
Studies of Conformational Changes of Tubulin Induced by Interaction with Kinesin Using Atomistic Molecular Dynamics Simulations


Abstract
:1. Introduction
2. Results
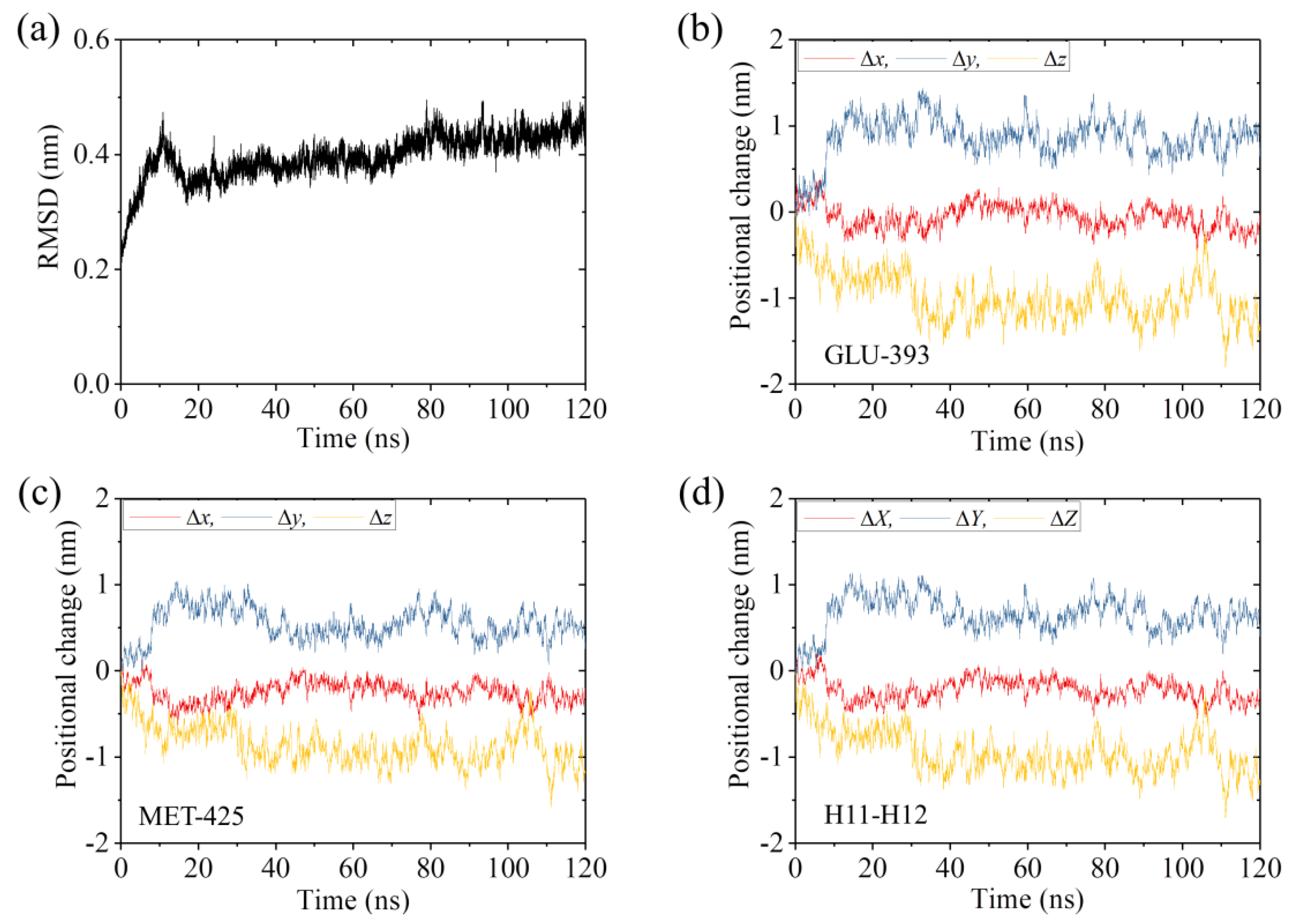
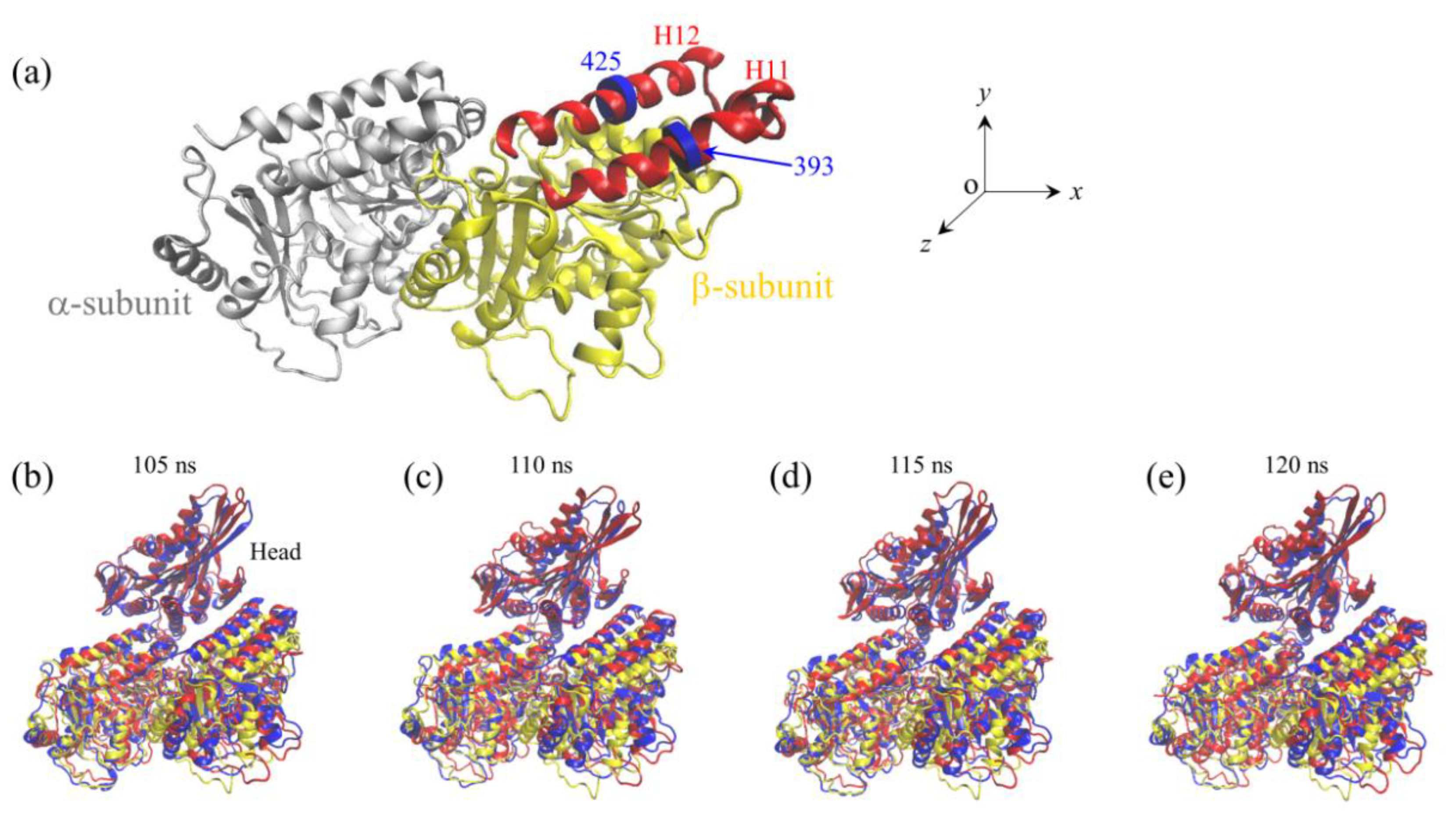
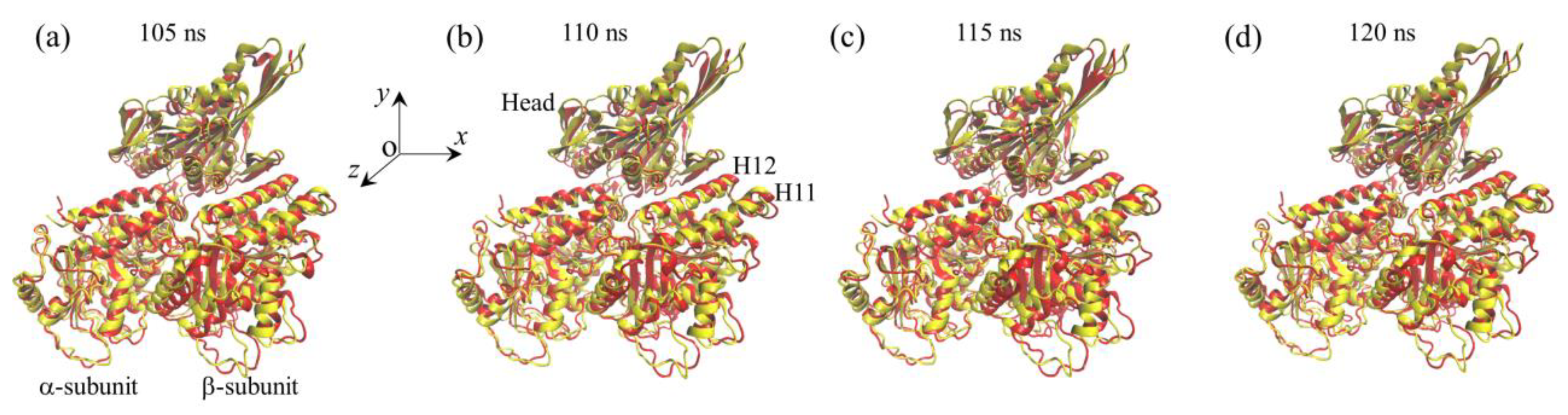
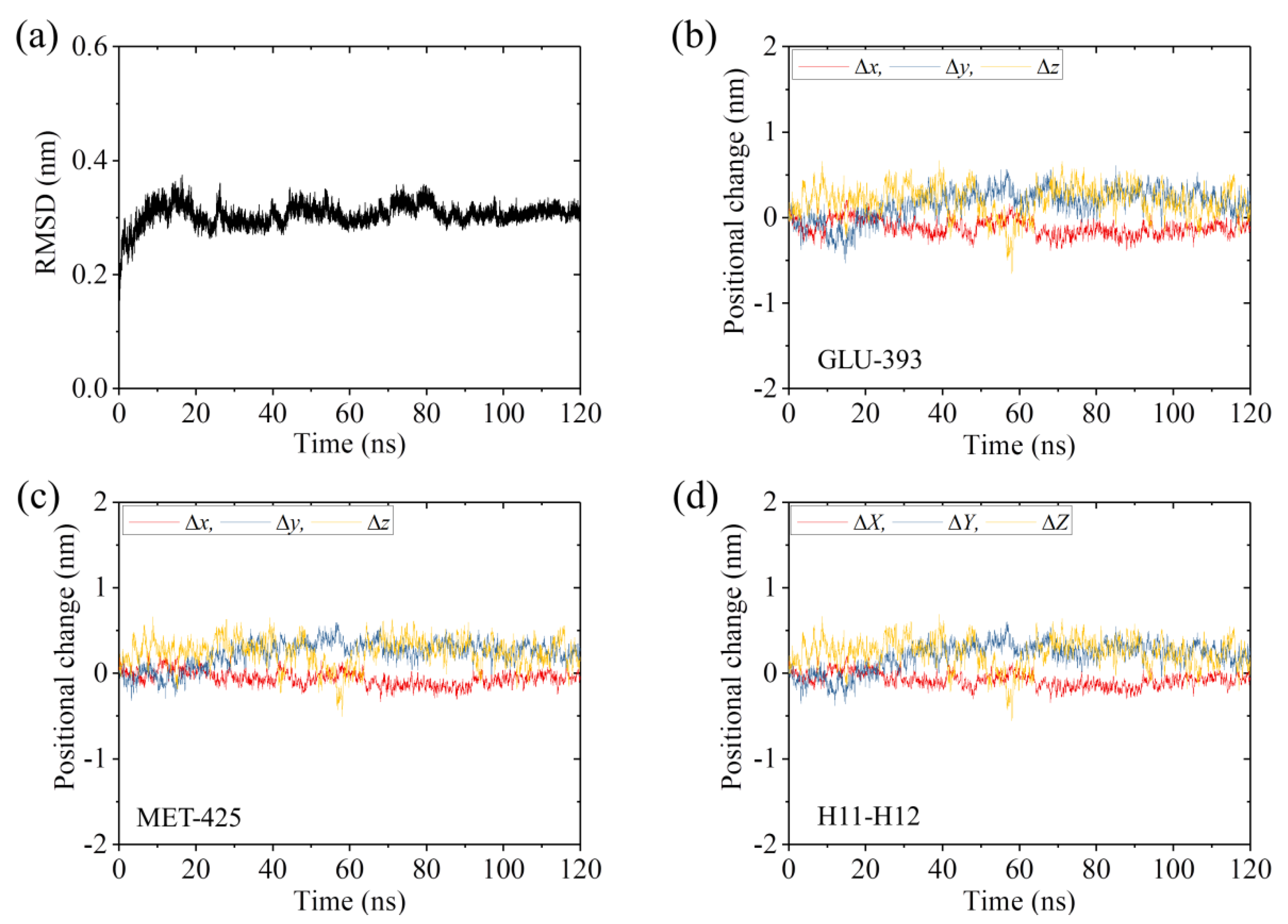
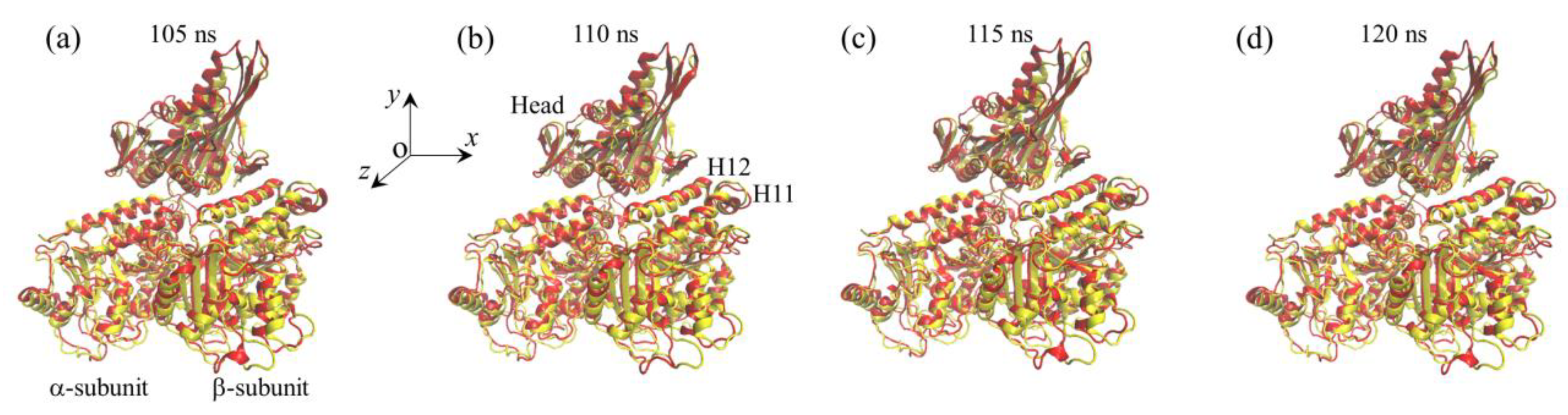
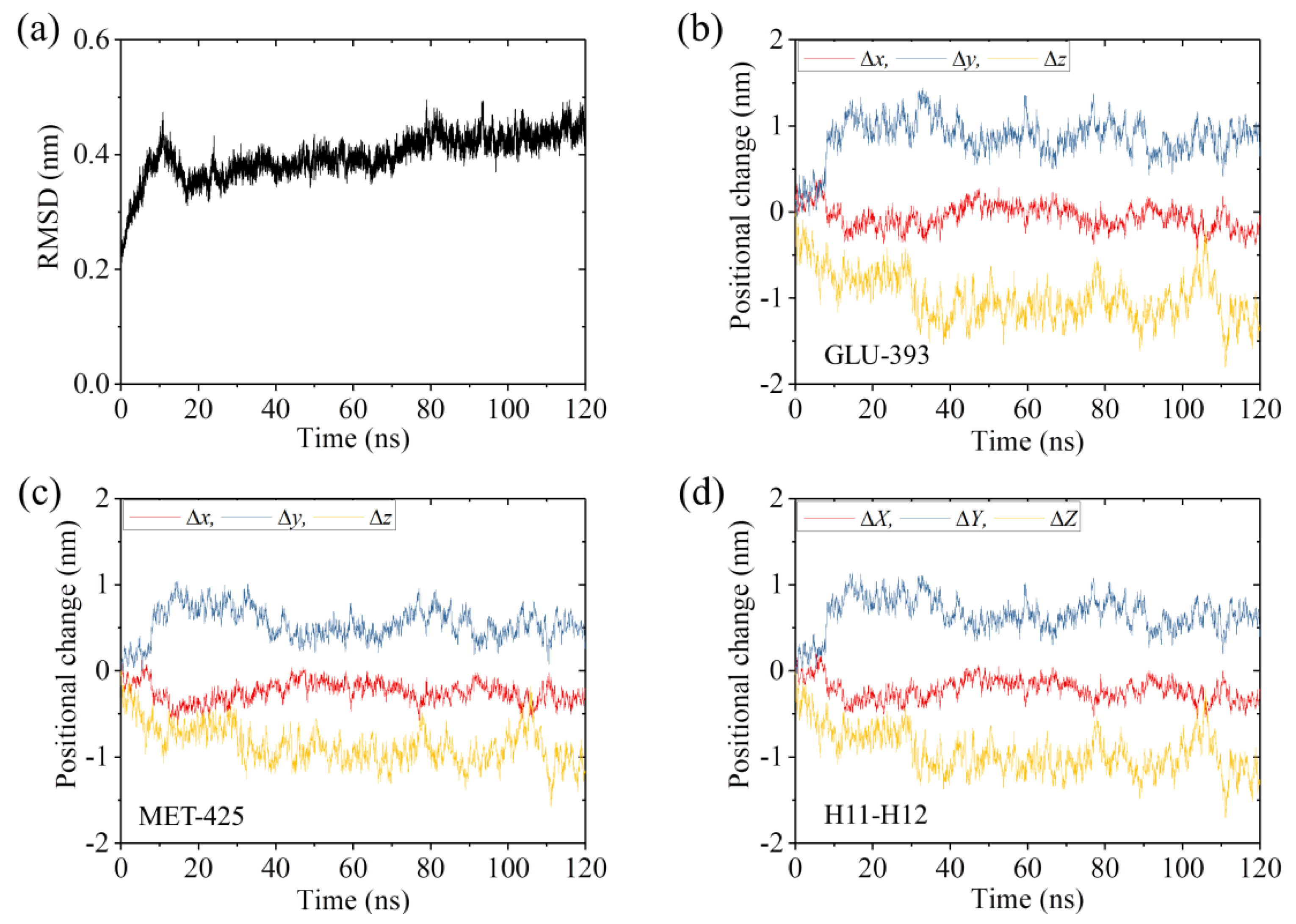
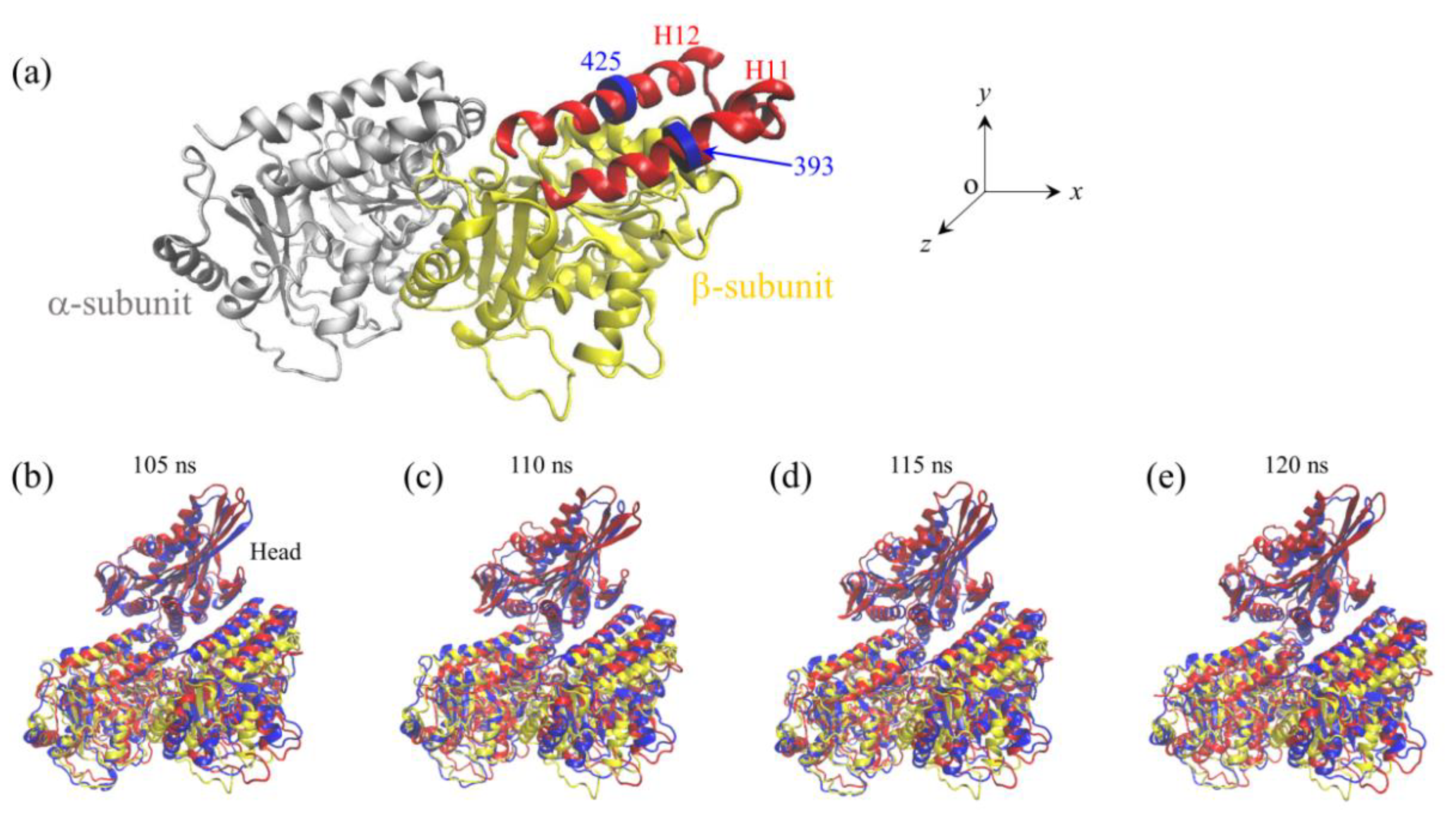
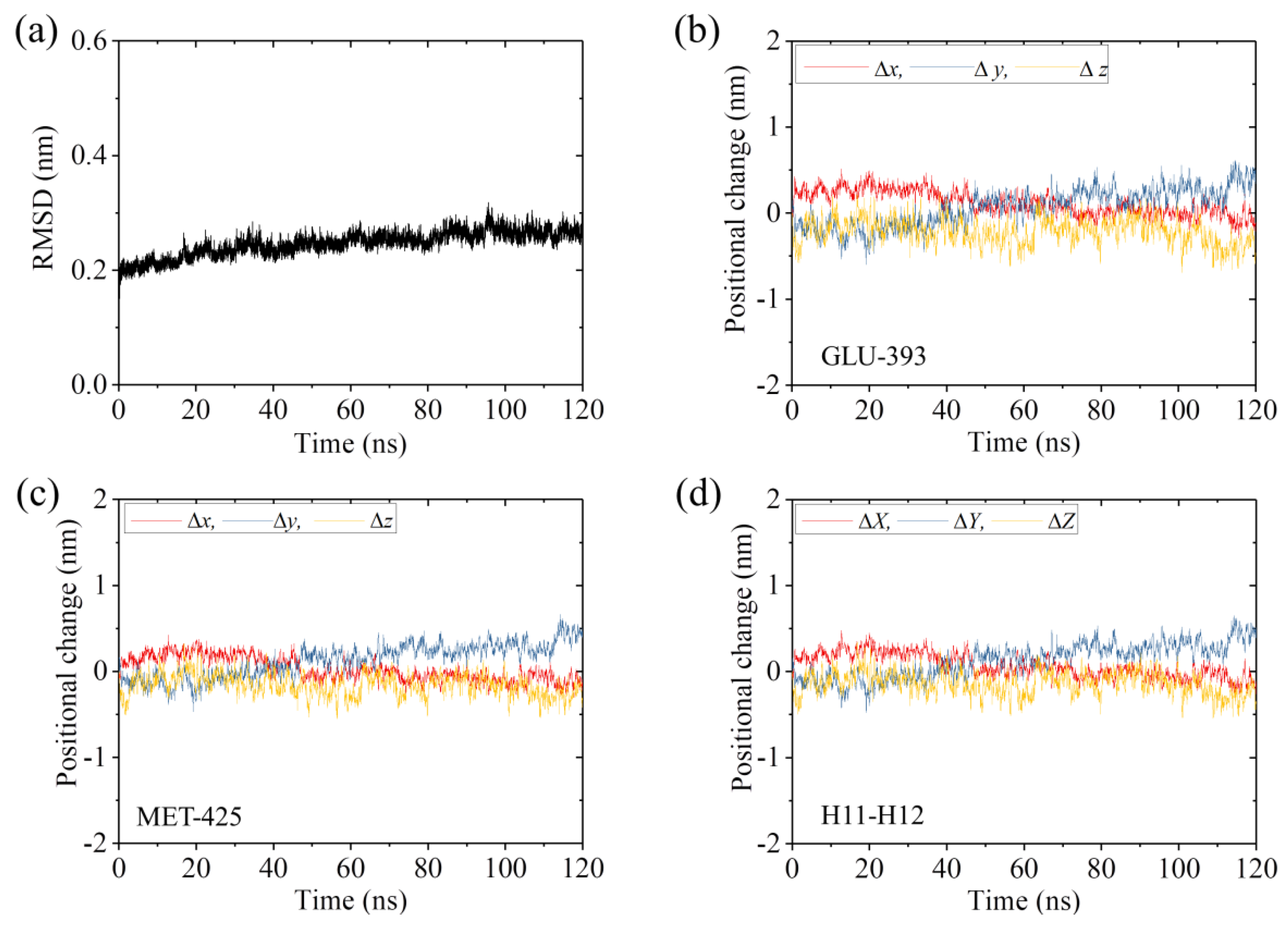
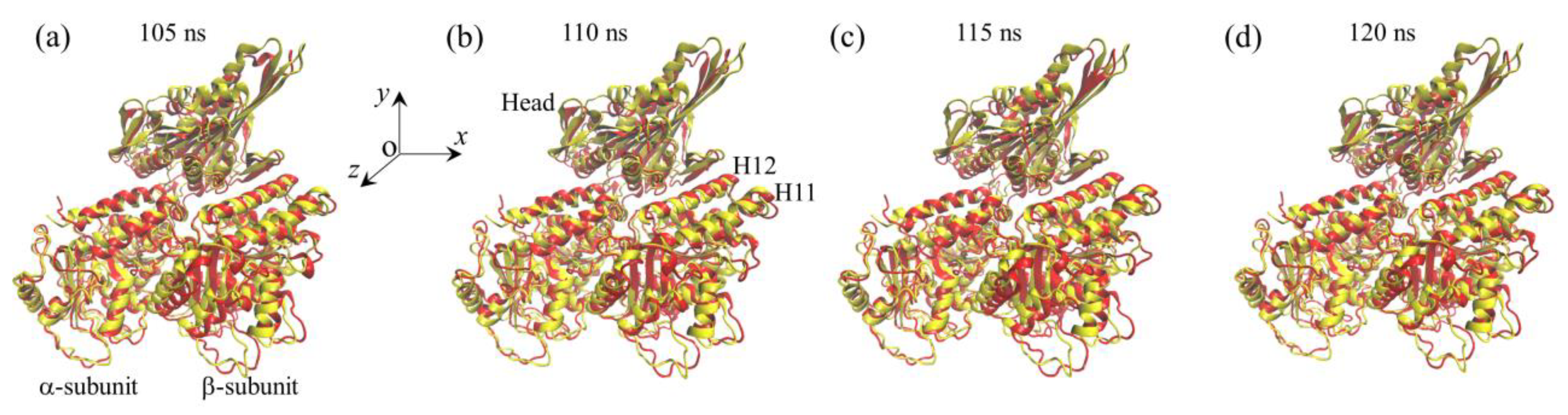
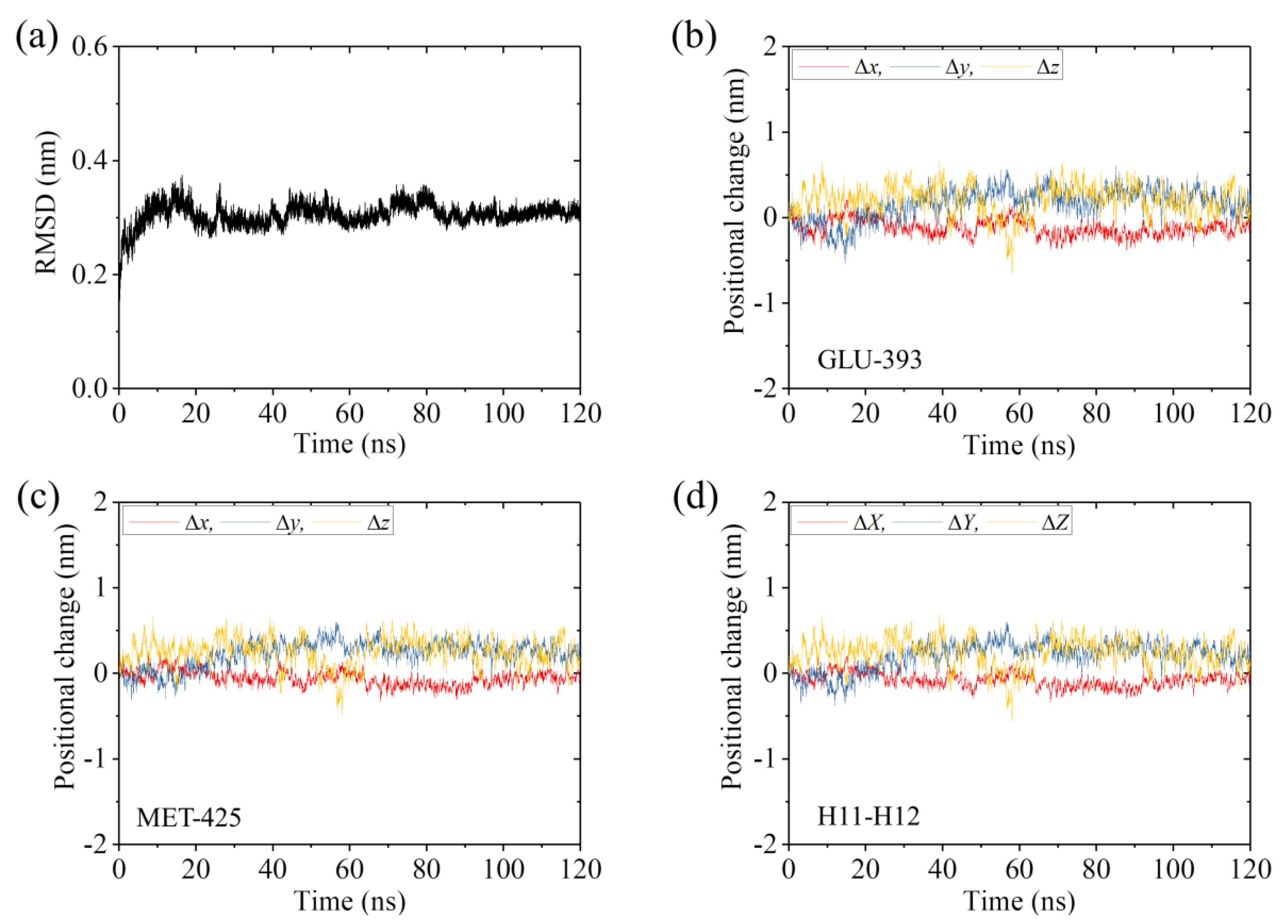
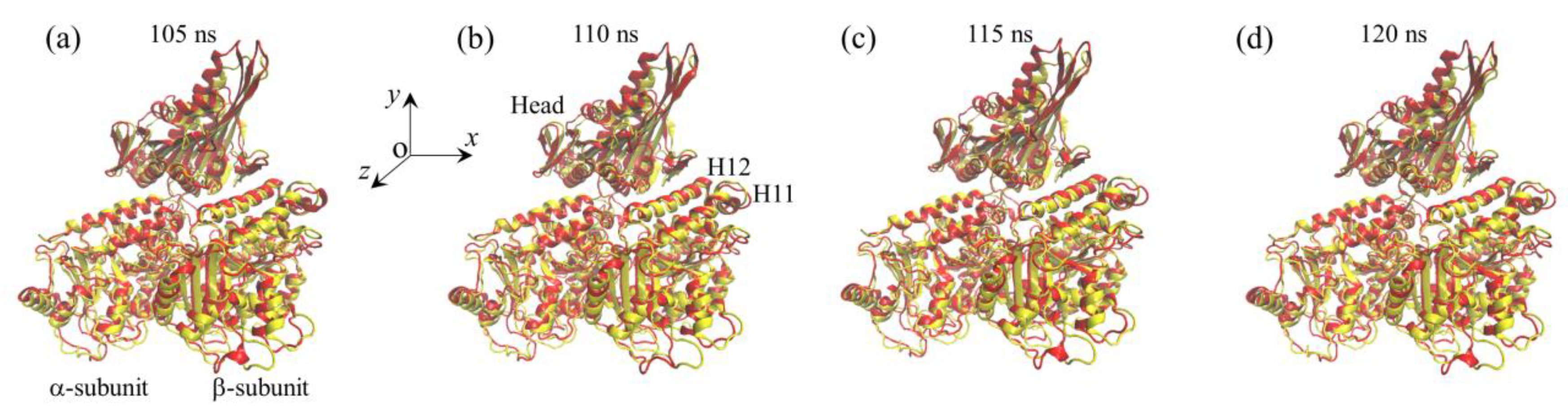
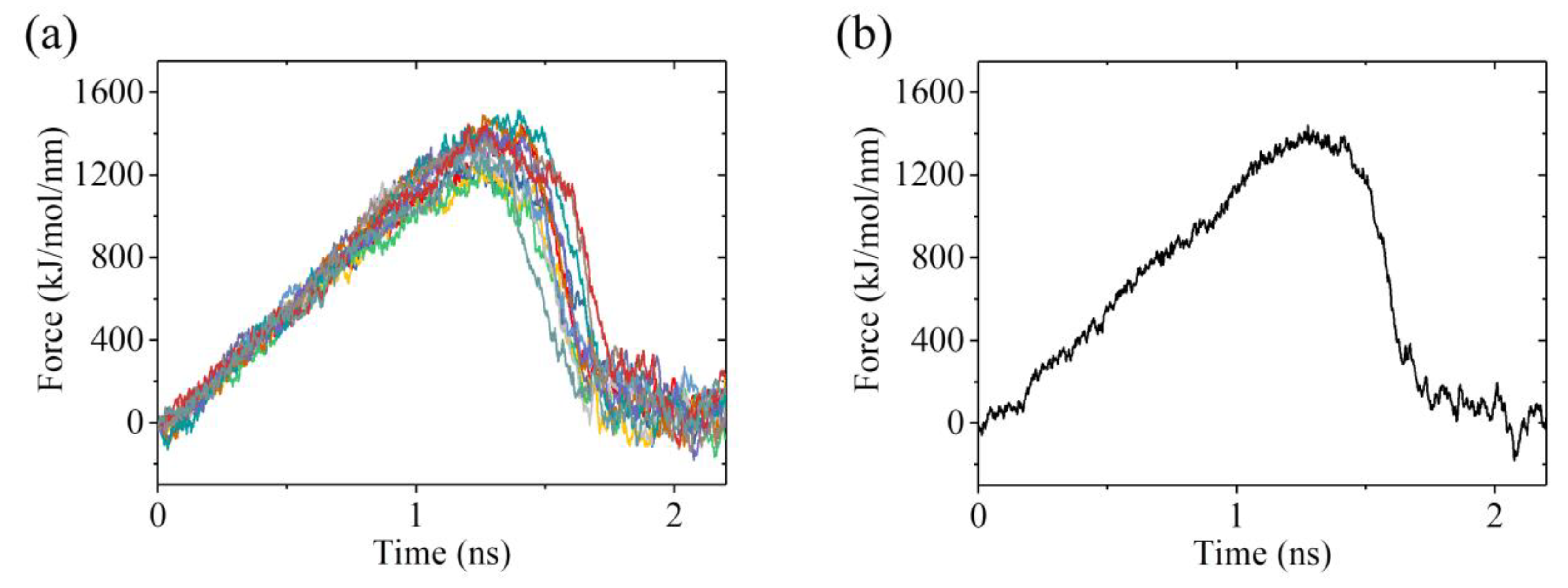
2.1. The Interaction of Kinesin -Head with α/β-Tubulin Induces Rapid Large Conformational Changes of the α/β-Tubulin
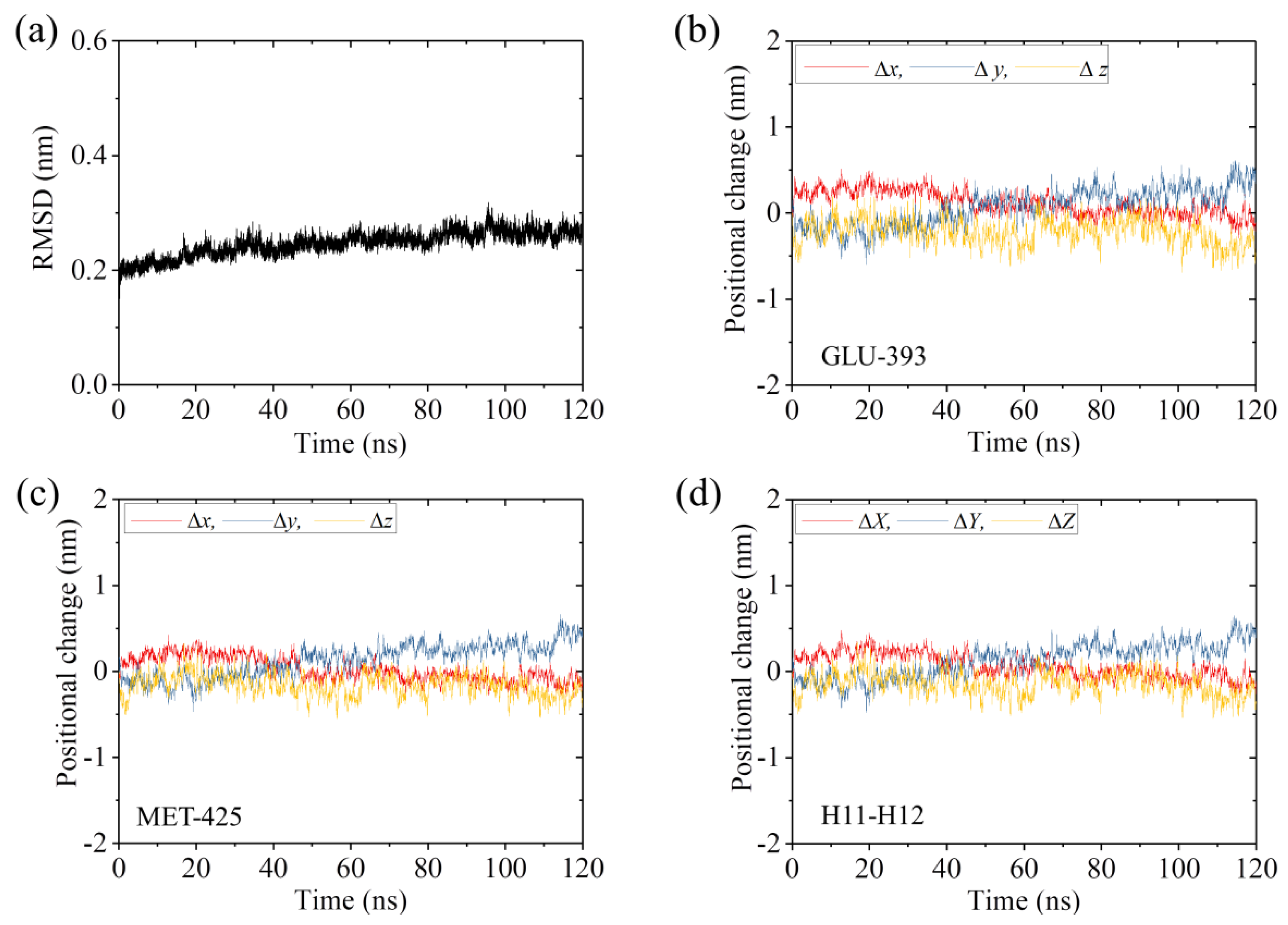
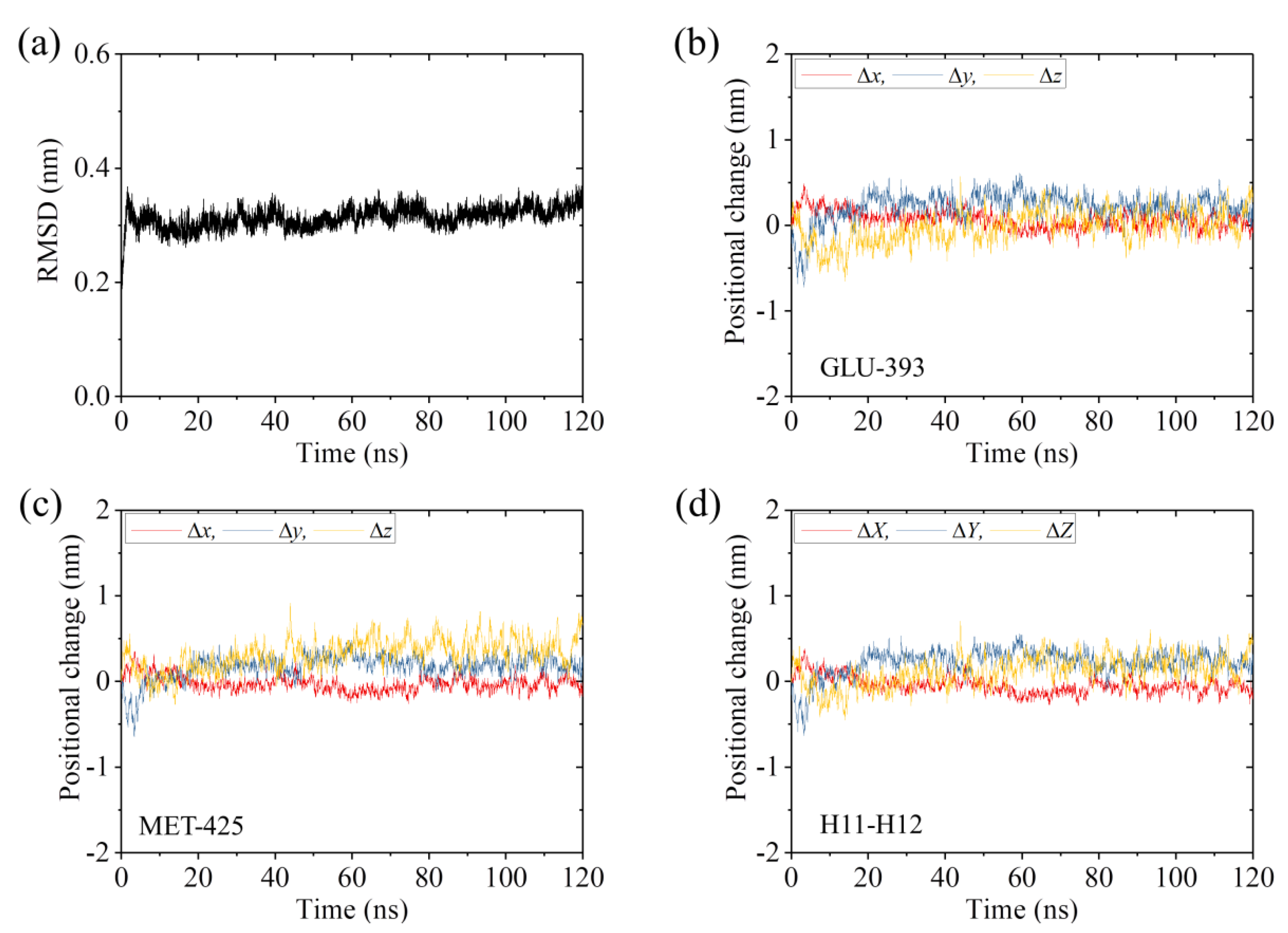
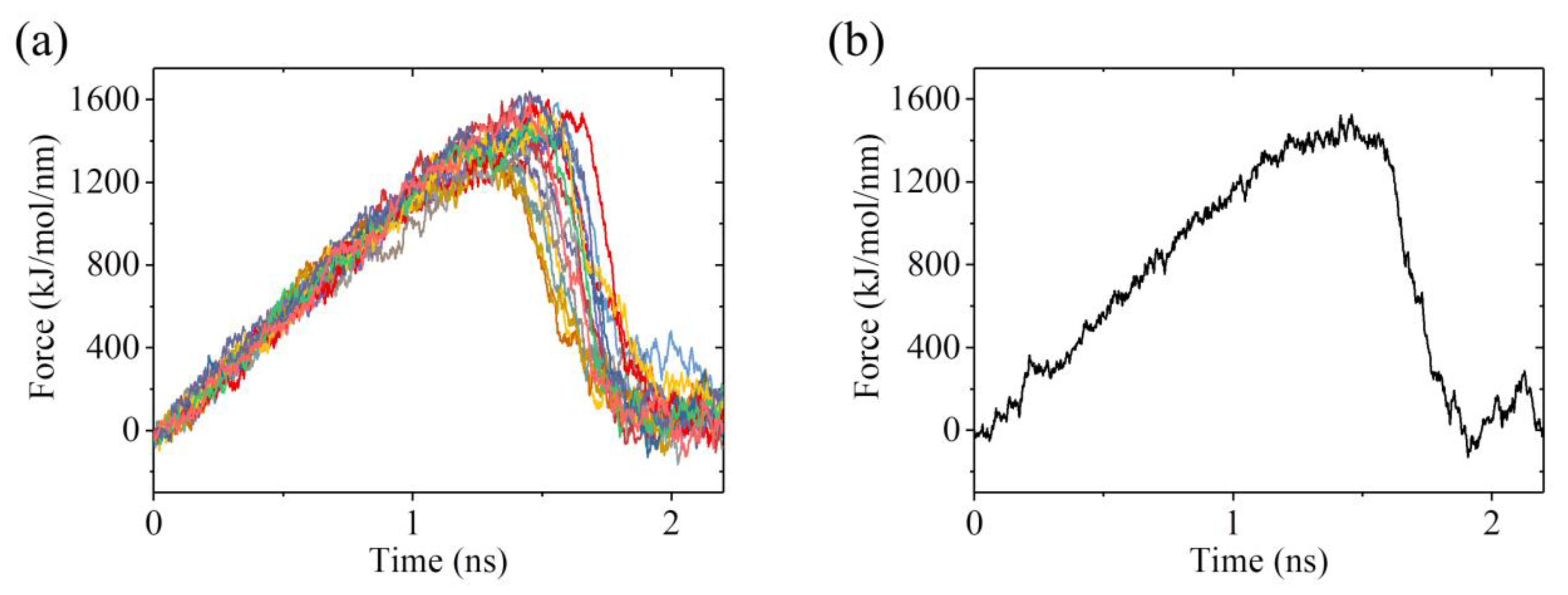
2.2. The Interaction of Kinesin ADP-Head with α/β-Tubulin Induces no Conformational Change of the α/β-Tubulin
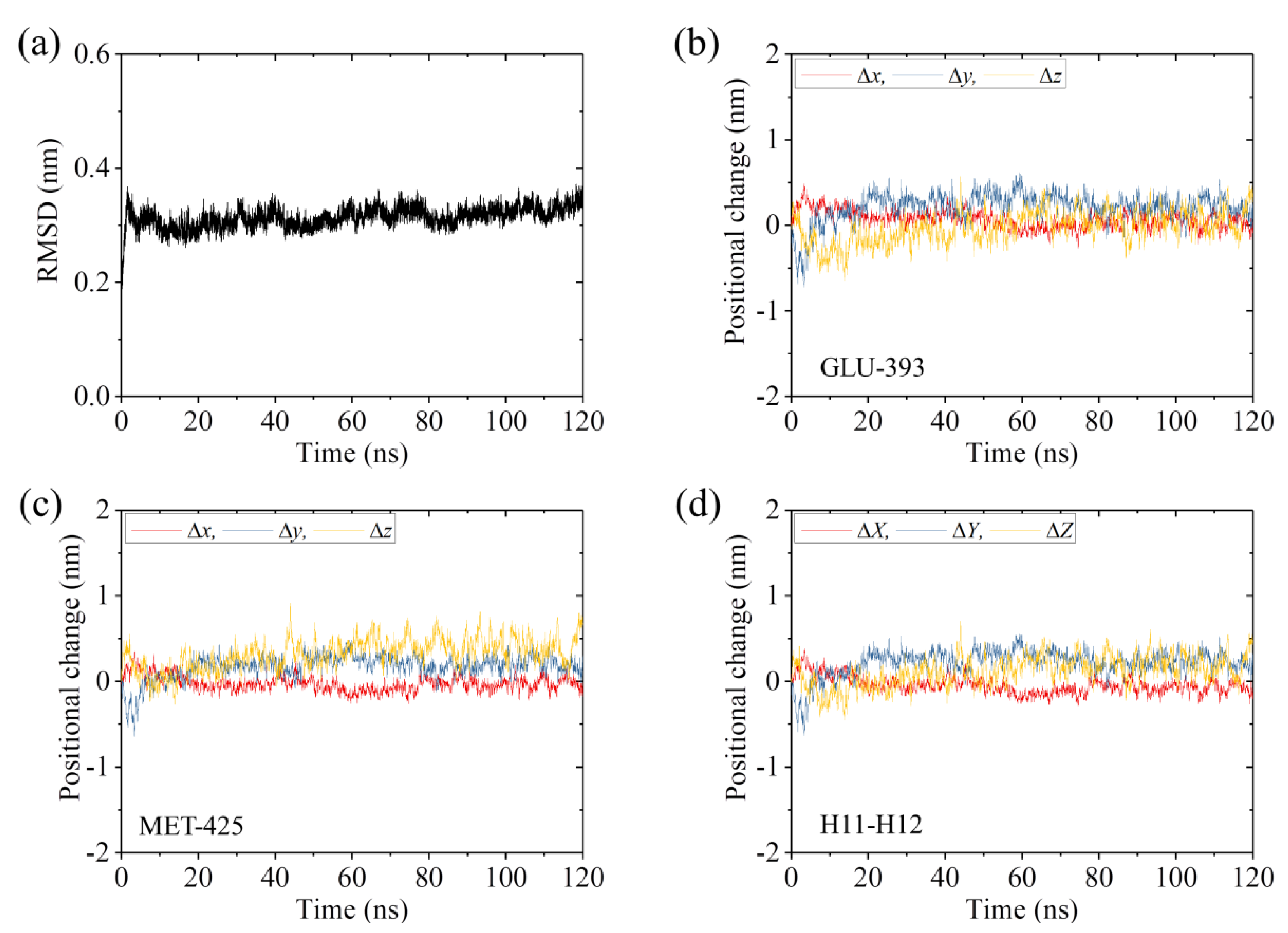
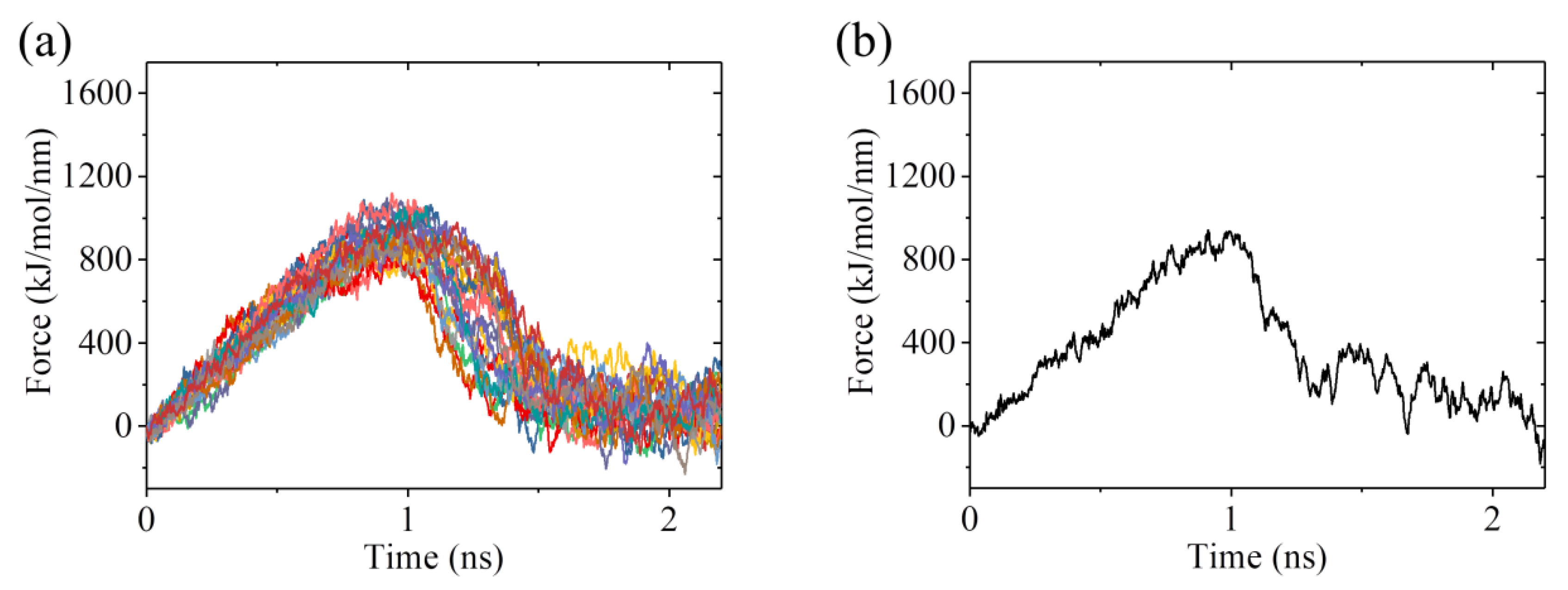
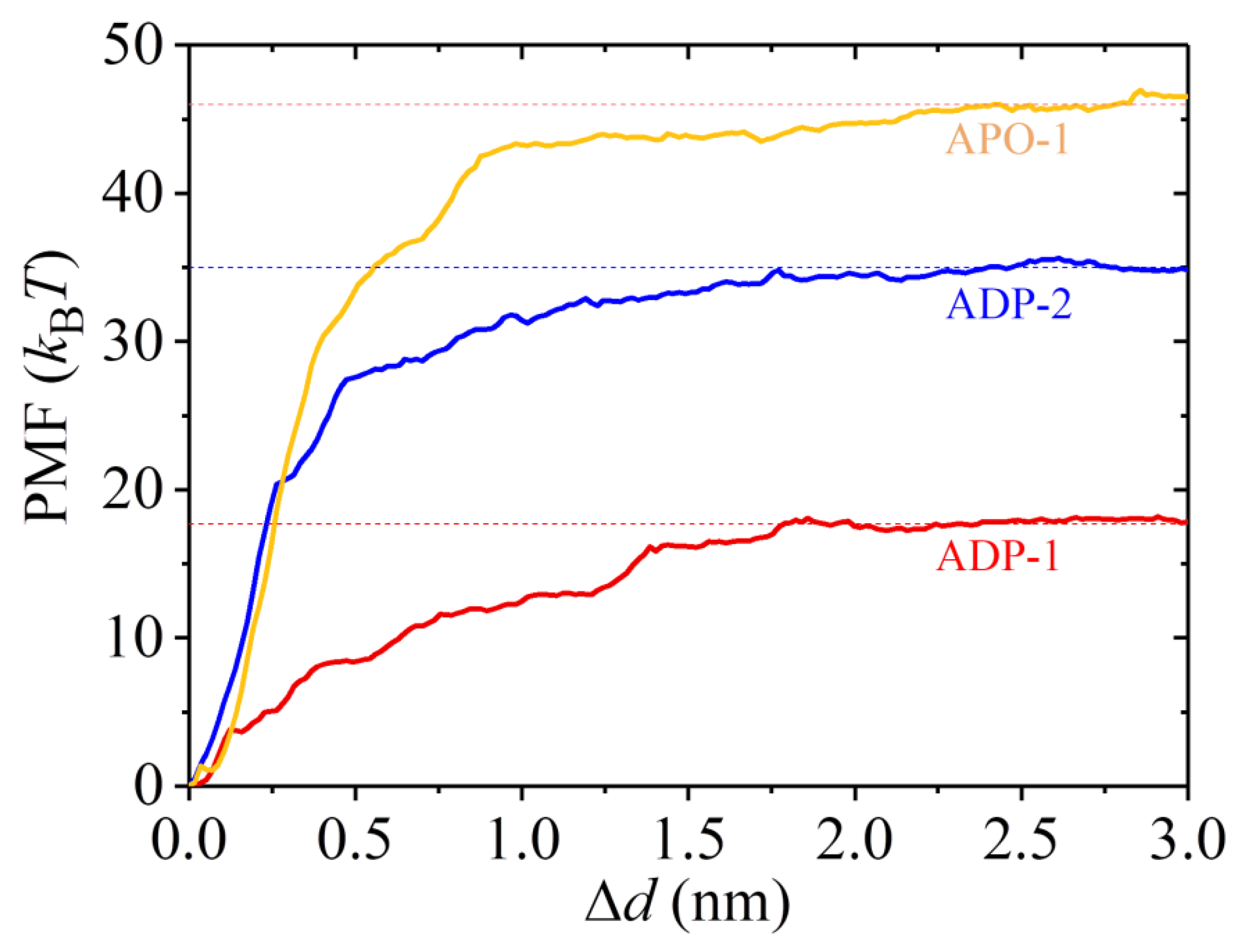
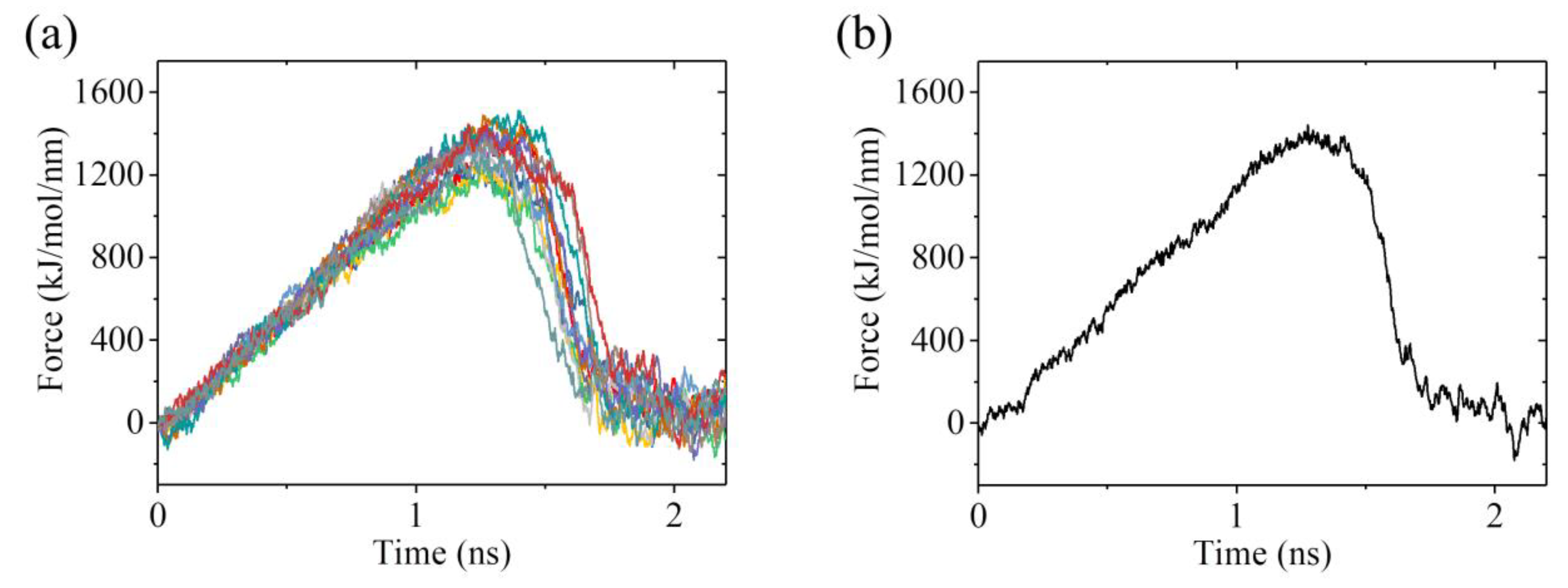
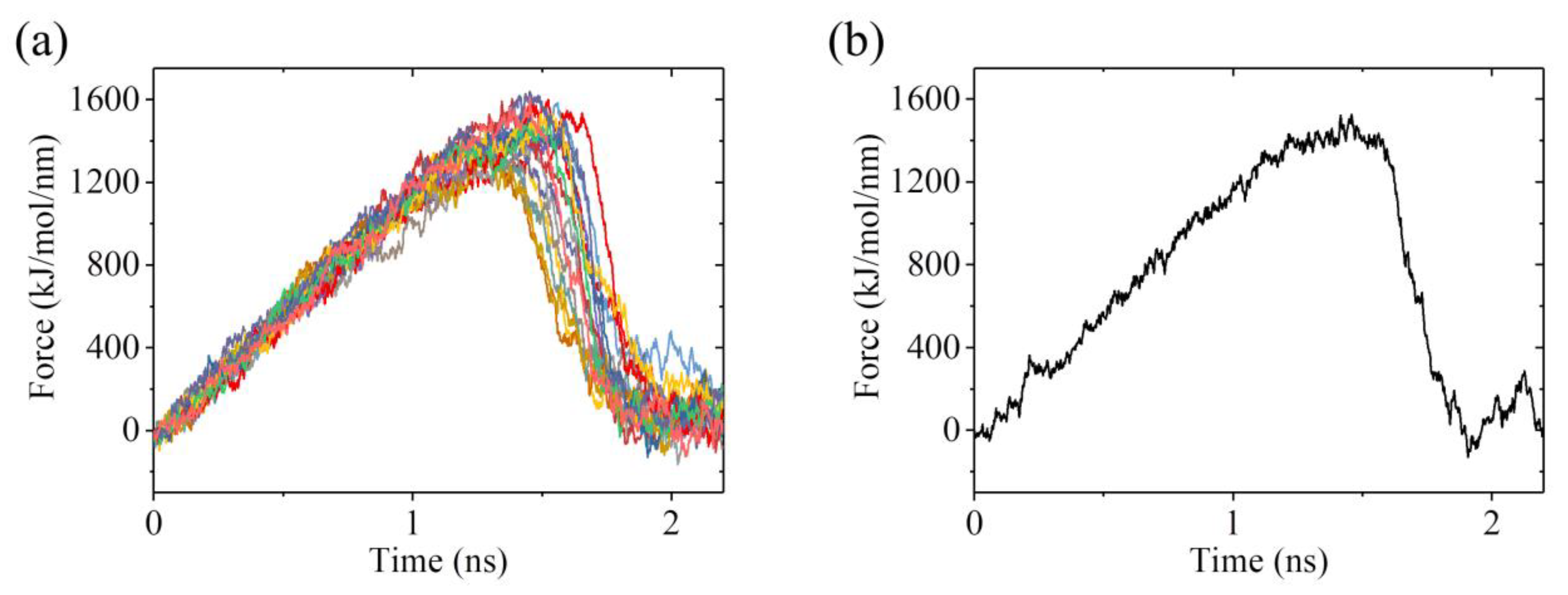
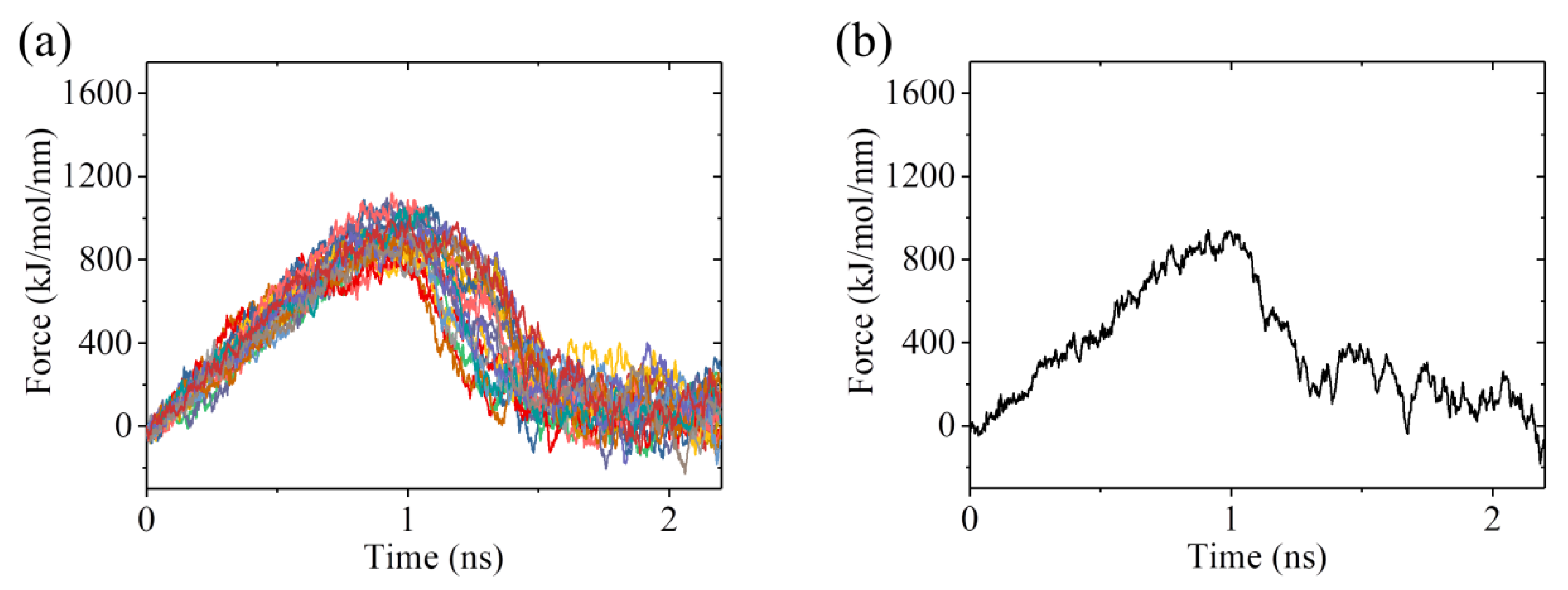
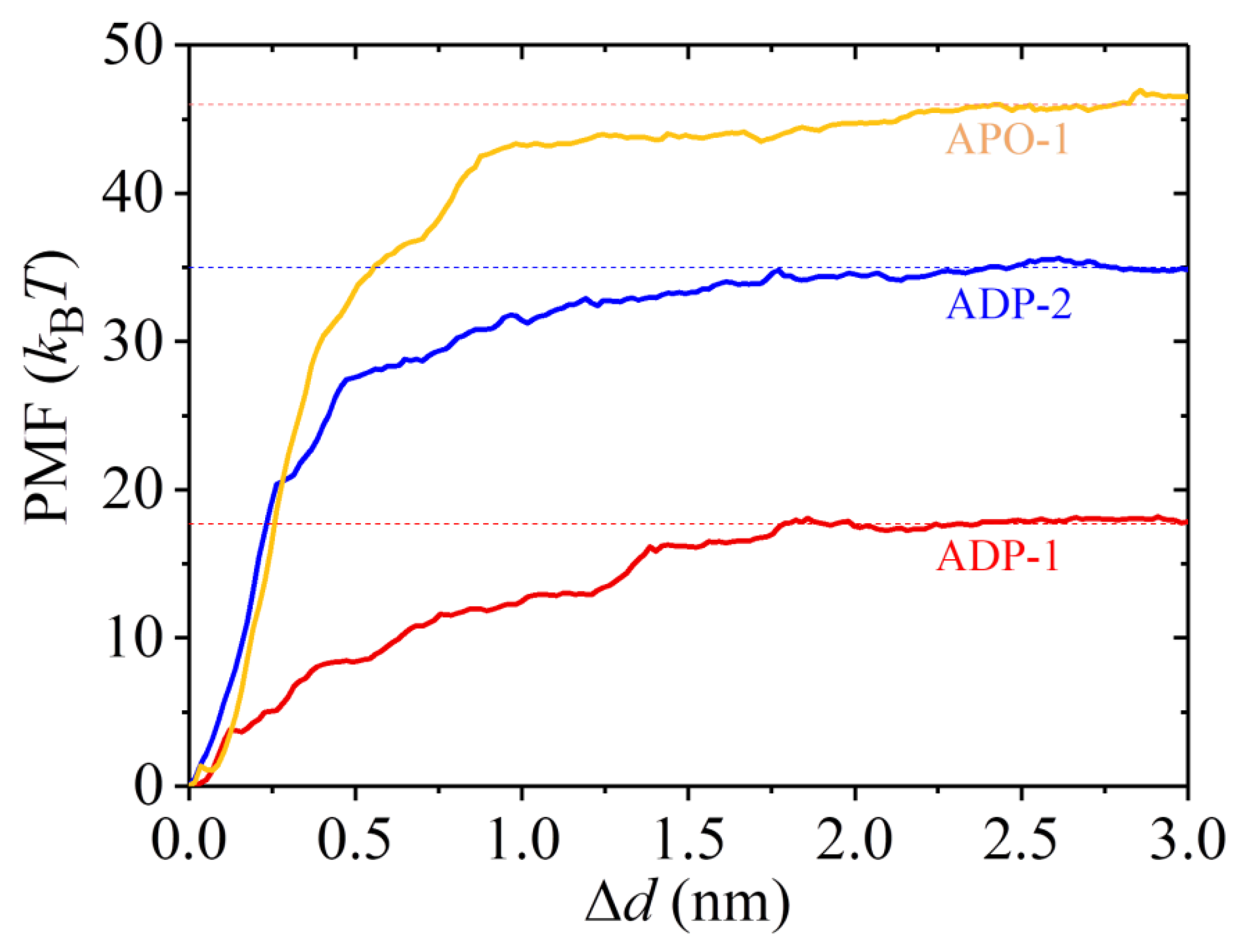
2.3. The ADP-Head Has a Much Weaker Affinity to α/β-Tubulin with Large Conformational Changes Than That with No Conformational Change
