
Epigenetic Regulation of Autophagy in Cardiovascular Pathobiology


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
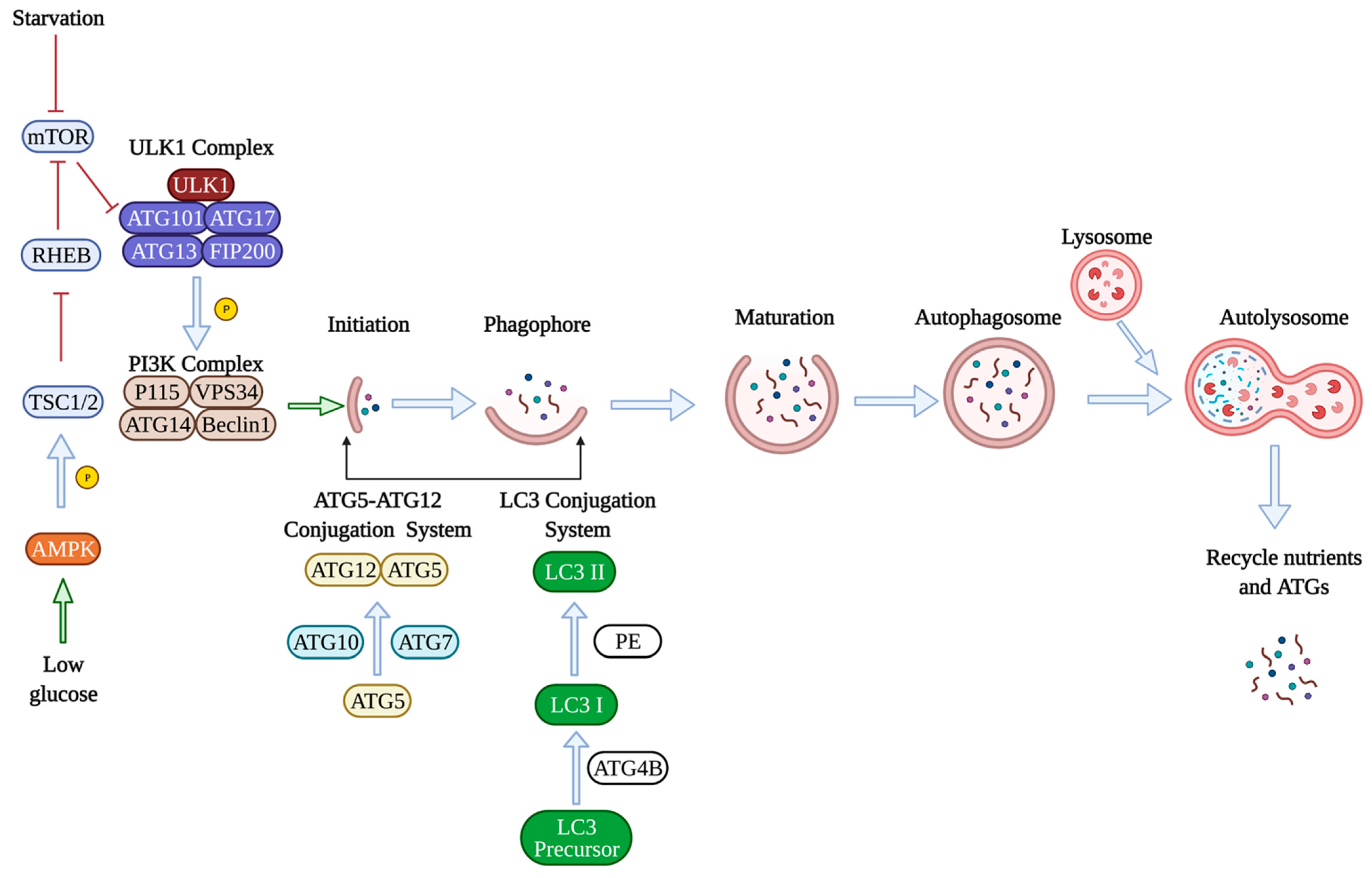
2. Molecular Mechanisms of Autophagy
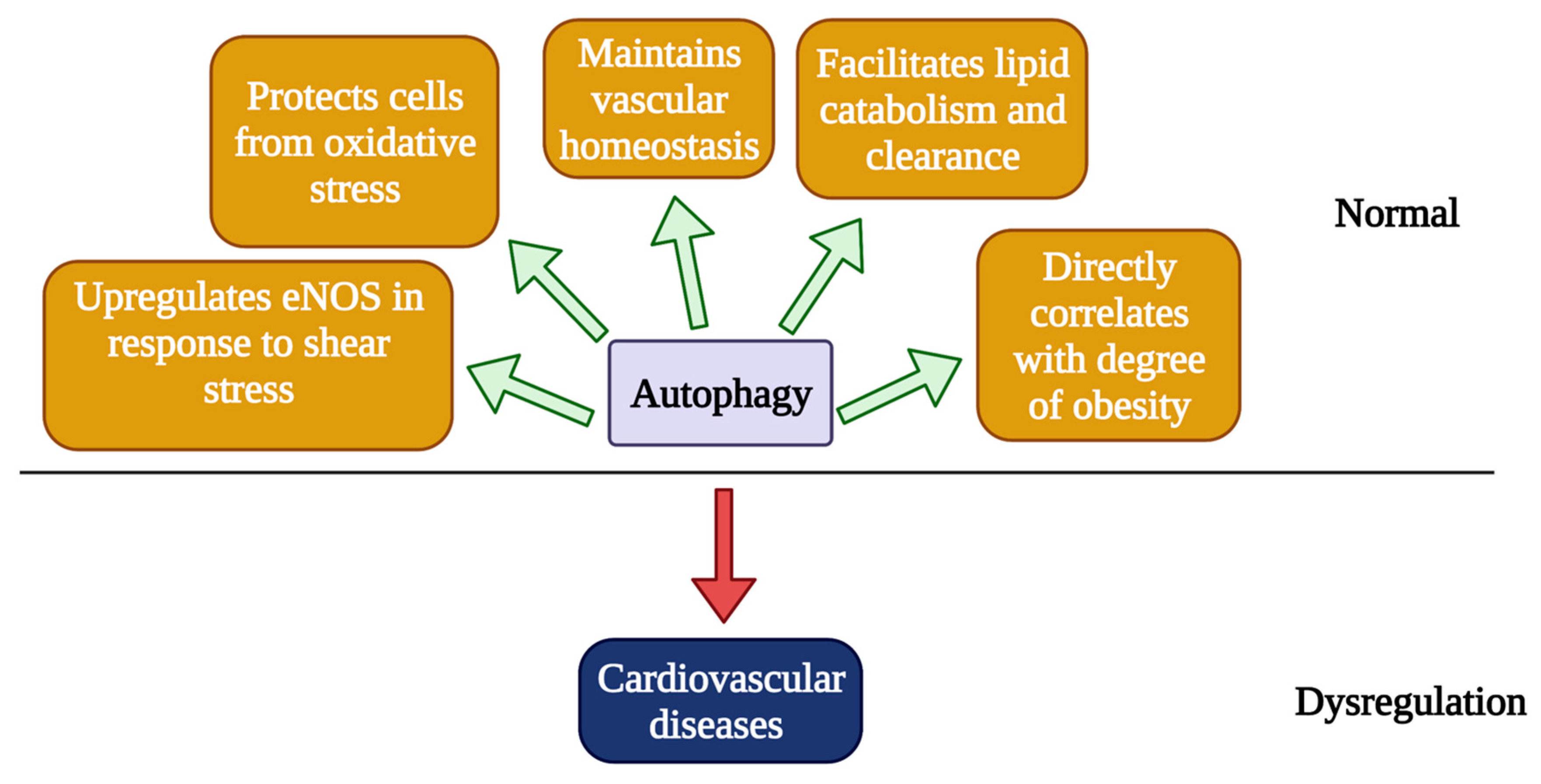
3. Autophagy in Cardiovascular Pathobiology
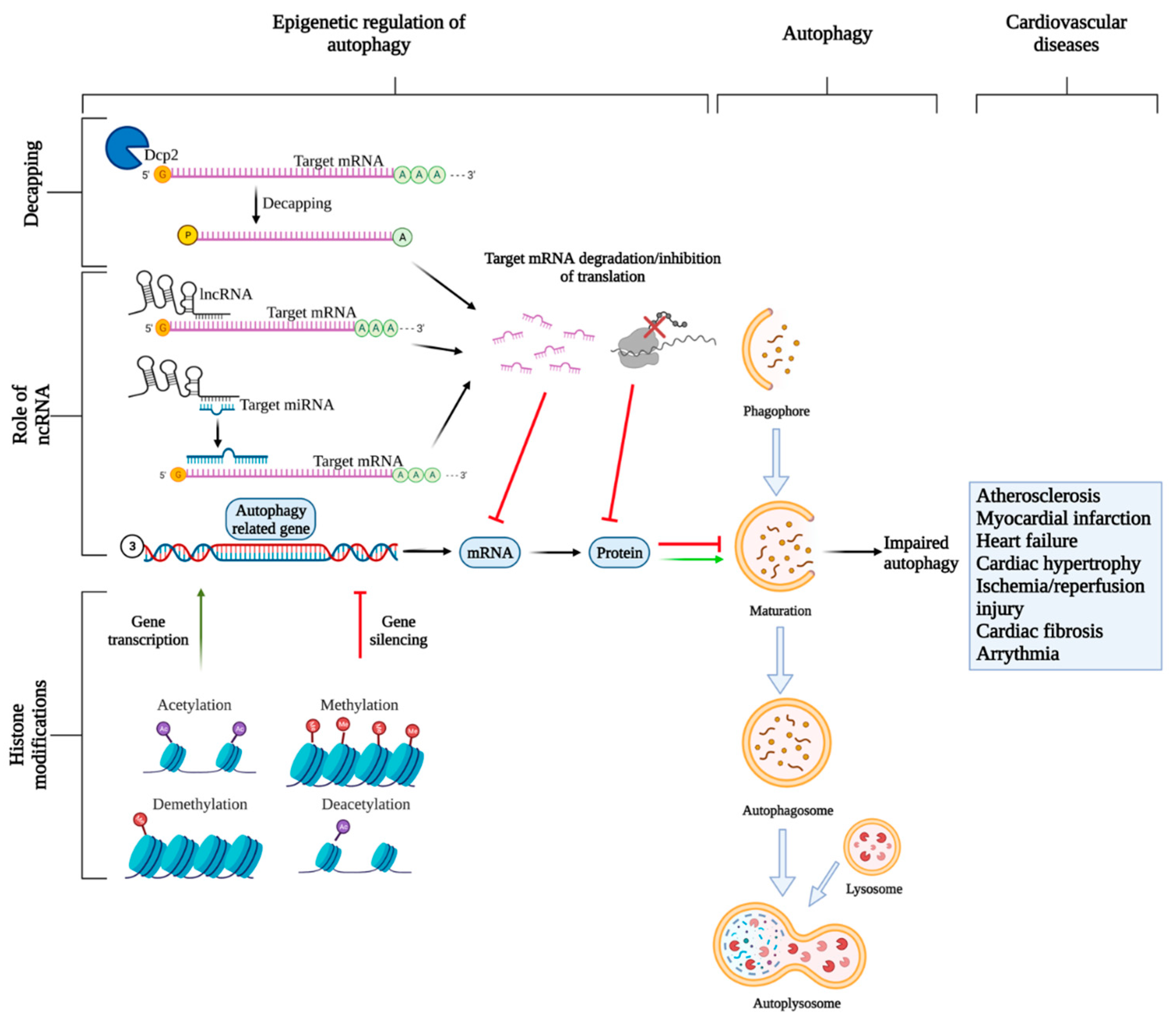
4. mRNA Transcript Degradation: Role of Non-Coding RNAs (ncRNAs)
5. mRNA Transcript Degradation: Role of Regulated mRNA Decapping/Degradation
6. mRNA Transcription: Role of Histone Modifications
7. Epigenetic Regulation in CVDs
7.1. Atherosclerosis
7.2. Cardiac Contractility Defects
7.3. Heart Failure
7.3.1. Myocardial Infarction
7.3.2. Cardiac Hypertrophy and Cardiac Fibrosis
8. Therapeutic Potential of ncRNA in CVDs
9. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An Overview of the Molecular Mechanism of Autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2014, 15, 65–74. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The Essential Autophagy Gene ATG7 Modulates Organ Fibrosis via Regulation of Endothelial-to-Mesenchymal Transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- World Health Organization (Ed.) Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2020, 10, 1568. [Google Scholar] [CrossRef] [PubMed]
- Lavandero, S.; Chiong, M.; Rothermel, B.A.; Hill, J.A. Autophagy in cardiovascular biology. J. Clin. Investig. 2015, 125, 55–64. [Google Scholar] [CrossRef]
- Freedman, J.E. Molecular Regulation of Platelet-Dependent Thrombosis. Circulation 2005, 112, 2725–2734. [Google Scholar] [CrossRef]
- Han, J.; Pan, X.-Y.; Xu, Y.; Xiao, Y.; An, Y.; Tie, L.; Pan, Y.; Li, X.-J. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012, 8, 812–825. [Google Scholar] [CrossRef]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell 2015, 15, 187–191. [Google Scholar] [CrossRef]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calò, L.A. The Role of Oxidized Low-Density Lipoproteins in Atherosclerosis: The Myths and the Facts. Mediat. Inflamm. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Altamimi, T.R.; Chowdhury, B.; Singh, K.K.; Zhang, L.; Mahmood, M.U.; Pan, Y.; Quan, A.; Teoh, H.; Verma, S.; Lopaschuk, G.D. A novel role of endothelial autophagy as a regulator of myocardial fatty acid oxidation. J. Thorac. Cardiovasc. Surg. 2019, 157, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Thompson, M.D.; Cohen, R.A.; Tong, X. Autophagy and oxidative stress in cardiovascular diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 243–251. [Google Scholar] [CrossRef]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.-H. 2-Deoxy-D-Glucose Treatment of Endothelial Cells Induces Autophagy by Reactive Oxygen Species-Mediated Activation of the AMP-Activated Protein Kinase. PLoS ONE 2011, 6, e17234. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 1–31. [Google Scholar] [CrossRef]
- Hill, B.; Haberzettl, P.; Ahmed, Y.; Srivastava, S.; Bhatnagar, A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem. J. 2008, 410, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Haberzettl, P.; Hill, B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013, 1, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Gavras, I.; Gavras, H. Angiotensin II as a cardiovascular risk factor. J. Hum. Hypertens. 2002, 16, S2–S6. [Google Scholar] [CrossRef]
