
Management of Congenital Diaphragmatic Hernia (CDH): Role of Molecular Genetics


Abstract
1. Introduction
2. Diaphragm Development
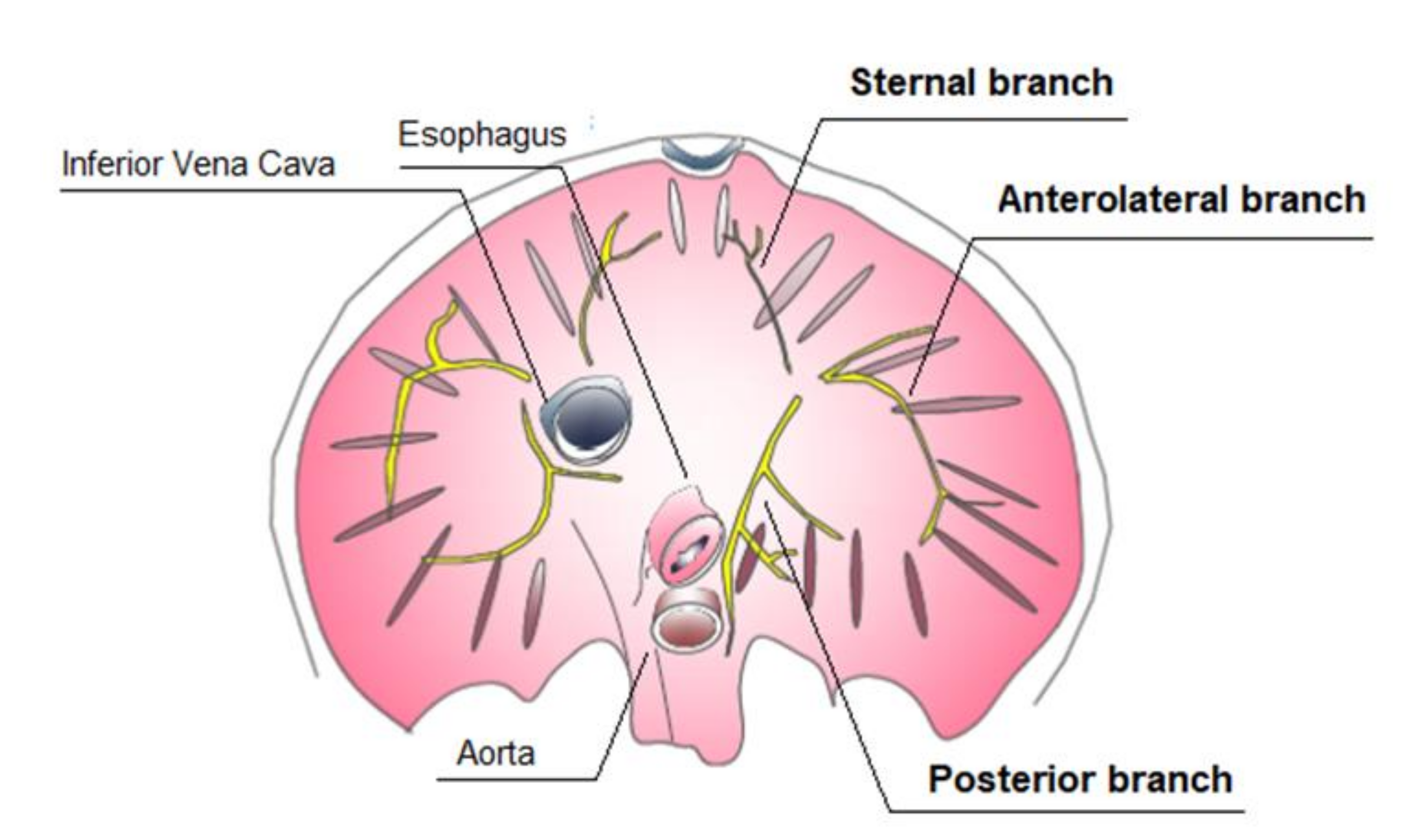
2.1. Diaphragm Basic Anatomy
2.2. Anatomic Findings
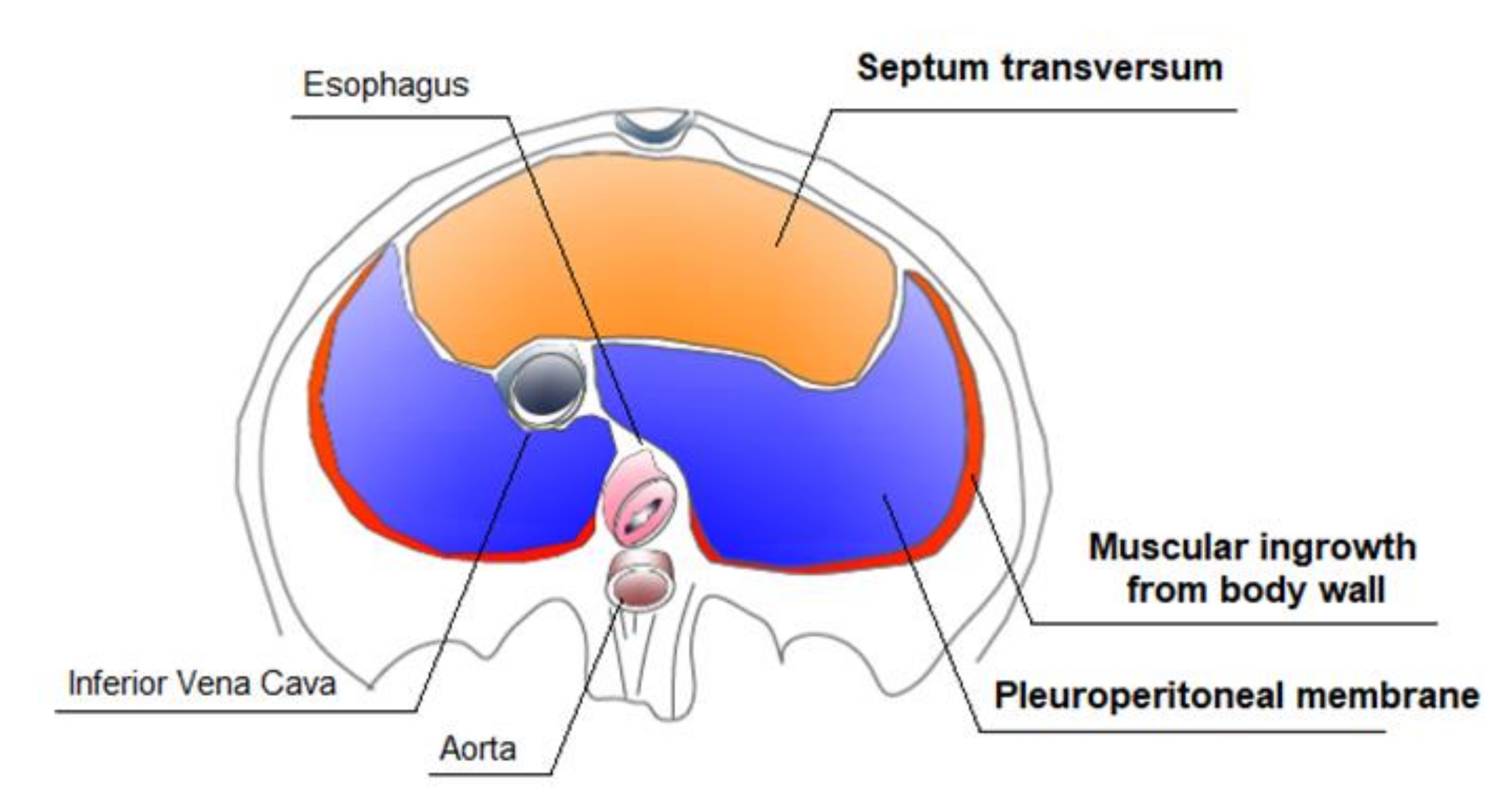
2.3. Diaphragm Embryogenesis
3. Prognostic Factors
3.1. Prenatal Prognostic Factors
3.2. Postnatal Prognostic Factors
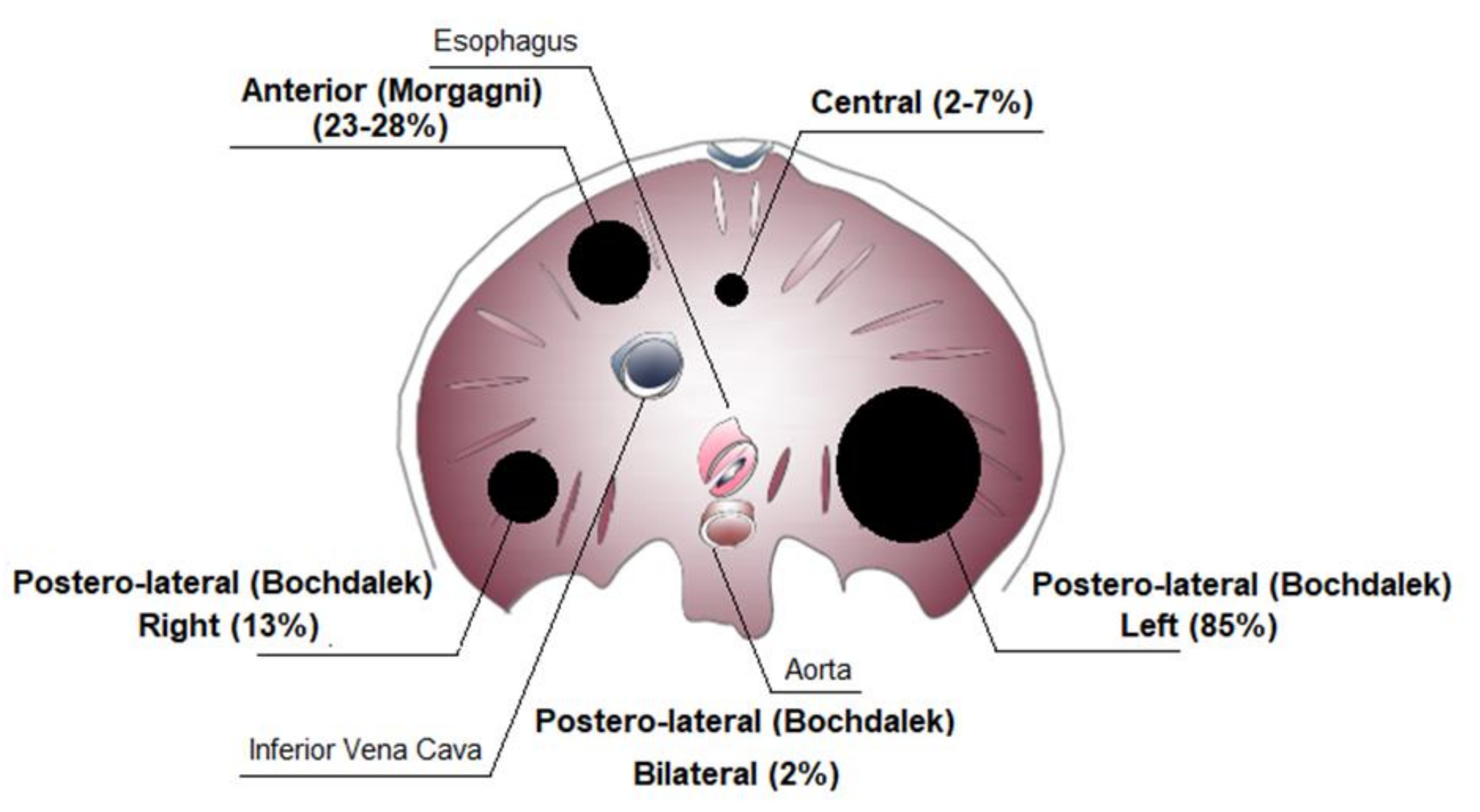
4. Classification of Congenital Diaphragmatic Hernia (CDH)
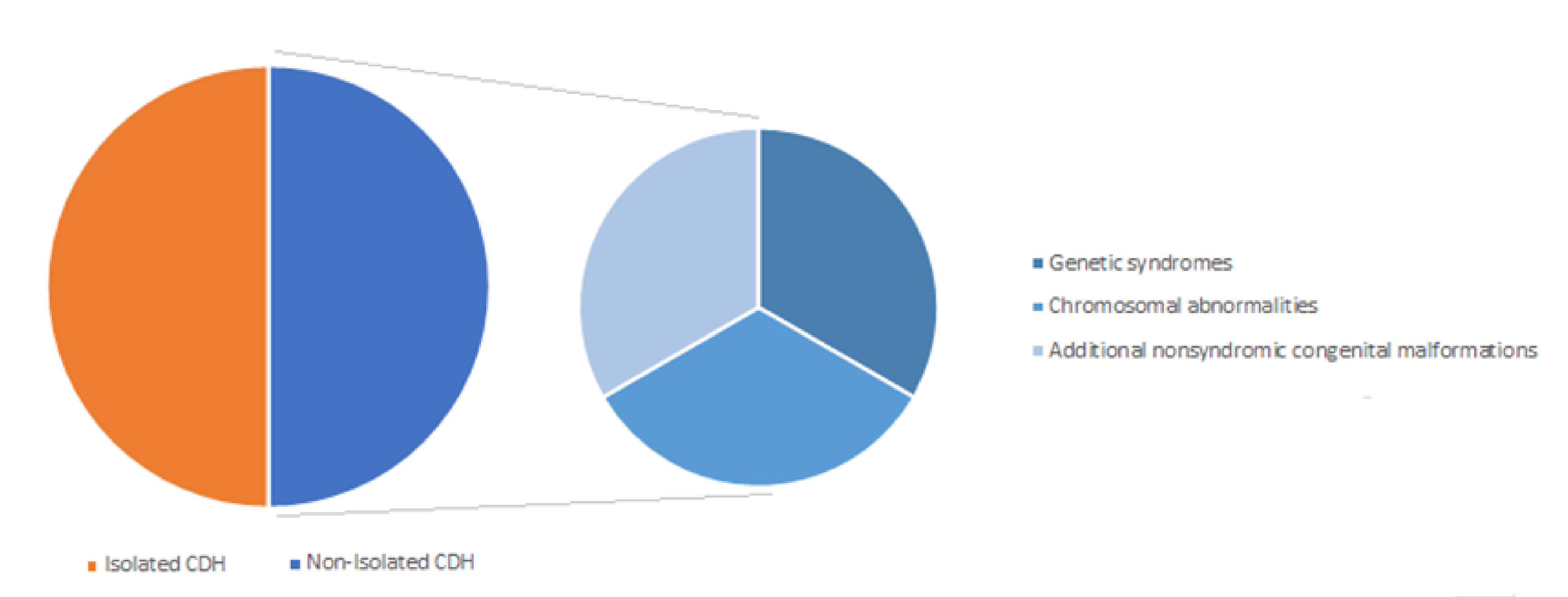
Isolated and Non-Isolated Congenital Diaphragmatic Hernia (CDH)
5. Genetic Contribution to Congenital Diaphragmatic Hernia (CDH)
5.1. Aneuploidies
5.1.1. Trisomy 21—Down Syndrome
5.1.2. Trisomy 18—Edwards Syndrome
5.1.3. Trisomy 13—Patau Syndrome
5.1.4. Turner syndrome (45, X) and Trisomy X (46, XXX)
5.1.5. Tetrasomy 12p—Mosaic Isochromosome 12p Syndrome—Pallister–Killian Syndrome (OMIM 601803)
5.2. Copy Number Variants (CNVs)
5.2.1. Deletion 1q41-q42 (OMIM 612530)
5.2.2. Duplication 1q25q31.2
5.2.3. Deletion 3q22
5.2.4. Deletion 4p16 (OMIM 194190)
5.2.5. Deletion 6p25
