
A Novel Model for the RNase MRP-Induced Switch between the Formation of Different Forms of 5.8S rRNA



Abstract
1. Introduction

2. Results
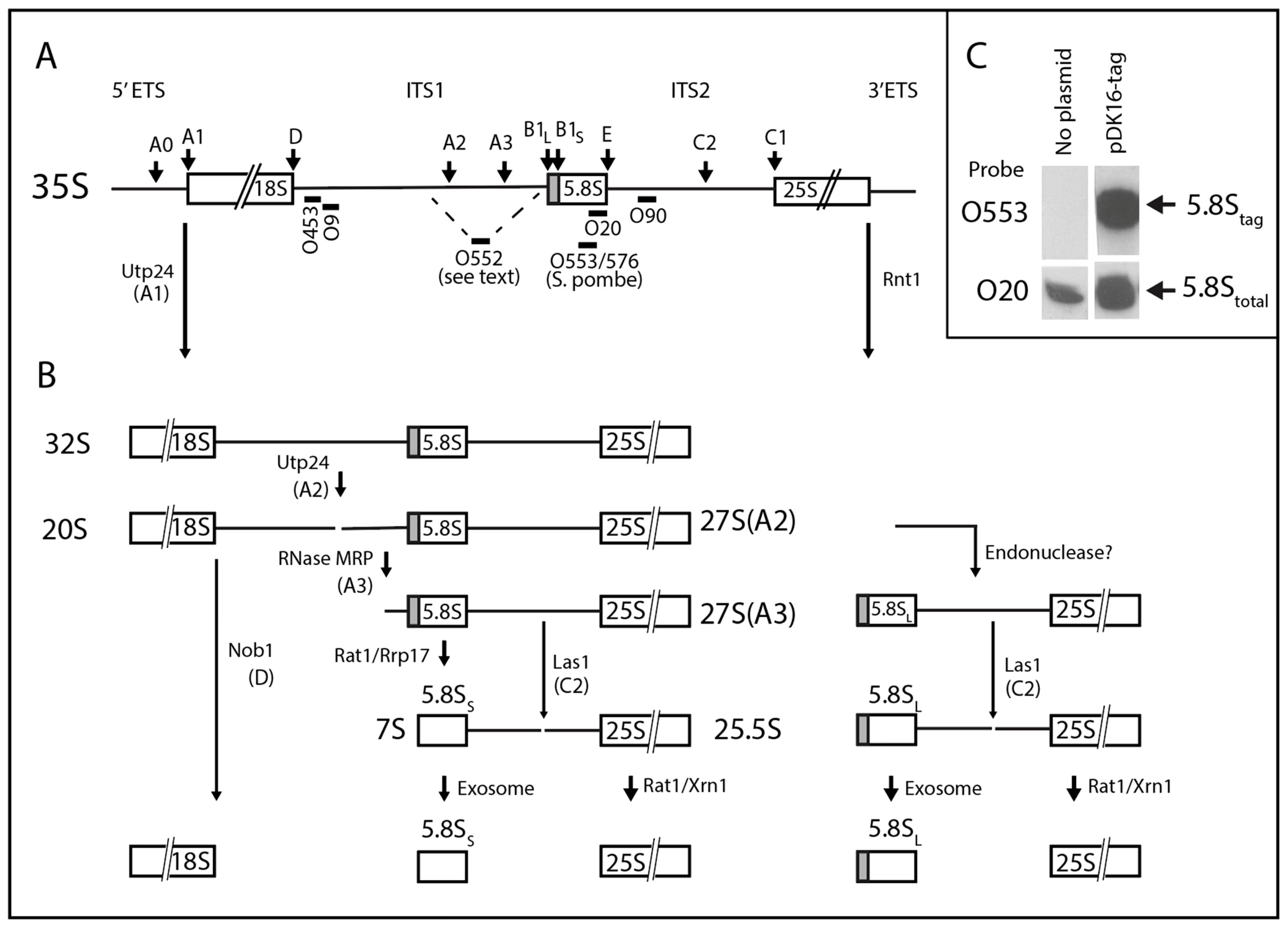
2.1. System for Genetic Analysis
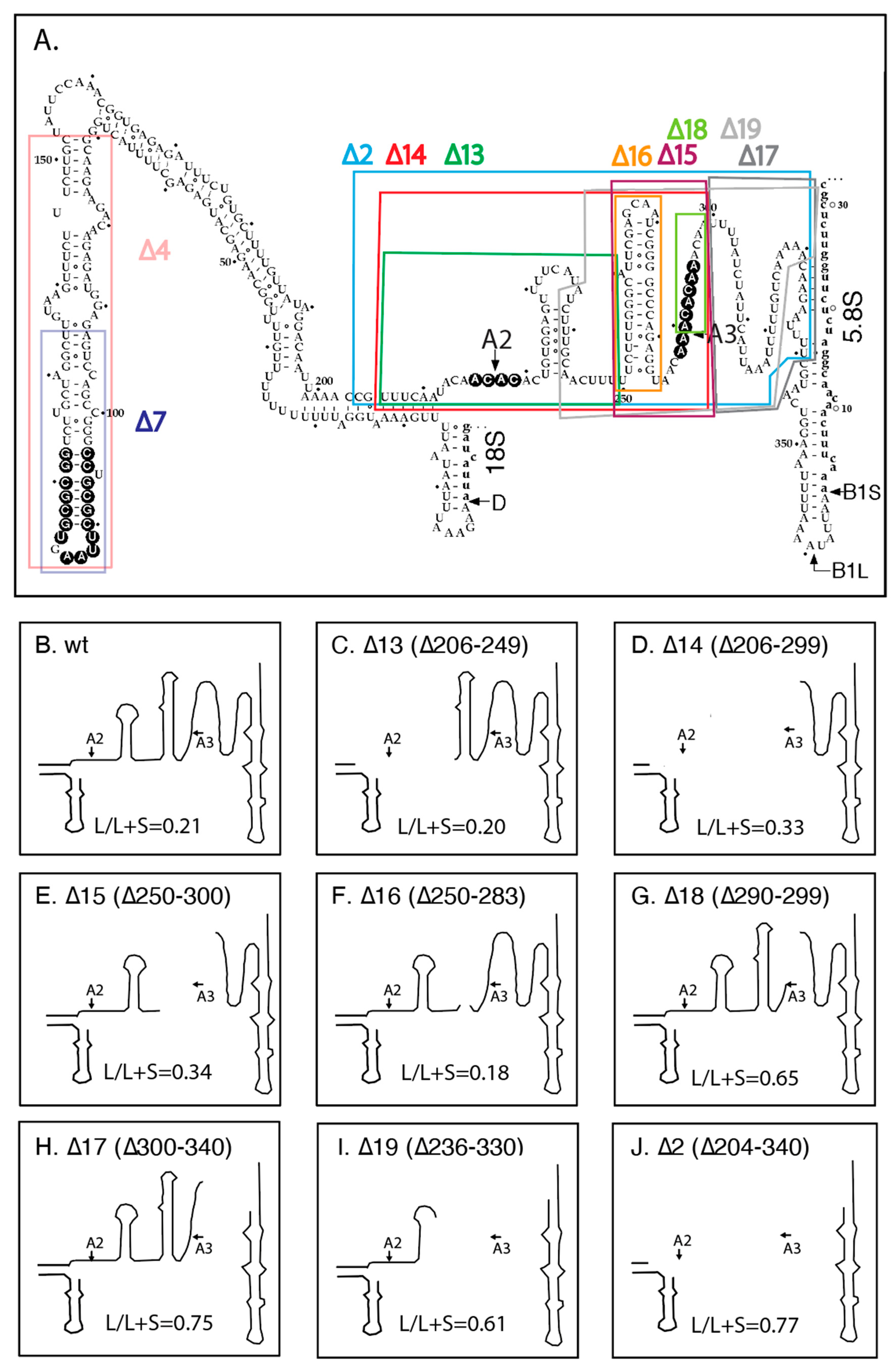
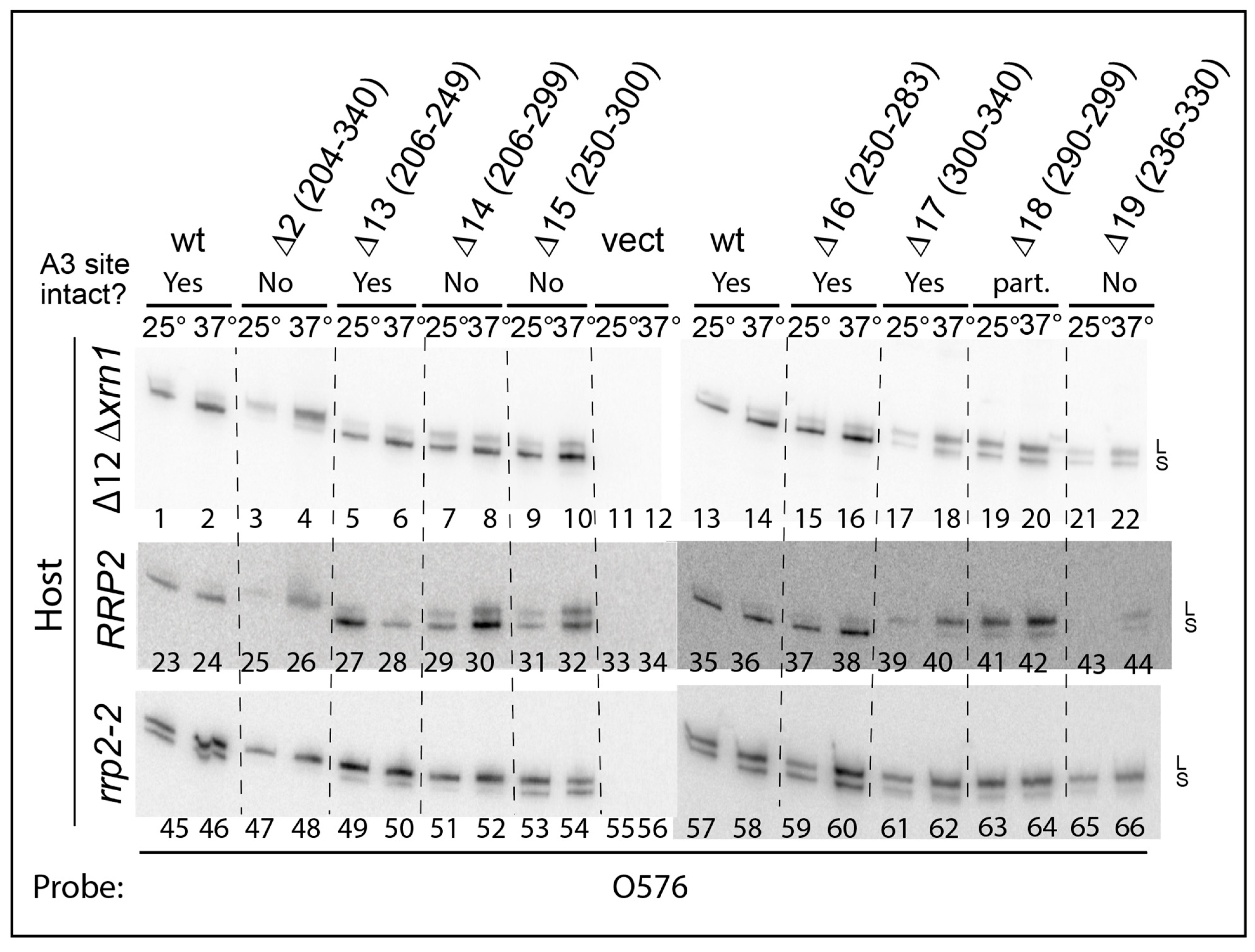
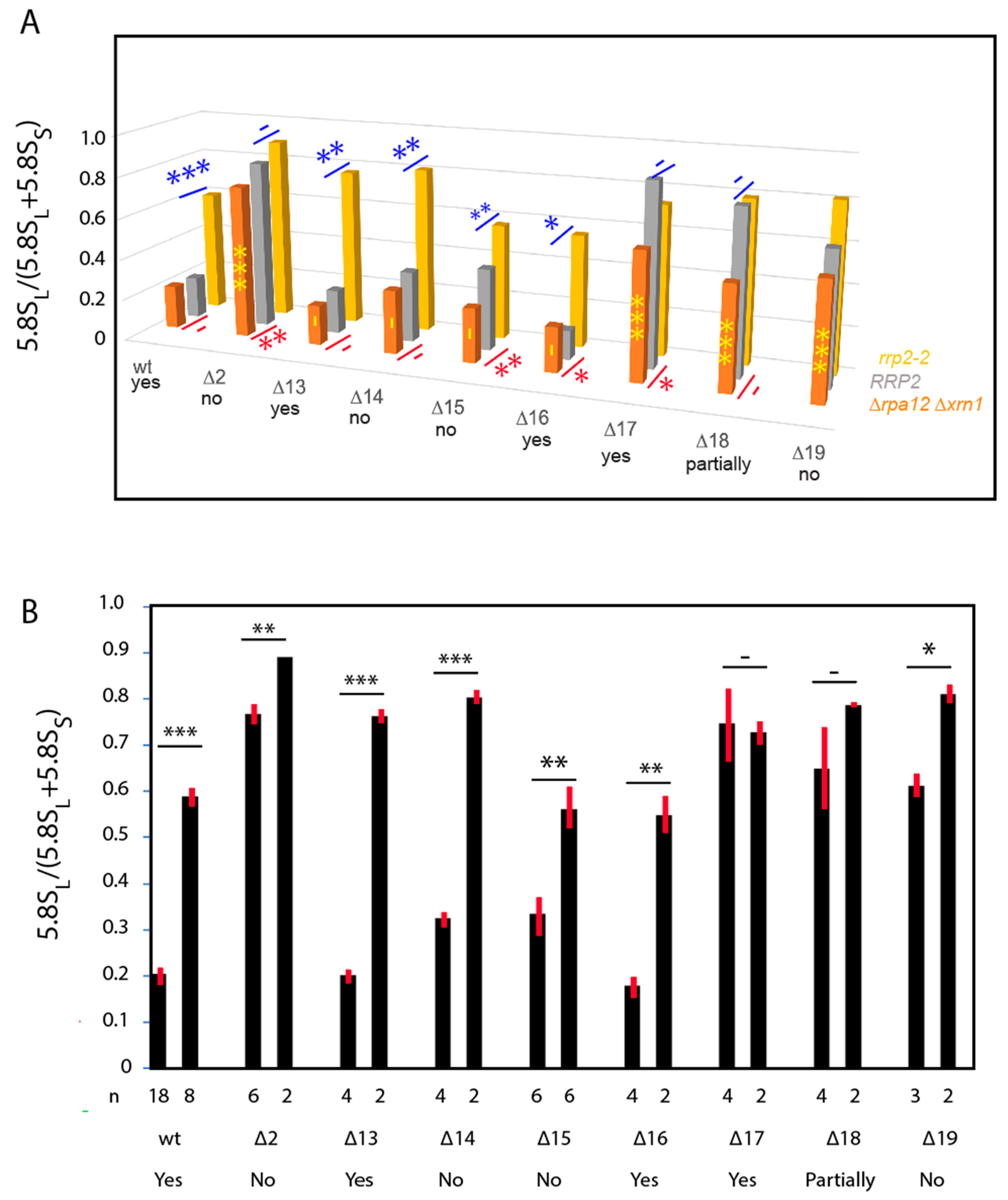
2.2. Effects of ITS1 Deletions on 5.8S rRNA Processing
2.3. pre-rRNA Processing Is Altered by an RNase MRP Mutation Even When the ITS1 A3 Site Is Deleted
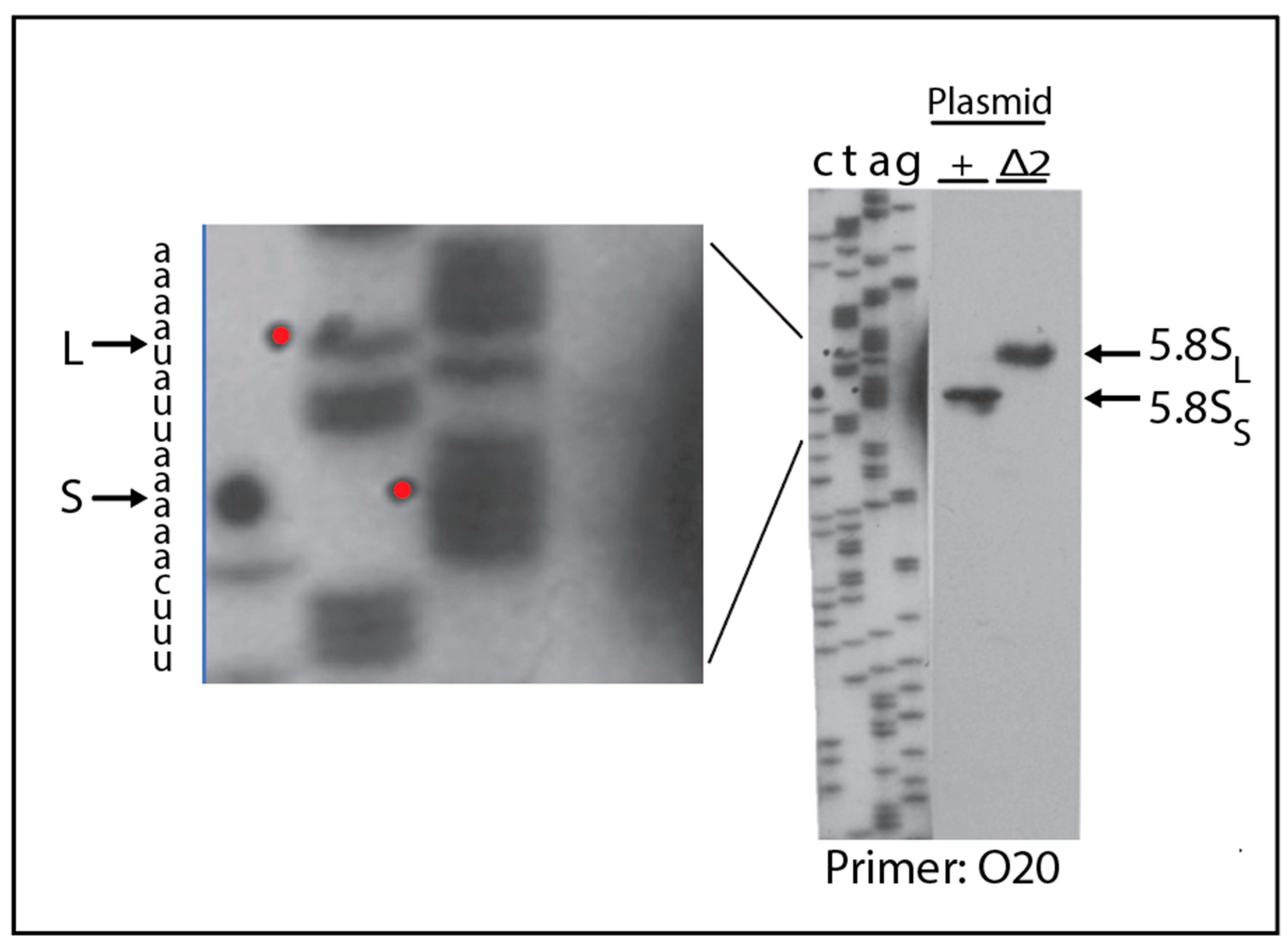
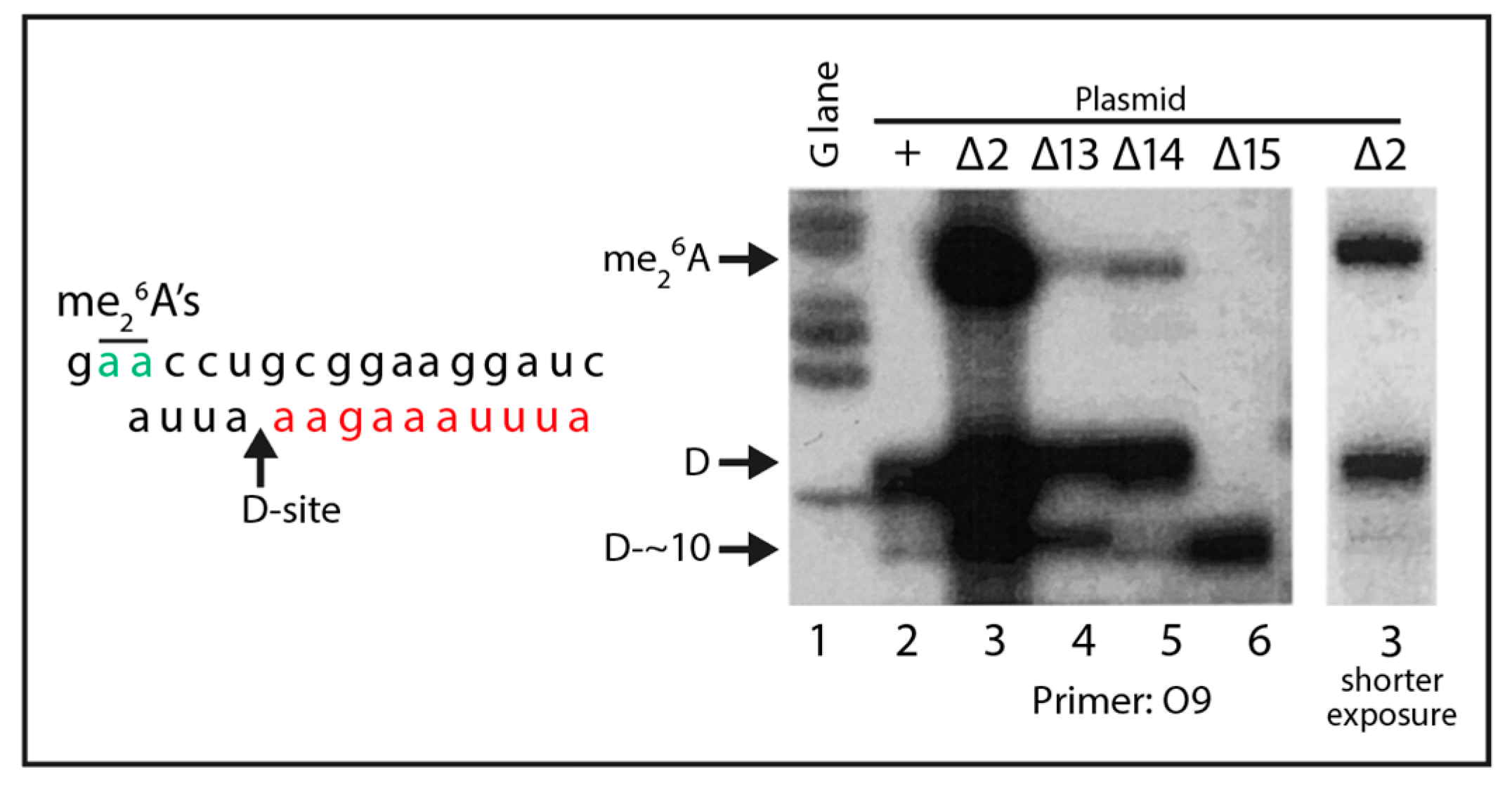
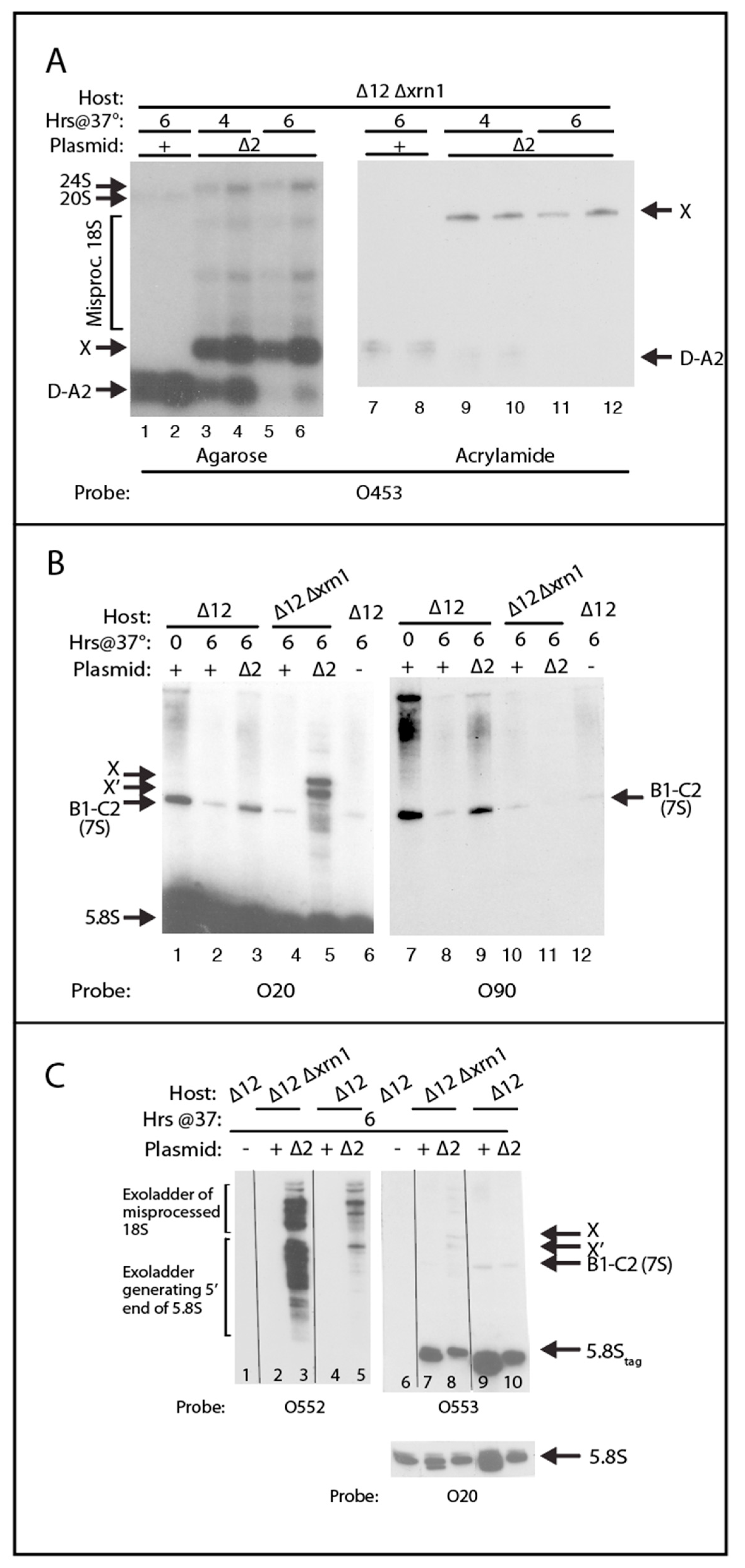
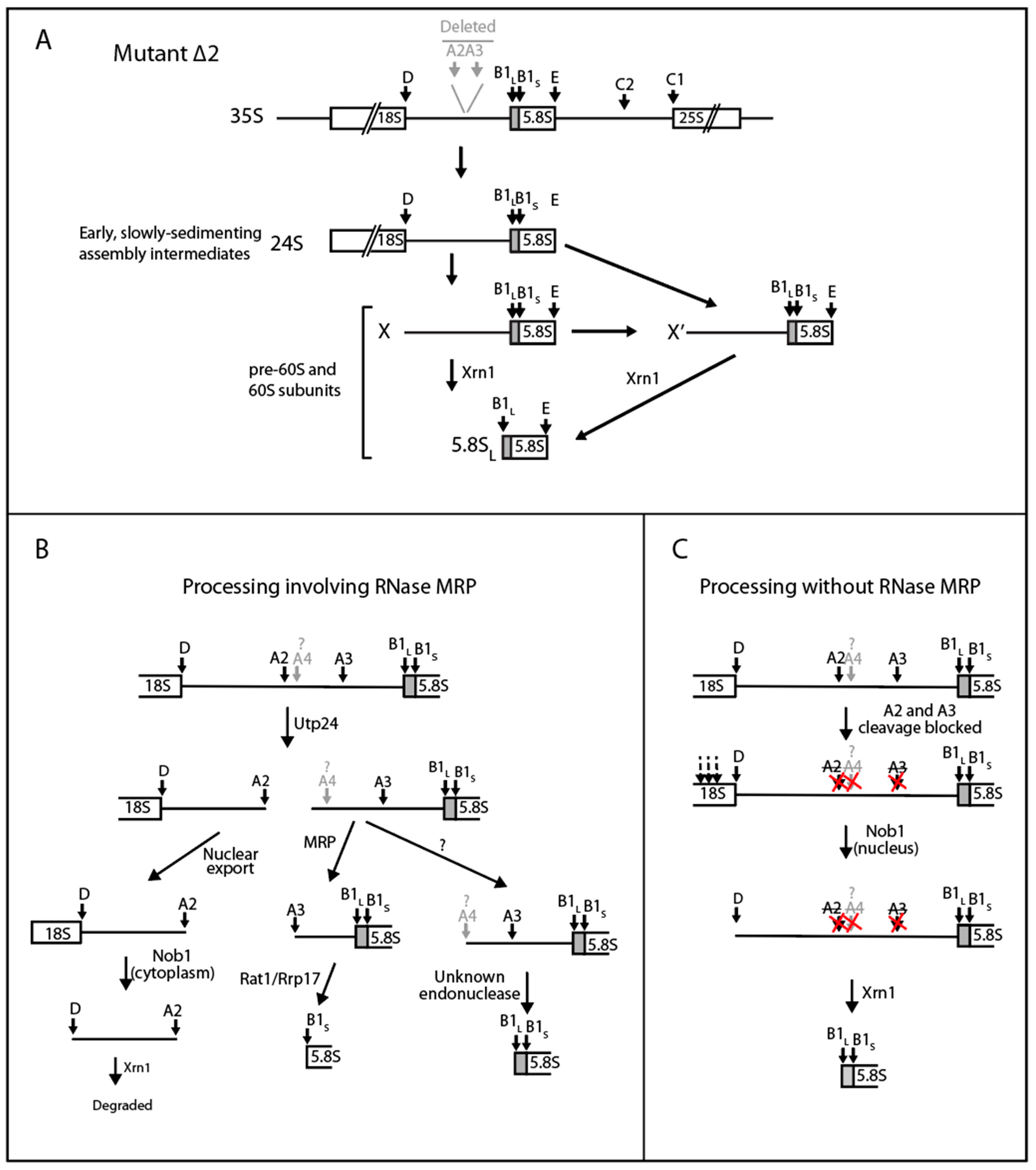
2.4. A Pathway to 5.8S rRNA That Bypasses the Canonical A2 and A3 Cleavage Sites in ITS1
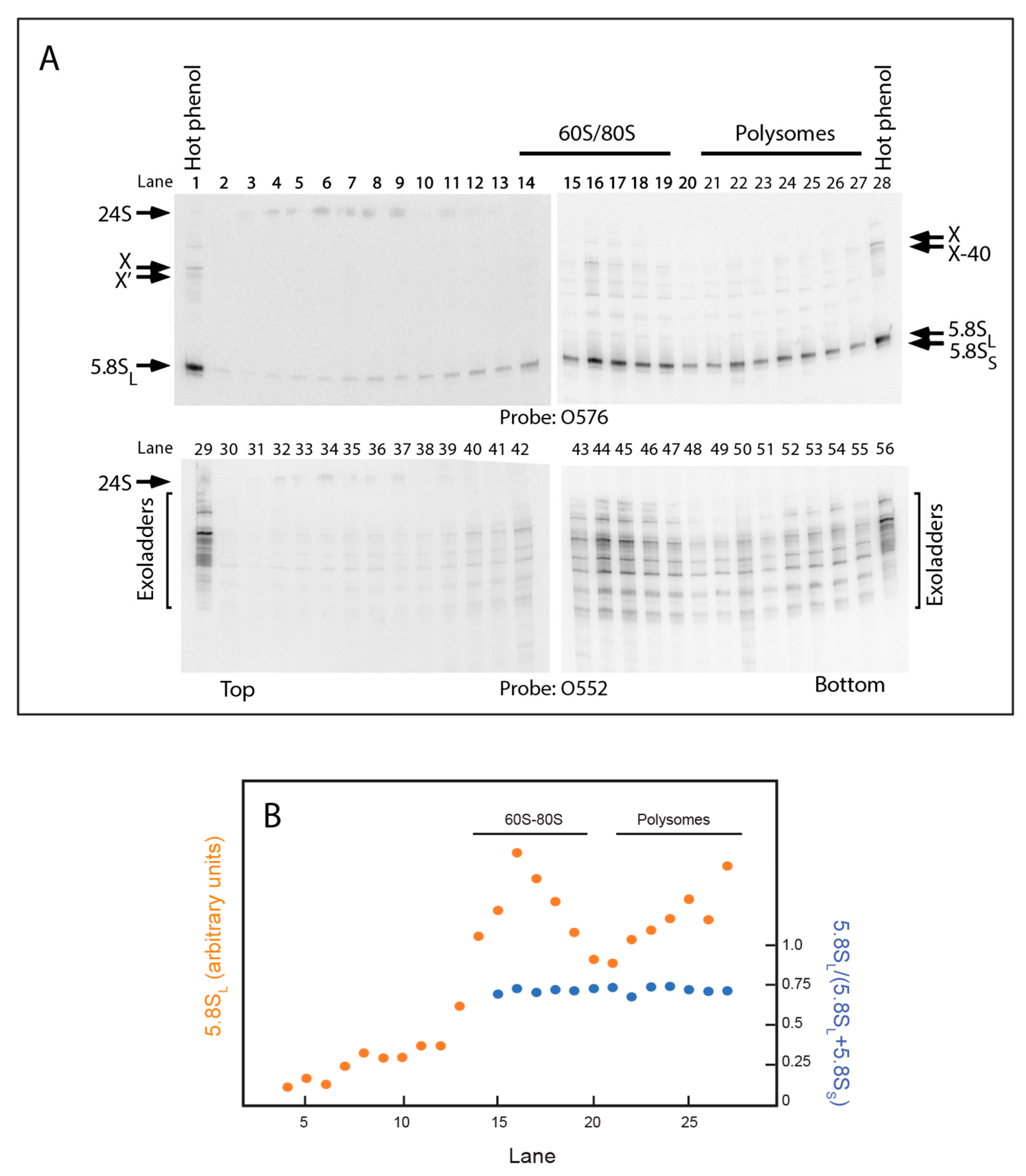
2.5. 5.8S rRNA with 5′ Extended Ends Are Incorporated into Functional Ribosomes
3. Discussion
3.1. Steps in the Xrn1-Dependent Path to the 5′ End of 5.8SL
3.2. The RNase MRP-Induced Switch between 5.8SL and 5.8SS Production Not Due to Changes in A3 Cleavage
3.3. The 3′ End of ITS1 Facilitates the Xrn1-Mediated Processing
3.4. The 3′ End Maturation of 18S rRNA
3.5. Diversity of rRNA Processing Pathways
4. Materials and Methods
4.1. Strains and Growth Conditions
4.2. Plasmids and Oligonucleotides
4.3. Other Procedures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maaløe, O.; Kjeldgaard, N.O. Control of Macromolecular Synthesis; Benjamin: New York, NY, USA, 1966. [Google Scholar]
- Kjeldgaard, N.O.; Maaloe, O.; Schaechter, M. The transition between different physiological states during balanced growth of Salmonella typhimurium. J. Gen. Microbiol. 1958, 19, 607–616. [Google Scholar] [CrossRef]
- Warner, J.R. The economics of ribosome biosynthesis in yeast. Trends. Biochem. Sci. 1999, 24, 437–440. [Google Scholar] [CrossRef]
- Cerezo, E.; Plisson-Chastang, C.; Henras, A.K.; Lebaron, S.; Gleizes, P.E.; O’Donohue, M.F.; Romeo, Y.; Henry, Y. Maturation of pre-40S particles in yeast and humans. Wiley. Interdiscip. Rev. RNA 2019, 10, e1516. [Google Scholar] [CrossRef] [PubMed]