- Shan, H.; Guo, D.; Li, X.; Zhao, X.; Li, W.; Bai, X. From autophagy to senescence and apoptosis in Angiotensin II-treated vascular endothelial cells. Acta Pathol. Microbiol. Immunol. Scand. APMIS 2014, 122, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, A.; Singh, K.K.; Quan, A.; Plant, P.J.; Al-Omran, M.; Teoh, H.; Verma, S. Loss of vascular smooth muscle cell autophagy exacerbates angiotensin II-associated aortic remodeling. J. Vasc. Surg. 2018, 68, 859–871. [Google Scholar] [CrossRef]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yao, Z.; Jin, M.; Namkoong, S.; Yin, Z.; Lee, J.H.; Klionsky, D.J. Dhh1 promotes autophagy-related protein translation during nitrogen starvation. PLoS Biol. 2019, 17, e3000219. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.-J.; Liu, G.-S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef]
- Guo, F.; Li, X.; Peng, J.; Tang, Y.; Yang, Q.; Liu, L.; Wang, Z.; Jiang, Z.; Xiao, M.; Ni, C.; et al. Autophagy Regulates Vascular Endothelial Cell eNOS and ET-1 Expression Induced by Laminar Shear Stress in an Ex Vivo Perfused System. Ann. Biomed. Eng. 2014, 42, 1978–1988. [Google Scholar] [CrossRef]
- Yang, Q.; Li, X.; Li, R.; Peng, J.; Wang, Z.; Jiang, Z.; Tang, X.; Peng, Z.; Wang, Y.; Wei, D. Low Shear Stress Inhibited Endothelial Cell Autophagy Through TET2 Downregulation. Ann. Biomed. Eng. 2015, 44, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Münzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Carbone, S.; Canada, J.M.; Billingsley, H.E.; Siddiqui, M.S.; Elagizi, A.; Lavie, C.J. Obesity paradox in cardiovascular disease: Where do we stand? Vasc. Health Risk Manag. 2019, 15, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.; et al. Altered Autophagy in Human Adipose Tissues in Obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hua, Y.; Sreejayan, N.; Zhang, Y.; Ren, J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J. Mol. Cell Biol. 2012, 5, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef]
- Singh, K.K.; Yanagawa, B.; Quan, A.; Wang, R.; Garg, A.; Khan, R.; Pan, Y.; Wheatcroft, M.D.; Lovren, F.; Teoh, H.; et al. Autophagy gene fingerprint in human ischemia and reperfusion. J. Thorac. Cardiovasc. Surg. 2014, 147, 1065–1072.e1. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 3. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Yao, R.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.-H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Babiarz, J.E.; Ruby, J.G.; Wang, Y.; Bartel, D.P.; Blelloch, R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008, 22, 2773–2785. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.H.; Shin, S.; Jung, S.-R.; Kim, E.; Song, J.-J.; Hohng, S. Human Argonaute 2 Has Diverse Reaction Pathways on Target RNAs. Mol. Cell 2015, 59, 117–124. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, H.; Liu, X.; Li, B.; Chen, Y.; Ren, X.; Liu, C.-G.; Yang, J.-M. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009, 5, 816–823. [Google Scholar] [CrossRef]
- Wang, X.; Liu, P.; Zhu, H.; Xu, Y.; Ma, C.; Dai, X.; Huang, L.; Liu, Y.; Zhang, L.; Qin, C. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res. Bull. 2009, 80, 268–273. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef]
- Yang, J.; Chen, D.; He, Y.; Meléndez, A.; Feng, Z.; Hong, Q.; Bai, X.; Li, Q.; Cai, G.; Wang, J.; et al. MiR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene atg9. Age 2011, 35, 11–22. [Google Scholar] [CrossRef]
- Khanna, A.; Muthusamy, S.; Liang, R.; Sarojini, H.; Wang, E. Gain of survival signaling by down-regulation of three key miRNAs in brain of calorie-restricted mice. Aging 2011, 3, 223–236. [Google Scholar] [CrossRef]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A Polycistronic MicroRNA Cluster, miR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed]
