; Kambayashi, Y.; et al. Cloning, Expression and Regulation of Angiotensin II Receptors. Eur. Hear. J. 1994, 15, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 Receptor Signaling Pathways Mediating Physiological and Pathogenic Actions of Angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef]
- Kawai, T.; Forrester, S.J.; O’Brien, S.; Baggett, A.; Rizzo, V.; Eguchi, S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017, 125, 4–13. [Google Scholar] [CrossRef]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International Union of Pharmacology. XXIII. The Angiotensin II Receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The Intrarenal Renin-Angiotensin System: From Physiology to the Pathobiology of Hypertension and Kidney Disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef]
- Coble, J.P.; Grobe, J.L.; Johnson, A.K.; Sigmund, C.D. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: Importance of the subfornical organ. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R238–R249. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Saavedra, J.M. Characterization of AT2 angiotensin II receptors in rat anterior cerebral arteries. Am. J. Physiol. Circ. Physiol. 1991, 261, H667–H670. [Google Scholar] [CrossRef]
- Touyz, R.M.; Schiffrin, E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000, 52, 639–672. [Google Scholar]
- Dimitropoulou, C.; White, R.E.; Fuchs, L.; Zhang, H.; Catravas, J.D.; Carrier, G.O. Angiotensin II Relaxes Microvessels Via the AT 2 Receptor and Ca 2+ -Activated K + (BK Ca ) Channels. Hypertension 2001, 37, 301–307. [Google Scholar] [CrossRef]
- Enzo, R.P. The angiotensin II type 2 (AT2) receptor: An enigmatic seven transmembrane receptor. Front. Biosci. 2009, 14, 958–972. [Google Scholar] [CrossRef]
- Matavelli, L.C.; Siragy, H.M. AT2 Receptor Activities and Pathophysiological Implications. J. Cardiovasc. Pharmacol. 2015, 65, 226–232. [Google Scholar] [CrossRef]
- Carey, R.M. AT2Receptors: Potential Therapeutic Targets for Hypertension. Am. J. Hypertens. 2016, 30, 339–347. [Google Scholar] [CrossRef]
- Povlsen, A.L.; Grimm, D.; Wehland, M.; Infanger, M.; Krüger, M. The Vasoactive Mas Receptor in Essential Hypertension. J. Clin. Med. 2020, 9, 267. [Google Scholar] [CrossRef]
- Dupuis, F.; Atkinson, J.; Limiñana, P.; Chillon, J.-M. Captopril improves cerebrovascular structure and function in old hypertensive rats. Br. J. Pharmacol. 2005, 144, 349–356. [Google Scholar] [CrossRef]
- Dupuis, F.; Atkinson, J.; Limiñana, P.; Chillon, J.-M. Comparative effects of the angiotensin II receptor blocker, telmisartan, and the angiotensin-converting enzyme inhibitor, ramipril, on cerebrovascular structure in spontaneously hypertensive rats. J. Hypertens. 2005, 23, 1061–1066. [Google Scholar] [CrossRef]
- Vincent, J.-M.; Kwan, Y.W.; Chan, S.-L.; Perrin-Sarrado, C.; Atkinson, J.; Chillon, J.-M. Constrictor and Dilator Effects of Angiotensin II on Cerebral Arterioles. Stroke 2005, 36, 2691–2695. [Google Scholar] [CrossRef]
- Kalra, J.; Prakash, A.; Kumar, P.; Majeed, A.B.A. Cerebroprotective effects of RAS inhibitors: Beyond their cardio-renal actions. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 459–468. [Google Scholar] [CrossRef]
- Gironacci, M.; Vicario, A.; Cerezo, G.; Silva, M.G. The depressor axis of the renin–angiotensin system and brain disorders: A translational approach. Clin. Sci. 2018, 132, 1021–1038. [Google Scholar] [CrossRef]
- Feigin, V.L.; Forouzanfar, M.H.; Krishnamurthi, R.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.; Truelsen, T.; et al. Global and regional burden of stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet 2014, 383, 245–255. [Google Scholar] [CrossRef]
- Peisker, T.; Koznar, B.; Stetkarova, I.; Widimsky, P. Acute stroke therapy: A review. Trends Cardiovasc. Med. 2017, 27, 59–66. [Google Scholar] [CrossRef]
- Näveri, L.; Strömberg, C.; Saavedra, J.M. Angiotensin II AT, receptor mediated contraction of the perfused rat cerebral artery. NeuroReport 1994, 5, 2278–2280. [Google Scholar] [CrossRef]
- Stenman, E.; Edvinsson, L. Cerebral Ischemia Enhances Vascular Angiotensin AT 1 Receptor–Mediated Contraction in Rats. Stroke 2004, 35, 970–974. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kagiyama, T.; Kagiyama, S.; Phillips, M.I. Expression of angiotensin type 1 and 2 receptors in brain after transient middle cerebral artery occlusion in rats. Regul. Pept. 2003, 110, 241–247. [Google Scholar] [CrossRef]