3. Discussion
4. Methods and Materials
4.1. System Setup
4.2. MD Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vale, R.D.; Reese, T.S.; Sheetz, M.P. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 1985, 42, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Howard, J. The movement of kinesin along microtubules. Annu. Rev. Physiol. 1996, 58, 703–729. [Google Scholar] [CrossRef]
- Hirokawa, N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 1998, 279, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.; Lin, A.W.; Safer, D.; Hart, C.L.; Naber, N.; Carragher, B.; Cain, S.M.; Pechatnikova, E.; Wilson-Kubalek, E.M.; Whittaker, M.; et al. A structural change in the kinesin motor protein that drives motility. Nature 1999, 402, 778–784. [Google Scholar] [CrossRef]
- Block, S.M. Kinesin motor mechanics: Binding, stepping, tracking, gating, and limping. Biophys. J. 2007, 92, 2986–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Zhang, H.; Geng, Y.; Ji, Q. How Kinesin-1 Utilize the Energy of Nucleotide: The Conformational Changes and Mechanochemical Coupling in the Unidirectional Motion of Kinesin-1. Int. J. Mol. Sci. 2020, 21, 6977. [Google Scholar] [CrossRef]
- Hwang, W.; Karplus, M. Structural basis for power stroke vs. Brownian ratchet mechanisms of motor proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 19777–19785. [Google Scholar] [CrossRef] [Green Version]