- Meenhuis, A.; van Veelen, P.; De Looper, H.; Van Boxtel, N.; van de Berge, I.J.; Sun, S.M.; Taskesen, E.; Stern, P.; De Ru, A.; Van Adrichem, A.; et al. MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1–regulated pathways in mice. Blood 2011, 118, 916–925. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.; Tan, X.; Jing, H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012, 8, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Hornstein, E.; Mansfield, J.H.; Yekta, S.; Hu, J.K.-H.; Harfe, B.D.; McManus, M.T.; Baskerville, S.; Bartel, D.P.; Tabin, C.J. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature 2005, 438, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.-F.; Hébuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165. [Google Scholar] [CrossRef]
- Zhai, Z.; Wu, F.; Dong, F.; Chuang, A.Y.; Messer, J.S.; Boone, D.L.; Kwon, J.H. Human autophagy geneATG16L1is post-transcriptionally regulated byMIR142-3p. Autophagy 2014, 10, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12–5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Chen, J.; Xu, H.; Zhou, X.; He, Q.; Li, Y.; Jiang, G.; Shan, Y.; Xue, B.; Zhao, R.; et al. MIR106B and MIR93 Prevent Removal of Bacteria from Epithelial Cells by Disrupting ATG16L1-Mediated Autophagy. Gastroenterology 2014, 146, 188–199. [Google Scholar] [CrossRef]
- Tekirdag-Kosar, A.; Korkmaz, G.; Öztürk, D.G.; Agami, R.; Gozuacik, D. MIR181Aregulates starvation- and rapamycin-induced autophagy through targeting ofATG5. Autophagy 2013, 9, 374–385. [Google Scholar] [CrossRef]
- Yeh, C.-F.; Chang, Y.-C.E.; Lu, C.-Y.; Hsuan, C.-F.; Chang, W.-T.; Yang, K.-C. Expedition to the missing link: Long noncoding RNAs in cardiovascular diseases. J. Biomed. Sci. 2020, 27, 1–16. [Google Scholar] [CrossRef]
- Wang, K.; Liu, C.-Y.; Zhou, L.-Y.; Wang, J.-X.; Wang, M.; Zhao, B.; Zhao, W.-K.; Jian-Xun, W.; Yan-Fang, Z.; Zhang, X.-J.; et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat. Commun. 2015, 6, 6779. [Google Scholar] [CrossRef]
- Yin, G.; Yang, X.; Li, Q.; Guo, Z. GATA1 activated lncRNA (Galont) promotes anoxia/reoxygenation-induced autophagy and cell death in cardiomyocytes by sponging miR-338. J. Cell. Biochem. 2018, 119, 4161–4169. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, F.; Zhou, L.-Y.; Long, B.; Yuan, S.-M.; Wang, Y.; Liu, C.-Y.; Sun, T.; Zhang, X.-J.; Li, P.-F. The Long Noncoding RNA CHRF Regulates Cardiac Hypertrophy by Targeting miR-489. Circ. Res. 2014, 114, 1377–1388. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, X.; Huang, J. Long Non-Coding RNA-ROR Mediates the Reprogramming in Cardiac Hypertrophy. PLoS ONE 2016, 11, e0152767. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Molkentin, J.D. Regulation of cardiac hypertrophy and remodeling through the dual-specificity MAPK phosphatases (DUSPs). J. Mol. Cell. Cardiol. 2016, 101, 44–49. [Google Scholar] [CrossRef]