- Walther, T.; Olah, L.; Harms, C.; Maul, B.; Bader, M.; Hörtnagl, H.; Schultheiss, H.-P.; Mies, G. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 2001, 16, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Cerebral Arteriolar Structure in Mice Overexpressing Human Renin and Angiotensinogen. Hypertension 2003, 41, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, F.; Vincent, J.-M.; Limiñana, P.; Chillon, J.-M.; Capdeville-Atkinson, C.; Atkinson, J. Effects of suboptimal doses of the AT1 receptor blocker, telmisartan, with the angiotensin-converting enzyme inhibitor, ramipril, on cerebral arterioles in spontaneously hypertensive rat. J. Hypertens. 2010, 28, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Chillon, J.-M.; Baumbach, G. Effects of an Angiotensin-Converting Enzyme Inhibitor and a β-Blocker on Cerebral Arterioles in Rats. Hypertension 1999, 33, 856–861. [Google Scholar] [CrossRef][Green Version]
- Zhang, M.; Mao, Y.; Ramirez, S.; Tuma, R.; Chabrashvili, T. Angiotensin II induced cerebral microvascular inflammation and increased blood–brain barrier permeability via oxidative stress. Neuroscience 2010, 171, 852–858. [Google Scholar] [CrossRef]
- Kazama, K.; Anrather, J.; Zhou, P.; Girouard, H.; Frys, K.; Milner, T.A.; Iadecola, C. Angiotensin II Impairs Neurovascular Coupling in Neocortex Through NADPH Oxidase–Derived Radicals. Circ. Res. 2004, 95, 1019–1026. [Google Scholar] [CrossRef]
- De Silva, T.M.; Faraci, F.M. Effects of angiotensin II on the cerebral circulation: Role of oxidative stress. Front. Physiol. 2013, 3, 484. [Google Scholar] [CrossRef]
- Wanderer, S.; Grüter, B.E.; Strange, F.; Sivanrupan, S.; Di Santo, S.; Widmer, H.R.; Fandino, J.; Marbacher, S.; Andereggen, L. The Role of Sartans in the Treatment of Stroke and Subarachnoid Hemorrhage: A Narrative Review of Preclinical and Clinical Studies. Brain Sci. 2020, 10, 153. [Google Scholar] [CrossRef]
- Inci, S.; Spetzler, R.F. Intracranial aneurysms and arterial hypertension: A review and hypothesis. Surg. Neurol. 2000, 53, 530–542. [Google Scholar] [CrossRef]
- Daugherty, A.; Cassis, L. Angiotensin II and abdominal aortic aneurysms. Curr. Hypertens. Rep. 2004, 6, 442–446. [Google Scholar] [CrossRef]
- Daugherty, A.; Rateri, D.L.; Cassis, L.A. Role of the Renin-Angiotensin System in the Development of Abdominal Aortic Aneurysms in Animals and Humans. Ann. N. Y. Acad. Sci. 2006, 1085, 82–91. [Google Scholar] [CrossRef]
- Ohkuma, H.; Suzuki, S.; Fujita, S.; Nakamura, W. Role of a Decreased Expression of the Local Renin-Angiotensin System in the Etiology of Cerebral Aneurysms. Circulation 2003, 108, 785–787. [Google Scholar] [CrossRef]
- Nishimura, M.; Aoki, T.; Kataoka, H.; Ishibashi, R.; Miyake, T.; Takagi, Y.; Morishita, R.; Hashimoto, N. Role of angiotensin II type 1 receptor in cerebral aneurysm formation in rats. Int. J. Mol. Med. 2009, 24, 353–359. [Google Scholar] [CrossRef]
- Ishibashi, R.; Aoki, T.; Nishimura, M.; Miyamoto, S. Imidapril Inhibits Cerebral Aneurysm Formation in an Angiotensin-Converting Enzyme–Independent and Matrix Metalloproteinase-9–Dependent Manner. Neurosurgery 2012, 70, 722–730. [Google Scholar] [CrossRef]
- Tada, Y.; Wada, K.; Shimada, K.; Makino, H.; Liang, E.I.; Murakami, S.; Kudo, M.; Kitazato, K.T.; Nagahiro, S.; Hashimoto, T. Roles of Hypertension in the Rupture of Intracranial Aneurysms. Stroke 2014, 45, 579–586. [Google Scholar] [CrossRef]
- Kehoe, A.D.; Eleftheriou, K.I.; Heron, M.; Coats, T.J.; Montgomery, H. Angiotensin-converting enzyme genotype may predict survival following major trauma. Emerg. Med. J. 2008, 25, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Zhou, J.; Macova, M.; Imboden, H.; Saavedra, J.M. Angiotensin II AT 1 Receptor Blockade Reverses Pathological Hypertrophy and Inflammation in Brain Microvessels of Spontaneously Hypertensive Rats. Stroke 2004, 35, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Iwai, N.; Inagami, T. Identification of two subtypes in the rat type I angiotensin II receptor. FEBS Lett. 1992, 298, 257–260. [Google Scholar] [CrossRef]
- Yamasaki, E.; Thakore, P.; Krishnan, V.; Earley, S. Differential expression of angiotensin II type 1 receptor subtypes within the cerebral microvasculature. Am. J. Physiol. Circ. Physiol. 2020, 318, H461–H469. [Google Scholar] [CrossRef] [PubMed]