- Xie, P. Insight into the chemomechanical coupling mechanism of kinesin molecular motors. Commun. Theor. Phys. 2021, 73, 057601. [Google Scholar] [CrossRef]
- Crevel, I.M.T.C.; Lockhart, A.; Cross, R.A. Weak and strong states of kinesin and Ncd. J. Mol. Biol. 1996, 257, 66–76. [Google Scholar] [CrossRef]
- Hancock, W.O.; Howard, J. Kinesin’s processivity results from mechanical and chemical coordination between the ATP hydrolysis cycles of the two motor domains. Proc. Natl. Acad. Sci. USA 1999, 96, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Sosa, H.; Peterman, E.J.G.; Moerner, W.E.; Goldstein, L.S.B. ADP-induced rocking of the kinesin motor domain revealed by single-molecule fluorescence polarization microscopy. Nat. Struct. Biol. 2001, 8, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Uemura, S.; Kawaguchi, K.; Yajima, J.; Edamatsu, M.; Toyoshima, Y.Y.; Ishiwata, S. Kinesin–microtubule binding depends on both nucleotide state and loading direction. Proc. Natl. Acad. Sci. USA 2002, 99, 5977–5981. [Google Scholar] [CrossRef] [Green Version]
- Cross, R.A. Mechanochemistry of the kinesin-1 ATPase. Biopolymers 2016, 105, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Gigant, B.; Wang, W.; Dreier, B.; Jiang, Q.; Pecqueur, L.; Plückthun, A.; Wang, C.; Knossow, M. Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat. Struct. Mol. Biol. 2013, 20, 1001–1007. [Google Scholar] [CrossRef]