- Tomecki, R.; Sikorski, P.J.; Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells—Focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 2017, 591, 1801–1850. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Bohnsack, M.T. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 2019, 38, e100278. [Google Scholar] [CrossRef] [PubMed]
- Klinge, S.; Woolford, J.L., Jr. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019, 20, 116–131. [Google Scholar] [CrossRef]
- Bassler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu. Rev. Biochem. 2019, 88, 8.1–8.26. [Google Scholar] [CrossRef]
- Woolford, J.L., Jr.; Baserga, S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef] [PubMed]
- Axt, K.; French, S.L.; Beyer, A.L.; Tollervey, D. Kinetic analysis demonstrates a requirement for the Rat1 exonuclease in cotranscriptional pre-rRNA cleavage. PLoS ONE 2014, 9, e85703. [Google Scholar] [CrossRef]
- Talkish, J.; Biedka, S.; Jakovljevic, J.; Zhang, J.; Tang, L.; Strahler, J.R.; Andrews, P.C.; Maddock, J.R.; Woolford, J.L., Jr. Disruption of ribosome assembly in yeast blocks cotranscriptional pre-rRNA processing and affects the global hierarchy of ribosome biogenesis. RNA 2016, 22, 852–866. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Du, Y.; Ye, K. Structural and functional analysis of Utp24, an endonuclease for processing 18S ribosomal RNA. PLoS ONE 2018, 13, e0195723. [Google Scholar] [CrossRef] [PubMed]
- Kufel, J.; Dichtl, B.; Tollervey, D. Yeast Rnt1p is required for cleavage of the pre-ribosomal RNA in the 3′ ETS but not the 5′ ETS. RNA 1999, 5, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Bleichert, F.; Granneman, S.; Osheim, Y.N.; Beyer, A.L.; Baserga, S.J. The PINc domain protein Utp24, a putative nuclease, is required for the early cleavage steps in 18S rRNA maturation. Proc. Natl. Acad. Sci. USA 2006, 103, 9464–9469. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.R.; Weichmann, F.; Colvin, D.; Sloan, K.E.; Kudla, G.; Tollervey, D.; Watkins, N.J.; Schneider, C. The PIN domain endonuclease Utp24 cleaves pre-ribosomal RNA at two coupled sites in yeast and humans. Nucleic Acids Res. 2016, 44, 9016. [Google Scholar] [CrossRef][Green Version]