- Zhao, Z.-H.; Hao, W.; Meng, Q.-T.; Du, X.-B.; Lei, S.-Q.; Xia, Z.-Y. Long non-coding RNA MALAT1 functions as a mediator in cardioprotective effects of fentanyl in myocardial ischemia-reperfusion injury. Cell Biol. Int. 2017, 41, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Deng, J.; Xu, J.; Wang, H.; Yuan, M.; Liu, N.; Jiang, Y.; Liu, J. High-mobility group box 1 (HMGB1) downregulates cardiac transient outward potassium current (Ito) through downregulation of Kv4.2 and Kv4.3 channel transcripts and proteins. J. Mol. Cell. Cardiol. 2010, 49, 438–448. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Tang, N. Long noncoding RNA Malat1 is a potent autophagy inducer protecting brain microvascular endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury by sponging miR-26b and upregulating ULK2 expression. Neuroscience 2017, 354, 1–10. [Google Scholar] [CrossRef]
- Guo, X.; Wu, X.; Han, Y.; Tian, E.; Cheng, J. LncRNA MALAT1 protects cardiomyocytes from isoproterenol-induced apoptosis through sponging miR-558 to enhance ULK1-mediated protective autophagy. J. Cell. Physiol. 2019, 234, 10842–10854. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, T.; Duan, J.; Mu, N.; Zhang, T. MALAT1/miR-15b-5p/MAPK1 mediates endothelial progenitor cells autophagy and affects coronary atherosclerotic heart disease via mTOR signaling pathway. Aging 2019, 11, 1089–1109. [Google Scholar] [CrossRef]
- Liang, H.; Su, X.; Wu, Q.; Shan, H.; Lv, L.; Yu, T.; Zhao, X.; Sun, J.; Yang, R.; Zhang, L.; et al. LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a. Autophagy 2020, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xu, W.; Zhang, W.; Wang, W.; Liu, T.; Zhou, X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics 2019, 9, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lu, W.; Ge, D.; Meng, N.; Li, Y.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. A new microRNA signal pathway regulated by long noncoding RNA TGFB2-OT1 in autophagy and inflammation of vascular endothelial cells. Autophagy 2015, 11, 2172–2183. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; de Stegmann, D.M.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 Regulates Autophagosome-Lysosome Fusion through HOPS Complex and LC3/GABARAP Proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef]
- Viereck, J.; Kumarswamy, R.; Foinquinos, A.; Xiao, K.; Avramopoulos, P.; Kunz, M.; Dittrich, M.; Maetzig, T.; Zimmer, K.; Remke, J.; et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl. Med. 2016, 8, 326ra22. [Google Scholar] [CrossRef]
- Hu, Y.-W.; Guo, F.-X.; Xu, Y.-J.; Li, P.; Lu, Z.-F.; McVey, D.G.; Zheng, L.; Wang, Q.; Ye, J.H.; Kang, C.-M.; et al. Long noncoding RNA NEXN-AS1 mitigates atherosclerosis by regulating the actin-binding protein NEXN. J. Clin. Investig. 2019, 129, 1115–1128. [Google Scholar] [CrossRef]
- Sallam, T.; Jones, M.; Thomas, B.J.; Wu, X.; Gilliland, T.; Qian, K.; Eskin, A.; Casero, D.; Zhang, Z.; Sandhu, J.; et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat. Med. 2018, 24, 304–312. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Zhang, Y.-H.; Li, R.-B.; Zhou, L.-Y.; Chan, S.; Zhang, R.-C.; Zhai, M.; Huang, Y.; Yan-Hui, Z.; Dong, Y.-H.; et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Zhuo, C.; Jiang, R.; Lin, X.; Shao, M. LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget 2016, 8, 1429–1437. [Google Scholar] [CrossRef]
- Ramanathan, A.; Robb, G.B.; Chan, S.-H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Hu, G.; McQuiston, T.; Bernard, A.; Park, Y.-D.; Qiu, J.; Vural, A.; Zhang, N.; Waterman, S.R.; Blewett, N.H.; Myers, T.G.; et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat. Cell. Biol. 2015, 17, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Panepinto, J.; Liu, L.; Ramos, J.; Zhu, X.; Valyi-Nagy, T.; Eksi, S.; Fu, J.; Jaffe, H.A.; Wickes, B.; Williamson, P.R. The DEAD-box RNA helicase Vad1 regulates multiple virulence-associated genes in Cryptococcus neoformans. J. Clin. Investig. 2005, 115, 632–641. [Google Scholar] [CrossRef]