5.2.6. Deletion 8q23 (OMIM 610187)
5.2.7. Deletion 8p23 (OMIM 222400)
5.2.8. Duplication of 8p21-p23.1
5.2.9. Deletion 9p24-pter
5.2.10. Deletion 11p13
5.2.11. Duplication 11q23.3-qter
5.2.12. Deletion 15q26
5.3. Monogenic Syndromes
5.3.1. Simpson–Golabi–Behmel Syndrome (OMIM 312870)
5.3.2. Craniofrontonasal Syndrome (OMIM 304110)
5.3.3. Myotubular Myopathy 1 (OMIM 310400)
5.3.4. Opitz G/BBB Syndrome (OMIM 300000)
5.3.5. Lowe Syndrome (OMIM 309000)
5.3.6. Focal Dermal Hypoplasia—Goltz Syndrome (OMIM 305600)
5.3.7. MIDAS Syndrome (OMIM 309801)
5.3.8. Cornelia de Lange Syndrome (OMIM 122470)
5.3.9. Denys–Drash Syndrome (OMIM 194080)
5.3.10. Marfan Syndrome (OMIM 154700)
5.3.11. CHARGE Syndrome (OMIM 214800)
5.3.12. Fryns Syndrome (OMIM 229850)
5.3.13. Donnai–Barrow Syndrome (OMIM 222448)
5.3.14. Matthew–Wood Syndrome (OMIM 601186, 615524)
5.3.15. Multiple Vertebral Segmentation Defects
5.4. Most Frequent Genes identified by Next-Generation Sequencing and Related Pathways
5.4.1. GATA4
5.4.2. GATA6
5.4.3. FOG2 (ZFPM2)
5.4.4. COUP-TFII (NRF2)
5.4.5. SIN3A
5.4.6. MYRF (MRF)