- Johren, O.; Saavedra, J.M. Expression of AT1A and AT1B angiotensin II receptor messenger RNA in forebrain of 2-wk-old rats. Am. J. Physiol. Metab. 1996, 271, E104–E112. [Google Scholar] [CrossRef]
- Villapol, S.; Balarezo, M.G.; Affram, K.; Saavedra, J.M.; Symes, A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 2015, 138, 3299–3315. [Google Scholar] [CrossRef]
- Villapol, S.; Saavedra, J.M. Neuroprotective Effects of Angiotensin Receptor Blockers. Am. J. Hypertens. 2015, 28, 289–299. [Google Scholar] [CrossRef]
- Benicky, J.; Hafko, R.; Sanchez-Lemus, E.; Aguilera, G.; Saavedra, J.M. Six Commercially Available Angiotensin II AT1 Receptor Antibodies are Non-specific. Cell. Mol. Neurobiol. 2012, 32, 1353–1365. [Google Scholar] [CrossRef]
- Herrera, M.; Sparks, M.A.; Alfonso-Pecchio, A.R.; Harrison-Bernard, L.M.; Coffman, T.M. Lack of Specificity of Commercial Antibodies Leads to Misidentification of Angiotensin Type 1 Receptor Protein. Hypertension 2013, 61, 253–258. [Google Scholar] [CrossRef]
- Bouressam, M.-L.; Lartaud, I.; Dupuis, F.; Lecat, S. No answer to the lack of specificity: Mouse monoclonal antibody targeting the angiotensin II type 1 receptor AT1 fails to recognize its target. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 883–889. [Google Scholar] [CrossRef]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50. [Google Scholar] [CrossRef]
- Zhou, J.; Pavel, J.; Macova, M.; Yu, Z.-X.; Imboden, H.; Ge, L.; Nishioku, T.; Dou, J.; Delgiacco, E.; Saavedra, J.M. AT 1 Receptor Blockade Regulates the Local Angiotensin II System in Cerebral Microvessels From Spontaneously Hypertensive Rats. Stroke 2006, 37, 1271–1276. [Google Scholar] [CrossRef]
- Lou, M.; Blume, A.; Zhao, Y.; Gohlke, P.; Deuschl, G.; Herdegen, T.; Culman, J. Sustained Blockade of Brain AT1 Receptors before and after Focal Cerebral Ischemia Alleviates Neurologic Deficits and Reduces Neuronal Injury, Apoptosis, and Inflammatory Responses in the Rat. Br. J. Pharmacol. 2004, 24, 536–547. [Google Scholar] [CrossRef]
- Nishimura, Y.; Ito, T.; Saavedra, J.M. Angiotensin II AT1Blockade Normalizes Cerebrovascular Autoregulation and Reduces Cerebral Ischemia in Spontaneously Hypertensive Rats. Stroke 2000, 31, 2478–2486. [Google Scholar] [CrossRef]
- Ito, T.; Yamakawa, H.; Bregonzio, C.; Terrón, J.A.; Falcón-Neri, A.; Saavedra, J.M. Protection Against Ischemia and Improvement of Cerebral Blood Flow in Genetically Hypertensive Rats by Chronic Pretreatment With an Angiotensin II AT 1 Antagonist. Stroke 2002, 33, 2297–2303. [Google Scholar] [CrossRef]
- Groth, W.; Blume, A.; Gohlke, P.; Unger, T.; Culman, J. Chronic pretreatment with candesartan improves recovery from focal cerebral ischaemia in rats. J. Hypertens. 2003, 21, 2175–2182. [Google Scholar] [CrossRef]
- Stier, C.T.; Adler, L.A.; Levine, S.; Chander, P.N. Stroke prevention by losartan in stroke-prone spontaneously hypertensive rats. J. Hypertens. 1993, 11, 37–42. [Google Scholar]
- Villapol, S.; Yaszemski, A.K.; Logan, T.T.; Sanchez-Lemus, E.; Saavedra, J.M.; Symes, A.J. Candesartan, an Angiotensin II AT1-Receptor Blocker and PPAR-γ Agonist, Reduces Lesion Volume and Improves Motor and Memory Function After Traumatic Brain Injury in Mice. Neuropsychopharmacology 2012, 37, 2817–2829. [Google Scholar] [CrossRef]
- Li, J.-M.; Mogi, M.; Iwanami, J.; Min, L.-J.; Tsukuda, K.; Sakata, A.; Fujita, T.; Iwai, M.; Horiuchi, M. Temporary Pretreatment With the Angiotensin II Type 1 Receptor Blocker, Valsartan, Prevents Ischemic Brain Damage Through an Increase in Capillary Density. Stroke 2008, 39, 2029–2036. [Google Scholar] [CrossRef]
- Arroja, M.M.C.; Reid, E.; McCabe, C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp. Transl. Stroke Med. 2016, 8, 1–14. [Google Scholar] [CrossRef]
- Hansson, L.; Lindholm, L.H.; Niskanen, L.; Lanke, J.; Hedner, T.; Niklason, A.; Luomanmäki, K.; Dahlöf, B.; de Faire, U.; Mörlin, C.; et al. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The Captopril Prevention Project (CAPPP) randomised trial. Lancet 1999, 353, 611–616. [Google Scholar] [CrossRef]
- Bosch, J.; Yusuf, S.; Pogue, J.; Sleight, P.; Lonn, E.; Rangoonwala, B.; Davies, R.; Ostergren, J.; Probstfield, J. Use of ramipril in preventing stroke: Double blind randomised trial. BMJ 2002, 324, 699. [Google Scholar] [CrossRef] [PubMed]