- Lucas, C.L.; Kuehn, H.S.; Zhao, F.; Niemela, J.E.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell. Signal. 2009, 21, 1356–1360. [Google Scholar] [CrossRef]
- Hajji, N.; Wallenborg, K.; Vlachos, P.; Füllgrabe, J.; Hermanson, O.; Joseph, B. Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II inhibitor etoposide. Oncogene 2010, 29, 2192–2204. [Google Scholar] [CrossRef]
- Shintani, T. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 Negatively Regulates the Mammalian Target of Rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.-Z.; Cao, Z.; Wang, X.; Wang, H.; Cai, M.-Y.; Li, T.; Hattori, N.; Wang, D.; Du, Y.; Song, B.; et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy 2015, 11, 2309–2322. [Google Scholar] [CrossRef] [PubMed]
- De Narvajas, A.A.-M.; Gomez, T.S.; Zhang, J.-S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.; Ellenrieder, V.; Bujanda, L.; et al. Epigenetic Regulation of Autophagy by the Methyltransferase G9a. Mol. Cell. Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.R.; Kim, H.; Oh, S.; Lee, J.-G.; Kee, M.; Ko, H.-J.; Kweon, M.-N.; Won, K.-J.; Baek, S.H. AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef]
- Shao, M.; Chen, G.; Lv, F.; Liu, Y.; Tian, H.; Tao, R.; Jiang, R.; Zhang, W.; Zhuo, C. LncRNA TINCR attenuates cardiac hypertrophy by epigenetically silencing CaMKII. Oncotarget 2017, 8, 47565–47573. [Google Scholar] [CrossRef]
- Kapoor-Vazirani, P.; Kagey, J.D.; Vertino, P.M. SUV420H2-Mediated H4K20 Trimethylation Enforces RNA Polymerase II Promoter-Proximal Pausing by Blocking hMOF-Dependent H4K16 Acetylation. Mol. Cell. Biol. 2011, 31, 1594–1609. [Google Scholar] [CrossRef]
- Hu, H.; Wu, J.; Yu, X.; Zhou, J.; Yu, H.; Ma, L. Long non-coding RNA MALAT1 enhances the apoptosis of cardiomyocytes through autophagy inhibition by regulating TSC2-mTOR signaling. Biol. Res. 2019, 52, 1–10. [Google Scholar] [CrossRef]
- Cardenas, C.L.L.; Kessinger, C.W.; Cheng, Y.; Macdonald, C.; MacGillivray, T.; Ghoshhajra, B.; Huleihel, L.; Nuri, S.; Yeri, A.S.; Jaffer, F.A.; et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; A Mengi, S.; Xu, Y.-J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.A.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Shang, C.; Jin, Z.; Yao, F.; Bai, L.; Wang, R.; Zhao, S.; Liu, E. miR-145 inhibits the proliferation and migration of vascular smooth muscle cells by regulating autophagy. J. Cell. Mol. Med. 2020, 24, 6658–6669. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Shukla, P.C.; Gupta, N.; Steer, B.M.; Ingram, A.J.; Gupta, M.; Al-Omran, M.; et al. MicroRNA-145 Targeted Therapy Reduces Atherosclerosis. Circulation 2012, 126 (Suppl. S1), S81–S90. [Google Scholar] [CrossRef]
- Guo, F.-X.; Wu, Q.; Li, P.; Zheng, L.; Ye, S.; Dai, X.-Y.; Kang, C.-M.; Lu, J.-B.; Xu, B.-M.; Xu, Y.-J.; et al. The role of the LncRNA-FA2H-2-MLKL pathway in atherosclerosis by regulation of autophagy flux and inflammation through mTOR-dependent signaling. Cell Death Differ. 2019, 26, 1670–1687. [Google Scholar] [CrossRef]
- Sugiura, S.; Kobayakawa, N.; Fujita, H.; Momomura, S.-I.; Chaen, S.; Sugi, H. Distinct kinetic properties of cardiac myosin isoforms revealed by in vitro studies. Adv. Exp. Med. Biol. 1998, 453, 125–130. [Google Scholar] [CrossRef]