- Cao, L.; Wang, W.; Jiang, Q.; Wang, C.; Knossow, M.; Gigant, B. The structure of apo-kinesin bound to tubulin links the nucleotide cycle to movement. Nat. Commun. 2014, 5, 5364. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Yajima, H.; Nitta, R.; Inoue, S.; Ogura, T.; Sato, C.; Hirokawa, N. X-ray and Cryo-EM structures reveal mutual conformational changes of kinesin and GTP-state microtubules upon binding. EMBO J. 2015, 34, 1270–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zheng, W. All-atom structural investigation of kinesin-microtubule complex constrained by high-quality cryoelectron-microscopy maps. Biochemistry 2012, 51, 5022–5032. [Google Scholar] [CrossRef]
- Chakraborty, S.; Zheng, W. Decrypting the Structural, Dynamic, and Energetic Basis of a Monomeric Kinesin Interacting with a Tubulin Dimer in Three ATPase States by All-Atom Molecular Dynamics Simulation. Biochemistry 2015, 54, 859–869. [Google Scholar] [CrossRef]
- Ma, Y.; Li, T.; Jin, Y.; Geng, Y.; Ji, Q. Shaft function of kinesin-1’s α4 helix in the processive movement. Cell. Mol. Bioeng. 2019, 12, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.-X.; Fu, Y.-B.; Guo, S.-K.; Wang, P.-Y.; Chen, H.; Xie, P. Investigating role of conformational changes of microtubule in regulating its binding affinity to kinesin by all-atom molecular dynamics simulation. Proteins 2018, 86, 1127–1139. [Google Scholar] [CrossRef]