- Mitchell, P.; Petfalski, E.; Shevchenko, A.; Mann, M.; Tollervey, D. The exosome: A conserved eukaryotic RNA processing complex containing multiple 3′→5′ exoribonucleases. Cell 1997, 91, 457–466. [Google Scholar] [CrossRef]
- Geerlings, T.H.; Vos, J.C.; Raue, H.A. The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5′→3′ exonucleases. RNA 2000, 6, 1698–1703. [Google Scholar] [CrossRef]
- Lygerou, Z.; Allmang, C.; Tollervey, D.; Seraphin, B. Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro. Science 1996, 272, 268–270. [Google Scholar] [CrossRef]
- Henry, Y.; Wood, H.; Morrissey, J.P.; Petfalski, E.; Kearsey, S.; Tollervey, D. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J. 1994, 13, 2452–2463. [Google Scholar] [CrossRef] [PubMed]
- Oeffinger, M.; Zenklusen, D.; Ferguson, A.; Wei, K.E.; El Hage, A.; Tollervey, D.; Chait, B.T.; Singer, R.H.; Rout, M.P. Rrp17p is a eukaryotic exonuclease required for 5′ end processing of Pre-60S ribosomal RNA. Mol. Cell 2009, 36, 768–781. [Google Scholar] [CrossRef]
- Allmang, C.; Henry, Y.; Morrissey, J.P.; Wood, H.; Petfalski, E.; Tollervey, D. Processing of the yeast pre-rRNA at sites A and A is linked. RNA 1996, 2, 63–73. [Google Scholar]
- Schmitt, M.E.; Clayton, D.A. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces. cerevisiae. Mol. Cell Biol. 1993, 13, 7935–7941. [Google Scholar] [PubMed]
- Chu, S.; Archer, R.H.; Zengel, J.M.; Lindahl, L. The RNA of RNase MRP is required for normal processing of ribosomal RNA. Proc. Natl. Acad. Sci. USA 1994, 91, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Granneman, S.; Petfalski, E.; Tollervey, D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. EMBO J. 2011. [Google Scholar] [CrossRef]
- Faber, A.W.; Vos, H.R.; Vos, J.C.; Raue, H.A. 5′-end formation of yeast 5.8SL rRNA is an endonucleolytic event. Biochem. Biophys. Res. Commun. 2006, 345, 796–802. [Google Scholar] [CrossRef]
- Shadel, G.S.; Buckenmeyer, G.A.; Clayton, D.A.; Schmitt, M.E. Mutational analysis of the RNA component of Saccharomyces cerevisiae RNase MRP reveals distinct nuclear phenotypes. Gene 2000, 245, 175–184. [Google Scholar] [CrossRef]
- Cai, T.; Reilly, T.R.; Cerio, M.; Schmitt, M.E. Mutagenesis of SNM1, which encodes a protein component of the yeast RNase MRP, reveals a role for this ribonucleoprotein endoribonuclease in plasmid segregation. Mol. Cell Biol. 1999, 19, 7857–7869. [Google Scholar] [CrossRef]