- Fujiwara, K.; Daido, S.; Yamamoto, A.; Kobayashi, R.; Yokoyama, T.; Aoki, H.; Iwado, E.; Shinojima, N.; Kondo, Y.; Kondo, S. Pivotal Role of the Cyclin-dependent Kinase Inhibitor p21WAF1/CIP1 in Apoptosis and Autophagy. J. Biol. Chem. 2008, 283, 388–397. [Google Scholar] [CrossRef]
- Chang, C.-P.; Yang, J.; Han, P.; Cheng, H.-L.; Shang, C.; Ashley, E.; Zhou, B. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2010, 466, 62–67. [Google Scholar] [CrossRef]
- Han, P.; Li, W.; Lin, C.-H.; Yang, J.; Shang, C.; Nuernberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.-Y.; Lin, C.-J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef]
- Nishi, H.; Ono, K.; Horie, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Watanabe, S.; Takaya, T.; Tamaki, Y.; Takanabe-Mori, R.; et al. MicroRNA-27a Regulates Beta Cardiac Myosin Heavy Chain Gene Expression by Targeting Thyroid Hormone Receptor 1 in Neonatal Rat Ventricular Myocytes. Mol. Cell. Biol. 2010, 31, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A Family of microRNAs Encoded by Myosin Genes Governs Myosin Expression and Muscle Performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef]
- Rawal, S.; Nagesh, P.T.; Coffey, S.; Van Hout, I.; Galvin, I.F.; Bunton, R.W.; Davis, P.; Williams, M.J.A.; Katare, R. Early dysregulation of cardiac-specific microRNA-208a is linked to maladaptive cardiac remodelling in diabetic myocardium. Cardiovasc. Diabetol. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Harada, M.; Luo, X.; Murohara, T.; Yang, B.; Dobrev, D.; Nattel, S. MicroRNA Regulation and Cardiac Calcium Signaling. Circ. Res. 2014, 114, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Lipskaia, L.; Chemaly, E.R.; Hadri, L.; Lompre, A.-M.; Hajjar, R.J. Sarcoplasmic reticulum Ca2+ATPase as a therapeutic target for heart failure. Expert Opin. Biol. Ther. 2009, 10, 29–41. [Google Scholar] [CrossRef]
- Wahlquist, C.; Jeong, D.; Rojas-Muñoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; Van Mil, A.; Park, W.J.; Sluijter, J.; Doevendans, P.A.F.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef]
- Ai, J.; Zhang, R.; Gao, X.; Niu, H.-F.; Wang, N.; Xu, Y.; Li, Y.; Ma, N.; Sun, L.-H.; Pan, Z.-W.; et al. Overexpression of microRNA-1 impairs cardiac contractile function by damaging sarcomere assembly. Cardiovasc. Res. 2012, 95, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Elton, T.S.; Györke, S. miR-1 Overexpression Enhances Ca2+ Release and Promotes Cardiac Arrhythmogenesis by Targeting PP2A Regulatory Subunit B56α and Causing CaMKII-Dependent Hyperphosphorylation of RyR2. Circ. Res. 2009, 104, 514–521. [Google Scholar] [CrossRef]
- Danielson, L.S.; Park, D.S.; Rotllan, N.; Jorganes, A.C.; Guijarro, M.V.; Fernandez-Hernando, C.; Fishman, G.I.; Phoon, C.K.L.; Hernando, E. Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 2013, 27, 1460–1467. [Google Scholar] [CrossRef]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.-L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Leonard, B.L.; Smaill, B.H.; LeGrice, I.J. Structural Remodeling and Mechanical Function in Heart Failure. Microsc. Microanal. 2012, 18, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Sygitowicz, G.; Tomaniak, M.; Błaszczyk, O.; Kołtowski, Ł.; Filipiak, K.J.; Sitkiewicz, D. Circulating microribonucleic acids miR-1, miR-21 and miR-208a in patients with symptomatic heart failure: Preliminary results. Arch. Cardiovasc. Dis. 2015, 108, 634–642. [Google Scholar] [CrossRef]