- Hoenger, A.; Sablin, E.P.; Vale, R.D.; Fletterick, R.J.; Milligan, R.A. Three-dimensional structure of a tubulin-motor-protein complex. Nature 1995, 376, 271–274. [Google Scholar] [CrossRef]
- Hoenger, A.; Milligan, R.A. Motor domains of kinesin and ncd interact with microtubule protofilaments with the same binding geometry. J. Mol. Biol. 1997, 265, 553–564. [Google Scholar] [CrossRef]
- Hirose, K.; Amos, W.B.; Lockhart, A.; Cross, R.A.; Amos, L.A. Three-dimensional cryoelectron microscopy of 16-protofilament microtubules: Structure, polarity, and interaction with motor proteins. J. Struct. Biol. 1997, 118, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.; Goldie, K.N.; Hoenger, A. Complex formation with kinesin motor domains affects the structure of microtubules. J. Mol. Biol. 2004, 335, 139–153. [Google Scholar] [CrossRef]
- Kull, F.J.; Sablin, E.P.; Lau, R.; Fletterick, R.J.; Vale, R.D. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature 1996, 380, 550–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, J.; Li, H.; Downing, K.H.; Nogales, E. Refined structure of αβ-tubulin at 3.5 Å resolution. J. Mol. Biol. 2001, 313, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Guo, S.-K.; Chen, H. A generalized kinetic model for coupling between stepping and ATP hydrolysis of kinesin molecular motors. Int. J. Mol. Sci. 2019, 20, 4911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, P. Theoretical analysis of dynamics of kinesin molecular motors. ACS Omega 2020, 5, 5721–5730. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.-K.; Shi, X.-X.; Wang, P.-Y.; Xie, P. Run length distribution of dimerized kinesin-3 molecular motors: Comparison with dimeric kinesin-1. Sci. Rep. 2019, 9, 16973. [Google Scholar] [CrossRef] [Green Version]
- Xie, P. A common ATP-dependent stepping model for kinesin-5 and kinesin-1: Mechanism of bi-directionality of kinesin-5. Biophys. Chem. 2021, 271, 106548. [Google Scholar] [CrossRef] [PubMed]
- Xie, P. A model of processive walking and slipping of kinesin-8 molecular motors. Sci. Rep. 2021, 11, 8081. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-K.; Xie, P. A common chemomechanical coupling model for orphan and conventional kinesin molecular motors. Biophys. Chem. 2020, 264, 106427. [Google Scholar] [CrossRef]
- Guo, S.-K.; Wang, P.-Y.; Xie, P. A model of processive movement of dimeric kinesin. J. Theor. Biol. 2017, 414, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Shi, X.; Wang, P.; Xie, P. Processivity of dimeric kinesin-1 molecular motors. FEBS Open Bio 2018, 8, 1332–1351. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-K.; Wang, P.-Y.; Xie, P. Dynamics of dimeric kinesins: Limping, effect of longitudinal force, effects of neck linker extension and mutation, and comparison between kinesin-1 and kinesin-2. Int. J. Biol. Macromol. 2017, 105, 1126–1137. [Google Scholar] [CrossRef]