- Chu, S.; Zengel, J.M.; Lindahl, L. A novel protein shared by RNase MRP and RNase, P. RNA 1997, 3, 382–391. [Google Scholar]
- Lygerou, Z.; Mitchell, P.; Petfalski, E.; Seraphin, B.; Tollervey, D. The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev. 1994, 8, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, L.; Bommankanti, A.; Li, X.; Hayden, L.; Jones, A.; Khan, M.; Oni, T.; Zengel, J.M. RNase MRP is required for entry of 35S precursor rRNA into the canonical processing pathway. RNA 2009, 15, 1407–1416. [Google Scholar] [CrossRef]
- Lindahl, L.; Archer, R.H.; Zengel, J.M. A new rRNA processing mutant of Saccharomyces cerevisiae. Nucleic Acids Res. 1992, 20, 295–301. [Google Scholar] [CrossRef]
- Schweizer, E.; MacKechnie, C.; Halvorson, H.O. The redundancy of ribosomal and transfer RNA genes in Saccharomyces cerevisiae. J. Mol. Biol. 1969, 40, 261–277. [Google Scholar] [CrossRef]
- Nogi, Y.; Yano, R.; Nomura, M. Synthesis of large rRNAs by RNA polymerase II in mutants of Saccharomyces. cerevisiae. defective in RNA polymerase I. Proc. Natl. Acad. Sci. USA 1991, 88, 3962–3966. [Google Scholar] [CrossRef] [PubMed]
- Nogi, Y.; Yano, R.; Dodd, J.; Carles, C.; Nomura, M. Gene RRN4 in Saccharomyces. cerevisiae. encodes the A12.2 subunit of RNA polymerase I and is essential only at high temperatures. Mol. Cell. Biol. 1993, 13, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, L.; Archer, R.H.; Zengel, J.M. Alternate pathways for processing in the internal transcribed spacer 1 in pre-rRNA of Saccharomyces. cerevisiae. Nucleic. Acids. Res. 1994, 22, 5399–5407. [Google Scholar] [CrossRef]
- Stevens, A.; Hsu, C.L.; Isham, K.R.; Larimer, F.W. Fragments of the internal transcribed spacer 1 of pre-rRNA accumulate in Saccharomyces. cerevisiae. lacking 5′-3′ exoribonuclease 1. J. Bacteriol. 1991, 173, 7024–7028. [Google Scholar] [CrossRef]
- Rubin, G.M. The nucleotide sequence of Saccharomyces. cerevisiae. 5.8 S ribosomal ribonucleic acid. J. Biol. Chem. 1973, 248, 3860–3875. [Google Scholar] [CrossRef]