- Tijsen, A.J.; Creemers, E.E.; Moerland, P.D.; De Windt, L.J.; Van Der Wal, A.C.; Kok, W.E.; Pinto, Y.M. MiR423-5p As a Circulating Biomarker for Heart Failure. Circ. Res. 2010, 106, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Van Boven, N.; Kardys, I.; Van Vark, L.C.; Akkerhuis, K.M.; De Ronde, M.W.J.; Khan, M.A.F.; Merkus, D.; Liu, Z.; Voors, A.A.; Asselbergs, F.W.; et al. Serially measured circulating microRNAs and adverse clinical outcomes in patients with acute heart failure. Eur. J. Heart Fail. 2018, 20, 89–96. [Google Scholar] [CrossRef]
- Lu, L.; Liu, M.; Sun, R.; Zheng, Y.; Zhang, P. Myocardial Infarction: Symptoms and Treatments. Cell Biophys. 2015, 72, 865–867. [Google Scholar] [CrossRef]
- Takemura, G.; Nakagawa, M.; Kanamori, H.; Minatoguchi, S.; Fujiwara, H. Benefits of reperfusion beyond infarct size limitation. Cardiovasc. Res. 2009, 83, 269–276. [Google Scholar] [CrossRef][Green Version]
- Liu, X.; Fan, Z.; Zhao, T.; Cao, W.; Zhang, L.; Li, H.; Xie, Q.; Tian, Y.; Wang, B. Plasma miR-1, miR-208, miR-499 as potential predictive biomarkers for acute myocardial infarction: An independent study of Han population. Exp. Gerontol. 2015, 72, 230–238. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Chen, W.; Xie, L.; Zhao, Z.-A.; Yang, J.; Chen, Y.; Lei, W.; Shen, Z. MicroRNA-133 overexpression promotes the therapeutic efficacy of mesenchymal stem cells on acute myocardial infarction. Stem Cell Res. Ther. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Hu, Y.; Hou, L.; Ju, J.; Li, X.; Du, N.; Guan, X.; Liu, Z.; Zhang, T.; Qin, W.; et al. β-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J. Mol. Cell. Cardiol. 2014, 75, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, H.; Yang, D.; He, F.; Yuan, Y.; Guo, J.; Hu, J.; Yu, J.; Yan, X.; Wang, S.; et al. Aloe-emodin attenuates myocardial infarction and apoptosis via up-regulating miR-133 expression. Pharmacol. Res. 2019, 146, 104315. [Google Scholar] [CrossRef]
- Wang, R.; Li, N.; Zhang, Y.; Ran, Y.; Pu, J. Circulating MicroRNAs are Promising Novel Biomarkers of Acute Myocardial Infarction. Intern. Med. 2011, 50, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dong, Y.; Chen, S.; Zhang, G.; Zhang, M.; Gong, Y.; Li, X. Circulating MicroRNA-146a and MicroRNA-21 Predict Left Ventricular Remodeling after ST-Elevation Myocardial Infarction. Cardiology 2015, 132, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Białek, S.; Górko, D.; Zajkowska, A.; Kołtowski, Ł.; Grabowski, M.; Stachurska, A.; Kochman, J.; Sygitowicz, G.; Małecki, M.; Opolski, G.; et al. Release Kinetics of Circulating MiRNA-208a in the Early Phase of Myocardial Infarction. Kardiol. Pol. 2015, 73, 613–619. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.; De Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef]
- Muller-Werdan, U.; Buerke, M.; Ebelt, H.; Heinroth, K.M.; Herklotz, A.; Loppnow, H.; Ruß, M.; Schlegel, F.; Schlitt, A.; Schmidt, H.B.; et al. Septic Cardiomyopathy—A Not yet Discovered Cardiomyopathy? Exp. Clin. Cardiol. 2006, 11, 226–236. [Google Scholar]
- Wu, H.; Liu, J.; Li, W.; Liu, G.; Li, Z. LncRNA-HOTAIR promotes TNF-α production in cardiomyocytes of LPS-induced sepsis mice by activating NF-κB pathway. Biochem. Biophys. Res. Commun. 2016, 471, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.-T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast–Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef]
- Pawar, K.G.; Hanisch, C.; Vera, S.E.P.; Einspanier, R.; Sharbati, S. Down regulated lncRNA MEG3 eliminates mycobacteria in macrophages via autophagy. Sci. Rep. 2016, 6, 19416. [Google Scholar] [CrossRef]