6. Implications of Congenital Diaphragmatic Hernia (CDH) Diagnosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greer, J.J. Current concepts on the pathogenesis and etiology of congenital diaphragmatic hernia. Respir. Physiol. Neurobiol. 2013, 189, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Grivell, R.M.; Andersen, C.; Dodd, J.M. Prenatal interventions for congenital diaphragmatic hernia for improving outcomes. Cochrane Database Syst. Rev. 2015, 11, CD008925. [Google Scholar] [CrossRef] [PubMed]
- Mohseni-Bod, H.; Bohn, D. Pulmonary hypertension in congenital diaphragmatic hernia. Semin. Pediatr. Surg. 2007, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, M.A.; Werner, N.L.; Gajarski, R.; Gadepalli, S.; Hirschl, R.; Barks, J.; Treadwell, M.C.; Ladino-Torres, M.; Kreutzman, J.; Mychaliska, G.B. Prenatally diagnosed severe CDH: Mortality and morbidity remain high. J. Pediatr. Surg. 2016, 51, 1091–1095. [Google Scholar] [CrossRef]
- Harrison, M.R.; Bjordal, R.I.; Langmark, F.; Knutrud, O. Congenital diaphragmatic hernia: The hidden mortality. J. Pediatr. Surg. 1978, 13, 227–230. [Google Scholar] [CrossRef]