- van Nues, R.W.; Rientjes, J.M.; van der Sande, C.A.; Zerp, S.F.; Sluiter, C.; Venema, J.; Planta, R.J.; Raue, H.A. Separate structural elements within internal transcribed spacer 1 of Saccharomyces cerevisiae precursor ribosomal RNA direct the formation of 17S and 26S rRNA. Nucleic Acids Res. 1994, 22, 912–919. [Google Scholar] [CrossRef]
- Li, X.; Zaman, S.; Langdon, Y.; Zengel, J.M.; Lindahl, L. Identification of a functional core in the RNA component of RNase MRP of budding yeasts. Nucleic Acids Res. 2004, 32, 3703–3711. [Google Scholar] [CrossRef][Green Version]
- Pertschy, B.; Schneider, C.; Gnadig, M.; Schafer, T.; Tollervey, D.; Hurt, E. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1. J. Biol. Chem. 2009, 284, 35079–35091. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.; Delcour, J.; Glasser, A.L.; Desgres, J.; Vandenhaute, J. The DIM1 gene responsible for the conserved m6Am6A dimethylation in the 3′-terminal loop of 18 S rRNA is essential in yeast. J. Mol. Biol. 1994, 241, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, L. Intermediates and time kinetics of the in vivo assembly of Escherichia coli ribosomes. J. Mol. Biol. 1975, 92, 15–37. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Shamsuzzaman, M.; Kondopaka, M.; Pascual, C.; Zengel, J.M.; Lindahl, L. The extended loops of ribosomal proteins uL4 and uL22 of Escherichia coli contribute to ribosome assembly and protein translation. Nucleic Acids Res. 2016, 44, 5798–5810. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Galan, O.; Garcia-Gomez, J.J.; Kressler, D.; de la Cruz, J. Immature large ribosomal subunits containing the 7S pre-rRNA can engage in translation in Saccharomyces cerevisiae. RNA Biol. 2015, 12, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Vos, H.R.; Faber, A.W.; de Gier, M.D.; Vos, J.C.; Raue, H.A. Deletion of the three distal S1 motifs of Saccharomyces cerevisiae Rrp5p abolishes pre-rRNA processing at site A without reducing the production of functional 40S subunits. Eukaryot. Cell. 2004, 3, 1504–1512. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Esakova, O.; Perederina, A.; Quan, C.; Berezin, I.; Krasilnikov, A.S. Substrate recognition by ribonucleoprotein ribonuclease MRP. RNA 2011, 17, 356–364. [Google Scholar] [CrossRef]