- Ma, M.; Hui, J.; Zhang, Q.-Y.; Zhu, Y.; He, Y.; Liu, X.-J. Long non-coding RNA nuclear-enriched abundant transcript 1 inhibition blunts myocardial ischemia reperfusion injury via autophagic flux arrest and apoptosis in streptozotocin-induced diabetic rats. Atherosclerosis 2018, 277, 113–122. [Google Scholar] [CrossRef]
- Li, Q.; Xie, J.; Li, R.; Shi, J.; Sun, J.; Gu, R.; Ding, L.; Wang, L.; Xu, B. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J. Cell. Mol. Med. 2014, 18, 919–928. [Google Scholar] [CrossRef]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Nguyen, S.S.; Winbanks, C.E.; Gao, X.; Boey, E.J.H.; Tham, Y.K.; Kiriazis, H.; Ooi, J.; Porrello, E.; Igoor, S.; et al. Therapeutic silencing of miR-652 restores heart function and attenuates adverse remodeling in a setting of established pathological hypertrophy. FASEB J. 2014, 28, 5097–5110. [Google Scholar] [CrossRef] [PubMed]
- Hinkel, R.; Penzkofer, D.; Zühlke, S.; Fischer, A.; Husada, W.; Xu, Q.-F.; Baloch, E.; van Rooij, E.; Zeiher, A.M.; Kupatt, C.; et al. Inhibition of MicroRNA-92a Protects Against Ischemia/Reperfusion Injury in a Large-Animal Model. Circulation 2013, 128, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.; Ray, T.D.; Sheedy, F.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.H.; Zamzam, A.; Valencia, J.; Khan, H.; Jain, S.; Singh, K.K.; Abdin, R.; Qadura, M. MicroRNA Profile of Patients with Chronic Limb-Threatening Ischemia. Diagnostics 2020, 10, 230. [Google Scholar] [CrossRef]
- Zhou, S.-S.; Jin, J.-P.; Wang, J.-Q.; Zhang, Z.-G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef]
- Mayr, M.; Zampetaki, A. Analytical challenges and technical limitations in assessing circulating MiRNAs. Thromb. Haemost. 2012, 108, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, X.; Shan, C.; Wang, Y.; Li, P.; Shao, K. Autophagy in cardiovascular diseases: Role of noncoding RNAs. Mol. Ther. Nucleic Acids 2021, 23, 101–118. [Google Scholar] [CrossRef]
- Singh, S.; Nguyen, H.C.; Ehsan, M.; Michels, D.C.R.; Singh, P.; Qadura, M.; Singh, K.K. Pravastatin-induced changes in expression of long non-coding and coding RNAs in endothelial cells. Physiol. Rep. 2021, 9, e14661. [Google Scholar] [CrossRef]
- Singh, K.; Mantella, L.; Pan, Y.; Quan, A.; Sabongui, S.; Sandhu, P.; Teoh, H.; Al-Omran, M.; Verma, S. A global profile of glucose-sensitive endothelial-expressed long non-coding RNAs. Can. J. Cardiol. 2016, 32, S232–S233. [Google Scholar] [CrossRef]
- Singh, K.K.; Matkar, P.N.; Quan, A.; Mantella, L.-E.; Teoh, H.; Al-Omran, M.; Verma, S. Investigation of TGFβ1-Induced Long Noncoding RNAs in Endothelial Cells. Int. J. Vasc. Med. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bu, S.; Singh, K.K. Epigenetic Regulation of Autophagy in Cardiovascular Pathobiology. Int. J. Mol. Sci. 2021, 22, 6544. https://doi.org/10.3390/ijms22126544
Bu S, Singh KK. Epigenetic Regulation of Autophagy in Cardiovascular Pathobiology. International Journal of Molecular Sciences. 2021; 22(12):6544. https://doi.org/10.3390/ijms22126544
Chicago/Turabian StyleBu, Shuhan, and Krishna K. Singh. 2021. "Epigenetic Regulation of Autophagy in Cardiovascular Pathobiology" International Journal of Molecular Sciences 22, no. 12: 6544. https://doi.org/10.3390/ijms22126544
APA StyleBu, S., & Singh, K. K. (2021). Epigenetic Regulation of Autophagy in Cardiovascular Pathobiology. International Journal of Molecular Sciences, 22(12), 6544. https://doi.org/10.3390/ijms22126544