- Guo, S.-K.; Wang, W.-C.; Wang, P.-Y.; Xie, P. Force dependence of velocity and run length of kinesin-1, kinesin-2 and kinesin-5 family molecular motors. Molecules 2019, 24, 287. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.-K.; Shi, X.-X.; Wang, P.-Y.; Xie, P. Force dependence of unbinding rate of kinesin motor during its processive movement on microtubule. Biophys. Chem. 2019, 253, 106216. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.-X.; Guo, S.-K.; Wang, P.-Y.; Chen, H.; Xie, P. All-atom molecular dynamics simulations reveal how kinesin transits from one-head-bound to two-heads-bound state. Proteins 2020, 88, 545–557. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, D.J.; Brooks, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Description | Initial Structural Data: pdb |
|---|---|---|
| APO-1 | ϕ-head bound with initially | head: 4LNU [16] |
| unperturbed α/β-tubulin | tubulin: 1JFF [27] | |
| APO-2 | ϕ-head bound with α/β-tubulin determined from | 4LNU [16] |
| X-ray crystallography | ||
| ADP-1 | ADP-head bound with α/β-tubulin having conformational changes | head: 1BG2 [26] tubulin: simulation for APO-1 |
| ADP-2 | ADP-head bound with | head: 1BG2 [26] |
| unperturbed α/β-tubulin | tubulin: 1JFF [27] | |
| α/β-tubulin | isolated unperturbed α/β-tubulin | 1JFF [27] |
| Positional Change ± Standard Deviation | |||
|---|---|---|---|
| System | |||