- Lan, P.; Zhou, B.; Tan, M.; Li, S.; Cao, M.; Wu, J.; Lei, M. Structural insight into precursor ribosomal RNA processing by ribonuclease MRP. Science 2020, 369, 656–663. [Google Scholar] [CrossRef]
- Perederina, A.; Li, D.; Lee, H.; Bator, C.; Berezin, I.; Hafenstein, S.L.; Krasilnikov, A.S. Cryo-EM structure of catalytic ribonucleoprotein complex RNase MRP. Nat. Commun. 2020, 11, 3474. [Google Scholar] [CrossRef]
- Goldfarb, K.C.; Cech, T.R. Targeted CRISPR disruption reveals a role for RNase MRP RNA in human preribosomal RNA processing. Genes Dev. 2017, 31, 59–71. [Google Scholar] [CrossRef]
- Fatica, A.; Oeffinger, M.; Dlakic, M.; Tollervey, D. Nob1p is required for cleavage of the 3′ end of 18S rRNA. Mol. Cell. Biol. 2003, 23, 1798–1807. [Google Scholar] [CrossRef][Green Version]
- Ameismeier, M.; Zemp, I.; van den Heuvel, J.; Thoms, M.; Berninghausen, O.; Kutay, U.; Beckmann, R. Structural basis for the final steps of human 40S ribosome maturation. Nature 2020, 587, 683–687. [Google Scholar] [CrossRef]
- Turowski, T.W.; Lebaron, S.; Zhang, E.; Peil, L.; Dudnakova, T.; Petfalski, E.; Granneman, S.; Rappsilber, J.; Tollervey, D. Rio1 mediates ATP-dependent final maturation of 40S ribosomal subunits. Nucleic Acids Res. 2014, 42, 12189–12199. [Google Scholar] [CrossRef]
- Zhang, J.; McCann, K.L.; Qiu, C.; Gonzalez, L.E.; Baserga, S.J.; Hall, T.M. Nop9 is a PUF-like protein that prevents premature cleavage to correctly process pre-18S rRNA. Nat. Commun. 2016, 7, 13085. [Google Scholar] [CrossRef] [PubMed]
- Udem, S.A.; Warner, J.R. The cytoplasmic maturation of a ribosomal precursor ribonucleic acid in yeast. J. Biol. Chem. 1973, 248, 1412–1416. [Google Scholar] [CrossRef]
- Moy, T.I.; Silver, P.A. Nuclear export of the small ribosomal subunit requires the ran-GTPase cycle and certain nucleoporins [In Process Citation]. Genes Dev. 1999, 13, 2118–2133. [Google Scholar] [CrossRef]
- Pena, C.; Hurt, E.; Panse, V.G. Eukaryotic ribosome assembly, transport and quality control. Nat. Struct. Mol. Biol. 2017, 24, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Pratte, D.; Singh, U.; Murat, G.; Kressler, D. Mak5 and Ebp2 act together on early pre-60S particles and their reduced functionality bypasses the requirement for the essential pre-60S factor Nsa1. PLoS ONE 2013, 8, e82741. [Google Scholar] [CrossRef]