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6105 individuals with previous stroke or transient ischaemic attack. Lancet 2001, 358, 1033–1041. [Google Scholar] [CrossRef]
- Dahlöf, B.; Devereux, R.B.; Kjeldsen, S.E.; Julius, S.; Beevers, G.; de Faire, U.; Fyhrquist, F.; Ibsen, H.; Kristiansson, K.; Lederballe-Pedersen, O.; et al. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): A randomised trial against atenolol. Lancet 2002, 359, 995–1003. [Google Scholar] [CrossRef]
- Lithell, H.; Hansson, L.; Skoog, I.; Elmfeldt, D.; Hofman, A.; Olofsson, B.; Trenkwalder, P.; Zanchetti, A. The Study on Cognition and Prognosis in the Elderly (SCOPE). J. Hypertens. 2003, 21, 875–886. [Google Scholar] [CrossRef]
- Schrader, J.; Lüders, S.; Kulschewski, A.; Hammersen, F.; Plate, K.; Berger, J.; Zidek, W.; Dominiak, P.; Diener, H.C. The MOSES Study Group Morbidity and Mortality After Stroke, Eprosartan Compared with Nitrendipine for Secondary Prevention. Stroke 2005, 36, 1218–1224. [Google Scholar] [CrossRef]
- Yusuf, S.; Teo, K.; Anderson, C.S.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: A randomised controlled trial. Lancet 2008, 372, 1174–1183. [Google Scholar] [CrossRef]
- Yusuf, S.; Diener, H.-C.; Sacco, R.L.; Cotton, D.; Ôunpuu, S.; Lawton, W.A.; Palesch, Y.; Martin, R.H.; Albers, G.W.; Bath, P.; et al. Telmisartan to Prevent Recurrent Stroke and Cardiovascular Events. N. Engl. J. Med. 2008, 359, 1225–1237. [Google Scholar] [CrossRef]
- Schrader, J.; Lüders, S.; Kulschewski, A.; Berger, J.; Zidek, W.; Treib, J.; Einhäupl, K.; Diener, H.C.; Dominiak, P. The ACCESS Study. Stroke 2003, 34, 1699–1703. [Google Scholar] [CrossRef]
- Sandset, E.C.; Bath, P.; Boysen, G.; Jatuzis, D.; Kõrv, J.; Lüders, S.; Murray, G.D.; Richter, P.S.; Roine, R.O.; Terént, A.; et al. The angiotensin-receptor blocker candesartan for treatment of acute stroke (SCAST): A randomised, placebo-controlled, double-blind trial. Lancet 2011, 377, 741–750. [Google Scholar] [CrossRef]
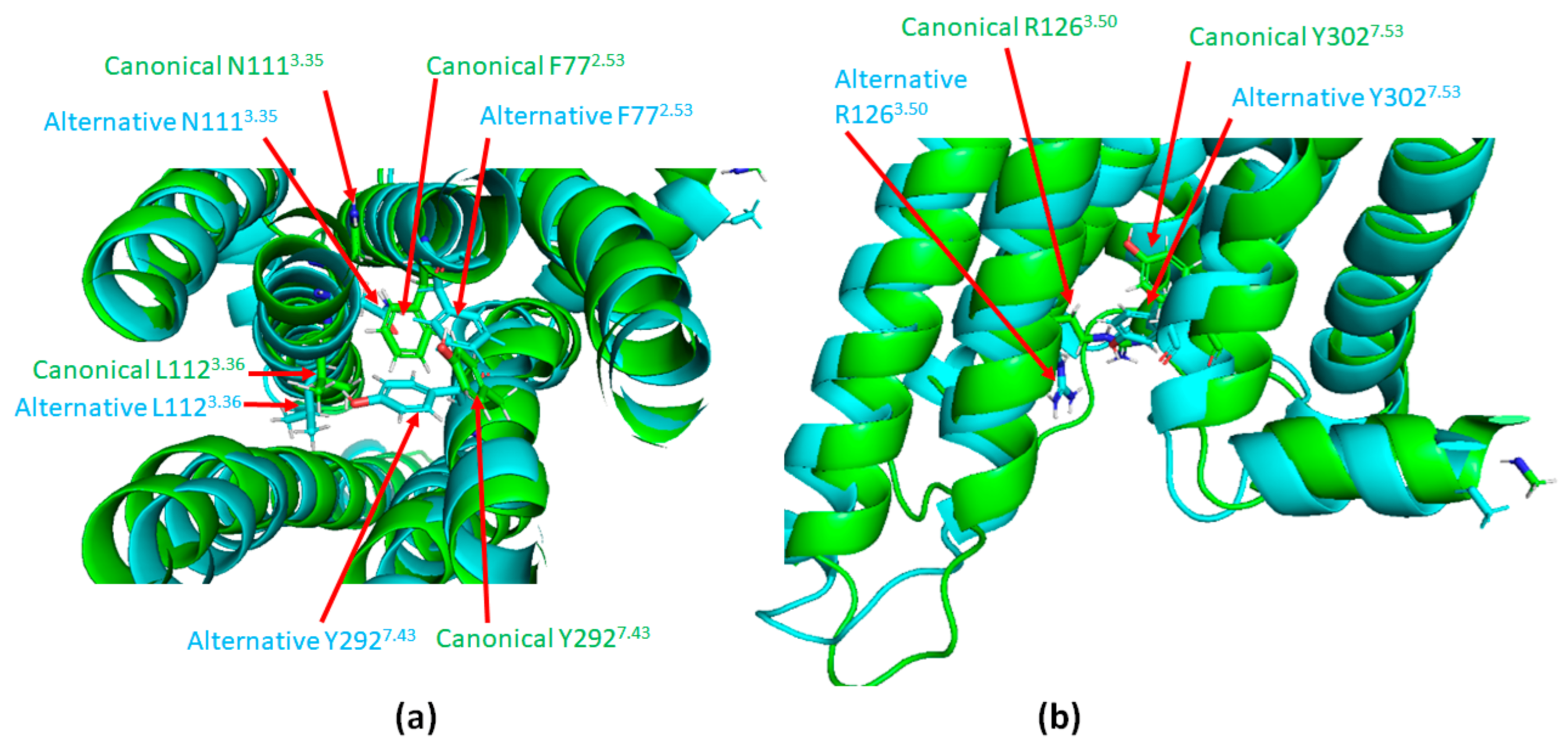
- Cabana, J.; Holleran, B.; Leduc, R.; Escher, E.; Guillemette, G.; Lavigne, P. Identification of Distinct Conformations of the Angiotensin-II Type 1 Receptor Associated with the Gq/11 Protein Pathway and the β-Arrestin Pathway Using Molecular Dynamics Simulations. J. Biol. Chem. 2015, 290, 15835–15854. [Google Scholar] [CrossRef]
- Guilluy, C.; Brégeon, J.; Toumaniantz, G.; Rolli-Derkinderen, M.; Retailleau, K.; Loufrani, L.; Henrion, D.; Scalbert, E.; Bril, A.; Torres, R.M.; et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 2010, 16, 183–190. [Google Scholar] [CrossRef]
- Miura, S.-I.; Zhang, J.; Boros, J.; Karnik, S.S. TM2-TM7 Interaction in Coupling Movement of Transmembrane Helices to Activation of the Angiotensin II Type-1 Receptor. J. Biol. Chem. 2003, 278, 3720–3725. [Google Scholar] [CrossRef]
- Galandrin, S.; Denis, C.; Boularan, C.; Marie, J.; M’Kadmi, C.; Pilette, C.; Dubroca, C.; Nicaise, Y.; Seguelas, M.-H.; N’Guyen, D.; et al. Cardioprotective Angiotensin-(1–7) Peptide Acts as a Natural-Biased Ligand at the Angiotensin II Type 1 Receptor. Hypertension 2016, 68, 1365–1374. [Google Scholar] [CrossRef]
- Namkung, Y.; LeGouill, C.; Kumar, S.; Cao, Y.; Teixeira, L.B.; Lukasheva, V.; Giubilaro, J.; Simões, S.C.; Longpré, J.-M.; Devost, D.; et al. Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signal. 2018, 11, eaat1631. [Google Scholar] [CrossRef]
- Connolly, A.; Holleran, B.J.; Simard, É.; Baillargeon, J.-P.; Lavigne, P.; Leduc, R. Interplay between intracellular loop 1 and helix VIII of the angiotensin II type 2 receptor controls its activation. Biochem. Pharmacol. 2019, 168, 330–338. [Google Scholar] [CrossRef]
- Wirth, A.; Benyó, Z.; Lukasova, M.; Leutgeb, B.; Wettschureck, N.; Gorbey, S.; Örsy, P.; Horváth, B.; Maser-Gluth, C.; Greiner, E.; et al. G12-G13–LARG–mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008, 14, 64–68. [Google Scholar] [CrossRef]
- Freeman, E.J.; Ruehr, M.L.; Dorman, R.V. ANG II-induced translocation of cytosolic PLA2 to the nucleus in vascular smooth muscle cells. Am. J. Physiol. Content 1998, 274, C282–C288. [Google Scholar] [CrossRef]
- Zafari, A.M.; Ushio-Fukai, M.; Minieri, C.A.; Akers, M.; Lassègue, B.; Griendling, K.K. Arachidonic Acid Metabolites Mediate Angiotensin II-Induced NADH/NADPH Oxidase Activity and Hypertrophy in Vascular Smooth Muscle Cells. Antioxid. Redox Signal. 1999, 1, 167–179. [Google Scholar] [CrossRef]