| ΔX (nm) | ΔY (nm) | ΔZ (nm) | |
| APO-1 | −0.24 ± 0.11 | 0.82 ± 0.14 | −1.04 ± 0.20 |
| APO-2 | −0.03 ± 0.09 | 0.28 ± 0.11 | 0.14 ± 0.13 |
| ADP-1 | −0.10 ± 0.07 | 0.24 ± 0.09 | 0.23 ± 0.15 |
| ADP-2 | −0.08 ± 0.07 | 0.23 ± 0.09 | 0.22 ± 0.14 |
| α/β-tubulin | −0.08 ± 0.09 | 0.38 ± 0.10 | −0.22 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, X.-X.; Wang, P.-Y.; Chen, H.; Xie, P. Studies of Conformational Changes of Tubulin Induced by Interaction with Kinesin Using Atomistic Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 6709. https://doi.org/10.3390/ijms22136709
Shi X-X, Wang P-Y, Chen H, Xie P. Studies of Conformational Changes of Tubulin Induced by Interaction with Kinesin Using Atomistic Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2021; 22(13):6709. https://doi.org/10.3390/ijms22136709
Chicago/Turabian StyleShi, Xiao-Xuan, Peng-Ye Wang, Hong Chen, and Ping Xie. 2021. "Studies of Conformational Changes of Tubulin Induced by Interaction with Kinesin Using Atomistic Molecular Dynamics Simulations" International Journal of Molecular Sciences 22, no. 13: 6709. https://doi.org/10.3390/ijms22136709
APA StyleShi, X.-X., Wang, P.-Y., Chen, H., & Xie, P. (2021). Studies of Conformational Changes of Tubulin Induced by Interaction with Kinesin Using Atomistic Molecular Dynamics Simulations. International Journal of Molecular Sciences, 22(13), 6709. https://doi.org/10.3390/ijms22136709