- Torchet, C.; Hermann-Le Denmat, S. Bypassing the rRNA processing endonucleolytic cleavage at site A2 in Saccharomyces cerevisiae. RNA 2000, 6, 1498–1508. [Google Scholar] [CrossRef][Green Version]
- Thoms, M.; Mitterer, V.; Kater, L.; Falquet, L.; Beckmann, R.; Kressler, D.; Hurt, E. Suppressor mutations in Rpf2-Rrs1 or Rpl5 bypass the Cgr1 function for pre-ribosomal 5S RNP-rotation. Nat. Commun. 2018, 9, 4094. [Google Scholar] [CrossRef] [PubMed]
- Larimer, F.W.; Stevens, A. Disruption of the gene XRN1, coding for a 5′→3′ exoribonuclease, restricts yeast cell growth. Gene 1990, 95, 85–90. [Google Scholar] [CrossRef]
- Sherman, F.; Lawrence, C.W.; Fink, G.R. Methods in Yeast Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1979; p. 98. [Google Scholar]
- Gregory, B.; Rahman, N.; Bommakanti, A.; Shamsuzzaman, M.; Thapa, M.; Lescure, A.; Zengel, J.M.; Lindahl, L. The small and large ribosomal subunits depend on each other for stability and accumulation. Life. Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- McCormick, J.R.; Zengel, J.M.; Lindahl, L. Intermediates in the degradation of mRNA from the lactose operon of Escherichia coli. Nucleic Acids Res. 1991, 19, 2767–2776. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligo | Sequence (5’→3’) | Complementary to |
|---|---|---|
| O9 | GCT CTT GCC AAA ACA AAA AAA TCC A | ITS1 24–53 |
| O20 | AAA TGA CGC TCA AAC AGG CAT GCC C | 5.8S 3′ end |
| O90 | GTA TCA CTC ACT ACC AAA CAG AAT G | ITS2 11–35 |
| O453 | AAC AAA AAA ATC CAT TTT CAA | ITS1 15–25 |
| O552 | CCA GTT ACG CGT TTT AAT TG | Spanning ∆2 deletion, see text |
| O553 | ATG CCT TTG GTA GAA CCC AAA GGC | S. pombe hairpin inserted in S. cerevisiae 5.8S |
| O576 | ATG CCT TTG GTA GAA CCC | S. pombe hairpin inserted in S. cerevisiae 5.8S |
| Name | Genotype | References |
|---|---|---|
| ∆rpa12 alias NOY504 | MATa rm4 (rpa12)::LEU2 ade2-101 ura3-1 trp1-1 leu2-3,112 his3-101 can1-100 | [34] |