- Cat, A.N.D.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef]
- Lohse, M.; Andexinger, S.; Pitcher, J.; Trukawinski, S.; Codina, J.; Faure, J.; Caron, M.; Lefkowitz, R. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. J. Biol. Chem. 1992, 267, 8558–8564. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Reiter, E.; Ahn, S.; Shukla, A.; Lefkowitz, R.J. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef]
- Bologna, Z.; Teoh, J.-P.; Bayoumi, A.S.; Tang, Y.; Kim, I.-M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017, 25, 12–25. [Google Scholar] [CrossRef]
- Galandrin, S.; Oligny-Longpré, G.; Bouvier, M. The evasive nature of drug efficacy: Implications for drug discovery. Trends Pharmacol. Sci. 2007, 28, 423–430. [Google Scholar] [CrossRef]
- Roth, B.L.; Chuang, D.-M. Multiple mechanisms of serotonergic signal transduction. Life Sci. 1987, 41, 1051–1064. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, 457–469.e13. [Google Scholar] [CrossRef]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.-Y.; Gauthier, C.; Lee, M.-H.; Pani, B.; et al. Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018, 11, eaat7650. [Google Scholar] [CrossRef]
- Gurevich, V.V. Arrestin-mediated signaling: Is there a controversy? World J. Biol. Chem. 2018, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Turu, G.; Balla, A.; Hunyady, L. The Role of β-Arrestin Proteins in Organization of Signaling and Regulation of the AT1 Angiotensin Receptor. Front. Endocrinol. 2019, 10, 519. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Lefkowitz, R.J. β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007, 28, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.; Elgeti, M.; Hilger, D.; Latorraca, N.R.; Lerch, M.T.; Staus, D.P.; Dror, R.O.; Kobilka, B.K.; Hubbell, W.L.; Lefkowitz, R.J. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 2019, 176, 468–478.e11. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 2019, 176, 479–490.e12. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.-M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef] [PubMed]
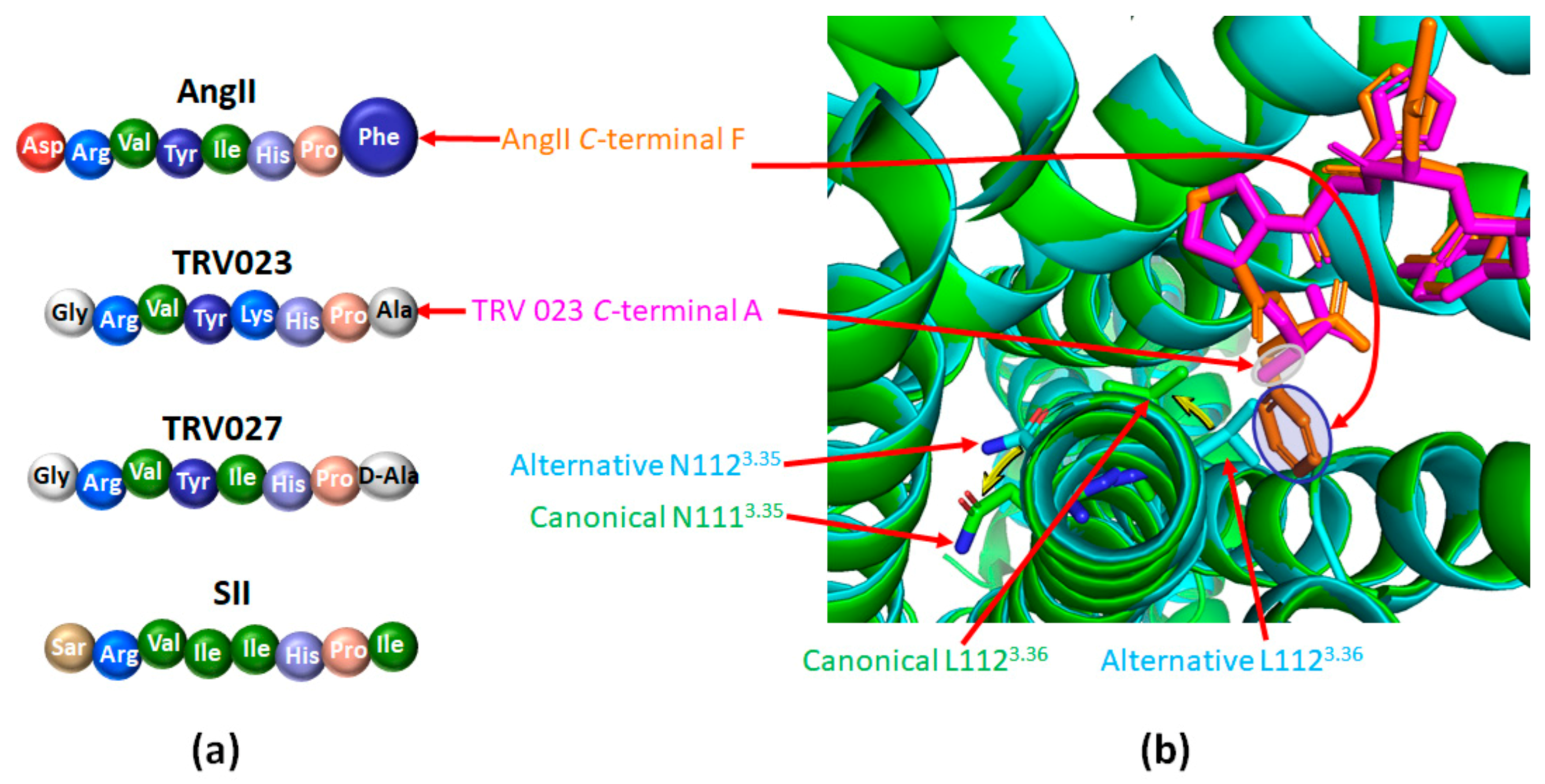
- Modestia, S.M.; De Sa, M.M.; Auger, E.; Trossini, G.H.G.; Krieger, J.E.; Rangel-Yagui, C.D.O.; Rangel-Yagui, C.O. Biased Agonist TRV027 Determinants in AT1R by Molecular Dynamics Simulations. J. Chem. Inf. Model. 2018, 59, 797–808. [Google Scholar] [CrossRef]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science 2020, 367, 881–887. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef]
- Valero, T.R.; Sturchler, E.; Jafferjee, M.; Rengo, G.; Magafa, V.; Cordopatis, P.; McDonald, P.; Koch, W.J.; Lymperopoulos, A. Structure–activity relationship study of angiotensin II analogs in terms of β -arrestin-dependent signaling to aldosterone production. Pharmacol. Res. Perspect. 2016, 4, e00226. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. β-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef]
- Galzi, J.-L.; Hachet-Haas, M.; Bonnet, D.; Daubeuf, F.; Lecat, S.; Hibert, M.; Haiech, J.; Frossard, N. Neutralizing endogenous chemokines with small molecules: Principles and potential therapeutic applications. Pharmacol. Ther. 2010, 126, 39–55. [Google Scholar] [CrossRef]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef]
- Ferro, A. S-Nitrosothiols as Nitric Oxide-Donors: Chemistry, Biology and Possible Future Therapeutic Applications. Curr. Med. Chem. 2004, 11, 2679–2690. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, B.; Zhang, L.; Zhang, Y.; Zhao, Y.; Guo, X.; Qiao, X.; Chen, C. SNObase, a database for S-nitrosation modification. Protein Cell 2012, 3, 929–933. [Google Scholar] [CrossRef][Green Version]
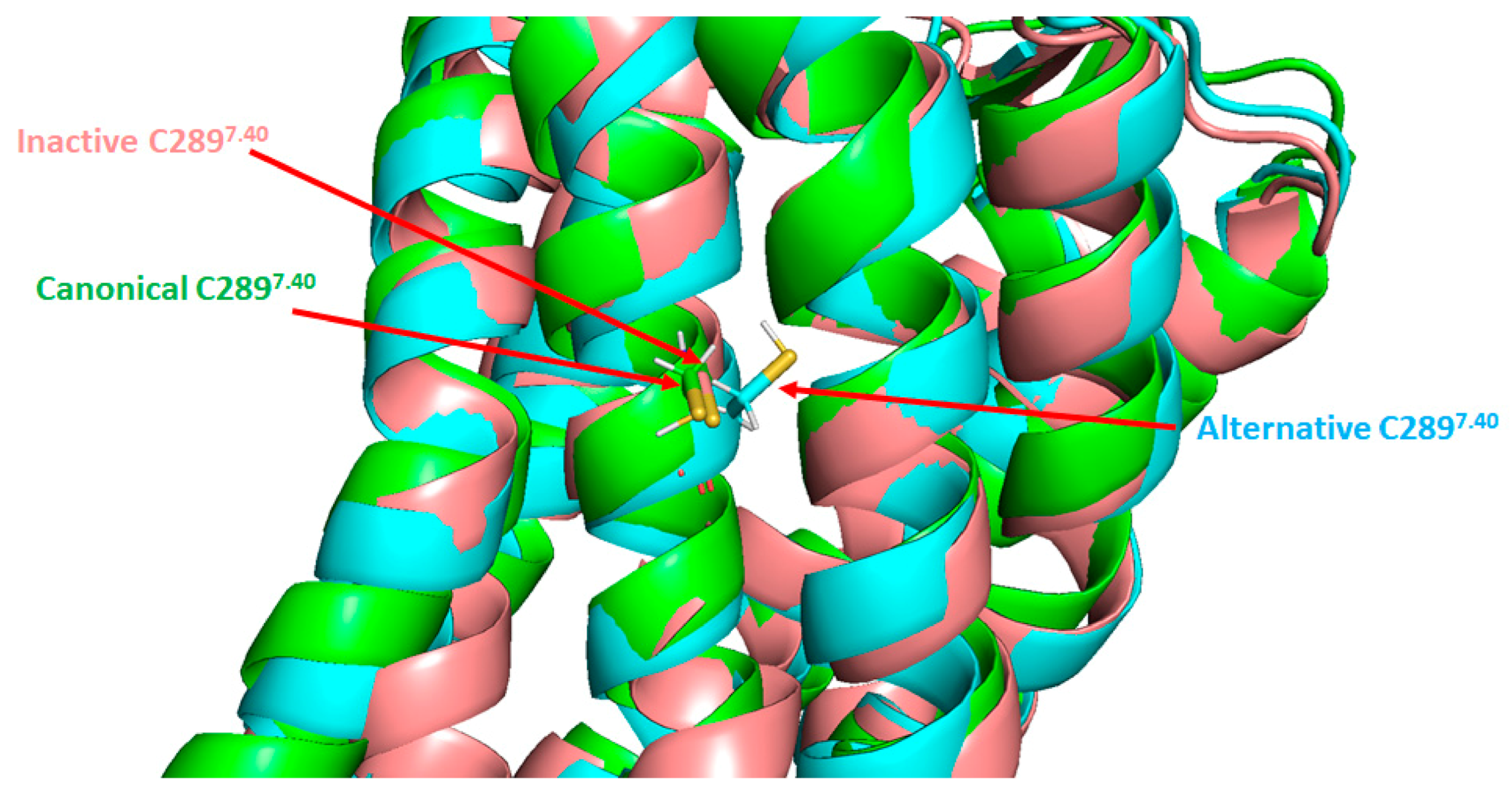
- Leclerc, P.C.; Lanctot, P.M.; Auger-Messier, M.; Escher, E.; LeDuc, R.; Guillemette, G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br. J. Pharmacol. 2006, 148, 306–313. [Google Scholar] [CrossRef]
- Bouressam, M.-L.; Lecat, S.; Raoul, A.; Gaucher, C.; Perrin-Sarrado, C.; Lartaud, I.; Dupuis, F. S-nitrosoglutathione inhibits cerebrovascular angiotensin II-dependent and -independent AT 1 receptor responses: A possible role of S -nitrosation. Br. J. Pharmacol. 2019, 176, 2049–2062. [Google Scholar] [CrossRef]