- Nobuhara, K.K.; Lund, D.P.; Mitchell, J.; Kharasch, V.; Wilson, J.M. Long-term Outlook for Survivors of Congenital Diaphragmatic Hernia. Clin. Perinatol. 1996, 23, 873–887. [Google Scholar] [CrossRef]
- Deprest, J.A.; Nicolaides, K.H.; Benachi, A.; Gratacos, E.; Ryan, G.; Persico, N.; Sago, H.; Johnson, A.; Wielgoś, M.; Berg, C.; et al. TOTAL Trial for Severe Hypoplasia Investigators.Randomized Trial of Fetal Surgery for Severe Left Diaphragmatic Hernia. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Graham, G.; Devine, P.C. Antenatal Diagnosis of Congenital Diaphragmatic Hernia. Semin. Perinatol. 2005, 29, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bagłaj, M. Late-presenting congenital diaphragmatic hernia in children: A clinical spectrum. Pediatr. Surg. Int. 2004, 20, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Chao, P.-H.; Chuang, J.-H.; Lee, S.-Y.; Huang, H.-C. Late-presenting congenital diaphragmatic hernia in childhood. Acta Paediatr. 2010, 100, 425–428. [Google Scholar] [CrossRef]
- Kitano, Y.; Lally, K.P.; Lally, P. A Congenital Diaphragmatic Hernia Study Group. Late-presenting congenital diaphragmatic hernia. J. Pediatr. Surg. 2005, 40, 1839–1843. [Google Scholar] [CrossRef]