| ∆rpa135 alias NOY408-la | MATa rpa135::LEU2 ade2-1 ura3-1 his3-10 1trp1-1 leu2-112 can1-100/pNOY102 | [33] |
| ∆rpa135/pDK16 | MATa rpa135::LEU2 ade2-l ura3-l his3-ll trpl-1 leu2-3,112 can1-100/pDK16 | This study |
| YLL53 | MATa ade2-101 his3∆200 ura3-52tyr1RRP2 | [23] |
| YLL54 | MATa ade2-101 his3∆200 ura3-52, lys2, rrp2-2 | [23] |
| pDK16 | YEplac112 carrying the rRNA transcription unit expressed from the CUP1 promoter | [35] |
| pDK16-tag | pDK16 with tagged 5,8S gene (see text) | This study |
| ITS1 Nucleotides | L-Fraction | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutant | A2 | A3 | 283–289 | 290–299 | 300–330 | 331–340 | ∆rpa12 ∆xrn1 | RRP2 (YLL53) | HI mw interm in ∆rpa12 ∆xrn1 | RRP2 sensitivity |
| wt | yes | yes | P | P | P | P | 0.21 | 0.20 | − | + |
| ∆13 | no | yes | P | P | P | P | 0.19 | 0.21 | − | + |
| ∆14 | no | no | A | A | P | P | 0.31 | 0.34 | − | + |
| ∆15 | yes | no | A | A | P | P | 0.26 | 0.40 | − | + |
| ∆16 | yes | yes | P | P | P | P | 0.22 | 0.14 | − | + |
| ∆17 | yes | yes | P | P | A | A | 0.61 | 0.88 | + | − |
| ∆18 | yes | half | P | A | P | P | 0.50 | 0.80 | + | − |
| ∆19 | yes | no | A | A | A | P | 0.56 | 0.64 | − | nd |
| ∆2 | no | no | A | A | A | A | 0.74 | 0.82 | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zengel, J.M.; Lindahl, L. A Novel Model for the RNase MRP-Induced Switch between the Formation of Different Forms of 5.8S rRNA. Int. J. Mol. Sci. 2021, 22, 6690. https://doi.org/10.3390/ijms22136690
Li X, Zengel JM, Lindahl L. A Novel Model for the RNase MRP-Induced Switch between the Formation of Different Forms of 5.8S rRNA. International Journal of Molecular Sciences. 2021; 22(13):6690. https://doi.org/10.3390/ijms22136690
Chicago/Turabian StyleLi, Xiao, Janice M. Zengel, and Lasse Lindahl. 2021. "A Novel Model for the RNase MRP-Induced Switch between the Formation of Different Forms of 5.8S rRNA" International Journal of Molecular Sciences 22, no. 13: 6690. https://doi.org/10.3390/ijms22136690
APA StyleLi, X., Zengel, J. M., & Lindahl, L. (2021). A Novel Model for the RNase MRP-Induced Switch between the Formation of Different Forms of 5.8S rRNA. International Journal of Molecular Sciences, 22(13), 6690. https://doi.org/10.3390/ijms22136690