- Khan, M.; Sekhon, B.; Giri, S.; Jatana, M.; Gilg, A.G.; Ayasolla, K.; Elango, C.; Singh, A.K.; Singh, I. S-Nitrosoglutathione Reduces Inflammation and Protects Brain against Focal Cerebral Ischemia in a Rat Model of Experimental Stroke. Br. J. Pharmacol. 2005, 25, 177–192. [Google Scholar] [CrossRef]
- Khan, M.; Dhammu, T.S.; Sakakima, H.; Shunmugavel, A.; Gilg, A.G.; Singh, A.K.; Singh, I. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J. Neurochem. 2012, 123, 86–97. [Google Scholar] [CrossRef]
- Parent, M.; Boudier, A.; Perrin, J.; Vigneron, C.; Maincent, P.; Violle, N.; Bisson, J.-F.; Lartaud, I.; Dupuis, F. In Situ Microparticles Loaded with S-Nitrosoglutathione Protect from Stroke. PLoS ONE 2015, 10, e0144659. [Google Scholar] [CrossRef]
- Pinheiro, L.C.; Oliveira-Paula, G.H.; Ferreira, G.C.; de Paula, T.D.-C.; Duarte, D.A.; Costa-Neto, C.M.; Tanus-Santos, J.E. Oral nitrite treatment increases S-nitrosylation of vascular protein kinase C and attenuates the responses to angiotensin II. Redox Biol. 2021, 38, 101769. [Google Scholar] [CrossRef]
- Schnitzler, M.M.Y.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar] [CrossRef]
- Hansen, J.L.; Haunsø, S.; Brann, M.R.; Sheikh, S.P.; Weiner, D.M. Loss-of-Function Polymorphic Variants of the Human Angiotensin II Type 1 Receptor. Mol. Pharmacol. 2004, 65, 770–777. [Google Scholar] [CrossRef]
- Cao, Y.; Kumar, S.; Namkung, Y.; Gagnon, L.; Cho, A.; Laporte, S.A. Angiotensin II type 1 receptor variants alter endosomal receptor–β-arrestin complex stability and MAPK activation. J. Biol. Chem. 2020, 295, 13169–13180. [Google Scholar] [CrossRef]
- Thomas, W.G.; Qian, H. Arresting angiotensin type 1 receptors. Trends Endocrinol. Metab. 2003, 14, 130–136. [Google Scholar] [CrossRef]
- Gáborik, Z.; Hunyady, L. Intracellular trafficking of hormone receptors. Trends Endocrinol. Metab. 2004, 15, 286–293. [Google Scholar] [CrossRef]
- Christensen, G.L.; Kelstrup, C.D.; Lyngsø, C.; Sarwar, U.; Bøgebo, R.; Sheikh, S.P.; Gammeltoft, S.; Olsen, J.V.; Hansen, J.L. Quantitative Phosphoproteomics Dissection of Seven-transmembrane Receptor Signaling Using Full and Biased Agonists. Mol. Cell. Proteom. 2010, 9, 1540–1553. [Google Scholar] [CrossRef]
- Kendall, R.T.; Strungs, E.G.; Rachidi, S.M.; Lee, M.-H.; El-Shewy, H.M.; Luttrell, D.K.; Janech, M.G.; Luttrell, L.M. The β-Arrestin Pathway-selective Type 1A Angiotensin Receptor (AT1A) Agonist [Sar1,Ile4,Ile8]Angiotensin II Regulates a Robust G Protein-independent Signaling Network. J. Biol. Chem. 2011, 286, 19880–19891. [Google Scholar] [CrossRef]
- Pfeiffer, C.T.; Wang, J.; Paulo, J.A.; Jiang, X.; Gygi, S.P.; Rockman, H.A. Mapping Angiotensin II Type 1 Receptor-Biased Signaling Using Proximity Labeling and Proteomics Identifies Diverse Actions of Biased Agonists. J. Proteome Res. 2021, 20, 3256–3267. [Google Scholar] [CrossRef]
- Sanni, S.; Hansen, J.; Bonde, M.; Speerschneider, T.; Christensen, G.L.; Munk, S.; Gammeltoft, S.; Hansen, J. β-Arrestin 1 and 2 stabilize the angiotensin II type I receptor in distinct high-affinity conformations. Br. J. Pharmacol. 2010, 161, 150–161. [Google Scholar] [CrossRef]
- Srivastava, A.; Gupta, B.; Gupta, C.; Shukla, A.K. Emerging Functional Divergence of β-Arrestin Isoforms in GPCR Function. Trends Endocrinol. Metab. 2015, 26, 628–642. [Google Scholar] [CrossRef] [PubMed]
- McCrink, K.A.; Maning, J.; Vu, A.; Jafferjee, M.; Marrero, C.; Brill, A.; Bathgate-Siryk, A.; Dabul, S.; Koch, W.J.; Lymperopoulos, A. β-Arrestin2 Improves Post–Myocardial Infarction Heart Failure via Sarco(endo)plasmic Reticulum Ca 2+ -ATPase–Dependent Positive Inotropy in Cardiomyocytes. Hypertension 2017, 70, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Vines, C.M.; Cimino, D.F.; Prossnitz, E.R. Arrestins Block G Protein-coupled Receptor-mediated Apoptosis. J. Biol. Chem. 2004, 279, 24578–24584. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Rajagopal, K.; Whalen, E.J. New Roles for β-Arrestins in Cell Signaling: Not Just for Seven-Transmembrane Receptors. Mol. Cell 2006, 24, 643–652. [Google Scholar] [CrossRef]
- DeWire, S.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-Arrestins and Cell Signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein–Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef]
- Watari, K.; Nakaya, M.; Nishida, M.; Kim, K.-M.; Kurose, H. β-arrestin2 in Infiltrated Macrophages Inhibits Excessive Inflammation after Myocardial Infarction. PLoS ONE 2013, 8, e68351. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, L.; Song, Y.; Zhang, M.; Shan, D.; Liu, Y.; Fang, M.; Lv, F.; Xiao, R.-P.; Zhang, Y. β-arrestin 2 mediates cardiac ischemia-reperfusion injury via inhibiting GPCR-independent cell survival signalling. Cardiovasc. Res. 2017, 113, 1615–1626. [Google Scholar] [CrossRef]
- Rajagopal, K.; Whalen, E.J.; Violin, J.D.; Stiber, J.A.; Rosenberg, P.B.; Premont, R.; Coffman, T.M.; Rockman, H.A.; Lefkowitz, R.J. β-Arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 16284–16289. [Google Scholar] [CrossRef]
- Kim, K.-S.; Abraham, D.; Williams, B.; Violin, J.D.; Mao, L.; Rockman, H.A. β-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am. J. Physiol. Circ. Physiol. 2012, 303, H1001–H1010. [Google Scholar] [CrossRef]
- Violin, J.D.; DeWire, S.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively Engaging β-Arrestins at the Angiotensin II Type 1 Receptor Reduces Blood Pressure and Increases Cardiac Performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, G.; Lark, M.W.; Whalen, E.J.; Soergel, D.G.; Violin, J.D.; Burnett, J.C.; Jr, J.C.B. Cardiorenal Actions of TRV120027, a Novel ß-Arrestin–Biased Ligand at the Angiotensin II Type I Receptor, in Healthy and Heart Failure Canines. Circ. Hear. Fail. 2011, 4, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, G.; Soergel, D.G.; Violin, J.D.; Lark, M.W.; Burnett, J.C. TRV120027, a Novel β-Arrestin Biased Ligand at the Angiotensin II Type I Receptor, Unloads the Heart and Maintains Renal Function When Added to Furosemide in Experimental Heart Failure. Circ. Hear. Fail. 2012, 5, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Soergel, D.; Subach, R.A.; James, I.E.; Cowan, C.L.; Gowen, M.; Lark, M. Trvo27, a beta-arrestin biased ligand at the angiotensin 2 type 1 receptor, produces rapid, reversible changes in hemodynamics in patients with stable systolic heart failure. J. Am. Coll. Cardiol. 2013, 61, E683. [Google Scholar] [CrossRef][Green Version]
- Felker, G.M.; Butler, J.; Collins, S.P.; Cotter, G.; Davison, B.A.; Ezekowitz, J.A.; Filippatos, G.; Levy, P.D.; Metra, M.; Ponikowski, P.; et al. Heart Failure Therapeutics on the Basis of a Biased Ligand of the Angiotensin-2 Type 1 Receptor. JACC Hear. Fail. 2015, 3, 193–201. [Google Scholar] [CrossRef]
- Pang, P.S.; Butler, J.; Collins, S.P.; Cotter, G.; Davison, B.A.; Ezekowitz, J.A.; Filippatos, G.; Levy, P.D.; Metra, M.; Ponikowski, P.; et al. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: A randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur. Hear. J. 2017, 38, 2364–2373. [Google Scholar] [CrossRef]
- Rakesh, K.; Yoo, B.; Kim, I.-M.; Salazar, N.C.; Kim, K.-S.; Rockman, H.A. β-Arrestin-Biased Agonism of the Angiotensin Receptor Induced by Mechanical Stress. Sci. Signal. 2010, 3, ra46. [Google Scholar] [CrossRef]
- Monasky, M.M.; Taglieri, D.M.; Henze, M.; Warren, C.M.; Utter, M.S.; Soergel, D.G.; Violin, J.D.; Solaro, R.J. The β-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am. J. Physiol. Circ. Physiol. 2013, 305, H856–H866. [Google Scholar] [CrossRef]
- Tarigopula, M.; Davis, R.T.; Mungai, P.T.; Ryba, D.M.; Wieczorek, D.F.; Cowan, C.L.; Violin, J.D.; Wolska, B.M.; Solaro, R.J. Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc. Res. 2015, 107, 226–234. [Google Scholar] [CrossRef]
- Zhang, H.; Han, G.W.; Batyuk, A.; Ishchenko, A.; White, K.L.; Patel, N.; Sadybekov, A.; Zamlynny, B.; Rudd, M.T.; Hollenstein, K.; et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nat. Cell Biol. 2017, 544, 327–332. [Google Scholar] [CrossRef]
- Teixeira, L.B.; Parreiras-E-Silva, L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simões, S.C.; Costa, R.M.; Rodríguez, D.Y.; Ferreira, P.A.B.; Silva, C.A.A.; Abrao, E.P.; et al. Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Ferreira, A.J.; Santos, R.A.; Almeida, A.P. Angiotensin-(1-7): Cardioprotective Effect in Myocardial Ischemia/Reperfusion. Hypertension 2001, 38, 665–668. [Google Scholar] [CrossRef]
- Grobe, J.L.; Mecca, A.P.; Lingis, M.; Shenoy, V.; Bolton, T.A.; Machado, J.M.; Speth, R.C.; Raizada, M.K.; Katovich, M.J. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7). Am. J. Physiol. Circ. Physiol. 2007, 292, H736–H742. [Google Scholar] [CrossRef]
- Ferreira, A.J.; Jacoby, B.A.; Araújo, C.A.A.; Macedo, F.A.F.F.; Silva, G.A.B.; Almeida, A.P.; Caliari, M.; Santos, R.A.S. The nonpeptide angiotensin-(1–7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am. J. Physiol. Circ. Physiol. 2007, 292, H1113–H1119. [Google Scholar] [CrossRef]
- Mecca, A.P.; Regenhardt, R.; O’Connor, T.E.; Joseph, J.P.; Raizada, M.K.; Katovich, M.J.; Sumners, C. Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp. Physiol. 2011, 96, 1084–1096. [Google Scholar] [CrossRef]
- Regenhardt, R.; Bennion, D.; Sumners, C. Cerebroprotective action of angiotensin peptides in stroke. Clin. Sci. 2014, 126, 195–205. [Google Scholar] [CrossRef]
- Jiang, T.; Gao, L.; Guo, J.; Lu, J.; Wang, Y.; Zhang, Y. Suppressing inflammation by inhibiting the NF-κB pathway contributes to the neuroprotective effect of angiotensin-(1-7) in rats with permanent cerebral ischaemia. Br. J. Pharmacol. 2012, 167, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Turu, G.; Szidonya, L.; Gáborik, Z.; Buday, L.; Spät, A.; Clark, A.J.; Hunyady, L. Differential β-arrestin binding of AT1and AT2angiotensin receptors. FEBS Lett. 2005, 580, 41–45. [Google Scholar] [CrossRef]
- Villela, D.; Leonhardt, J.; Patel, N.; Joseph, J.; Kirsch, S.; Hallberg, A.; Unger, T.; Bader, M.; Santos, R.A.; Sumners, C.; et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: A complex liaison. Clin. Sci. 2014, 128, 227–234. [Google Scholar] [CrossRef]
- Lyngsø, C.; Erikstrup, N.; Hansen, J.L. Functional interactions between 7TM receptors in the Renin-Angiotensin System—Dimerization or crosstalk? Mol. Cell. Endocrinol. 2009, 302, 203–212. [Google Scholar] [CrossRef]
- Porrello, E.R.; Pfleger, K.; Seeber, R.M.; Qian, H.; Oro, C.; Abogadie, F.; Delbridge, L.M.; Thomas, W.G. Heteromerization of angiotensin receptors changes trafficking and arrestin recruitment profiles. Cell. Signal. 2011, 23, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Szalai, B.; Barkai, L.; Turu, G.; Szidonya, L.; Várnai, P.; Hunyady, L. Allosteric interactions within the AT1 angiotensin receptor homodimer: Role of the conserved DRY motif. Biochem. Pharmacol. 2012, 84, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Erol, I.; Salmas, R.E.; Aksoydan, B.; Kantarcioglu, I. Oligomerization and cooperativity in GPCRs from the perspective of the angiotensin AT1 and dopamine D2 receptors. Neurosci. Lett. 2019, 700, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, T.-Y.; Wei, K.; Guan, Y.-F.; Wang, X.; Xu, H.; Su, D.-F.; Pei, G.; Miao, C.-Y. ARRB1/β-arrestin-1 mediates neuroprotection through coordination of BECN1-dependent autophagy in cerebral ischemia. Autophagy 2014, 10, 1535–1548. [Google Scholar] [CrossRef]
- Wang, H.; Deng, Q.-W.; Peng, A.-N.; Xing, F.-L.; Zuo, L.; Li, S.; Gu, Z.-T.; Yan, F.-L. β-arrestin2 functions as a key regulator in the sympathetic-triggered immunodepression after stroke. J. Neuroinflamm. 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Kanki, H.; Sasaki, T.; Matsumura, S.; Yokawa, S.; Yukami, T.; Shimamura, M.; Sakaguchi, M.; Furuno, T.; Suzuki, T.; Mochizuki, H. β-arrestin-2 in PAR-1-biased signaling has a crucial role in endothelial function via PDGF-β in stroke. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delaitre, C.; Boisbrun, M.; Lecat, S.; Dupuis, F. Targeting the Angiotensin II Type 1 Receptor in Cerebrovascular Diseases: Biased Signaling Raises New Hopes. Int. J. Mol. Sci. 2021, 22, 6738. https://doi.org/10.3390/ijms22136738
Delaitre C, Boisbrun M, Lecat S, Dupuis F. Targeting the Angiotensin II Type 1 Receptor in Cerebrovascular Diseases: Biased Signaling Raises New Hopes. International Journal of Molecular Sciences. 2021; 22(13):6738. https://doi.org/10.3390/ijms22136738
Chicago/Turabian StyleDelaitre, Céline, Michel Boisbrun, Sandra Lecat, and François Dupuis. 2021. "Targeting the Angiotensin II Type 1 Receptor in Cerebrovascular Diseases: Biased Signaling Raises New Hopes" International Journal of Molecular Sciences 22, no. 13: 6738. https://doi.org/10.3390/ijms22136738
APA StyleDelaitre, C., Boisbrun, M., Lecat, S., & Dupuis, F. (2021). Targeting the Angiotensin II Type 1 Receptor in Cerebrovascular Diseases: Biased Signaling Raises New Hopes. International Journal of Molecular Sciences, 22(13), 6738. https://doi.org/10.3390/ijms22136738