- Beurskens, L.W.J.E.; Tibboel, D.; Lindemans, J.; Duvekot, J.J.; Cohen-Overbeek, T.E.; Veenma, D.C.M.; De Klein, A.; Greer, J.J.; Steegers-Theunissen, R.P.M. Retinol Status of Newborn Infants Is Associated with Congenital Diaphragmatic Hernia. Pediatry 2010, 126, 712–720. [Google Scholar] [CrossRef]
- Beurskens, L.W.J.E.; Tibboel, D.; Steegers-Theunissen, R.P. Role of nutrition, lifestyle factors, and genes in the pathogenesis of congenital diaphragmatic hernia: Human and animal studies. Nutr. Rev. 2009, 67, 719–730. [Google Scholar] [CrossRef]
- Longoni, M.; High, F.A.; Russell, M.K.; Kashani, A.; Tracy, A.A.; Coletti, C.M.; Hila, R.; Shamia, A.; Wells, J.; Ackerman, K.G.; et al. Molecular pathogenesis of congenital diaphragmatic hernia revealed by exome sequencing, developmental data, and bioinformatics. Proc. Natl. Acad. Sci. USA 2014, 111, 12450–12455. [Google Scholar] [CrossRef]
- Russell, M.K.; Longoni, M.; Wells, J.; Maalouf, F.; Tracy, A.A.; Loscertales, M.; Ackerman, K.G.; Pober, B.R.; Lage, K.; Bult, C.J.; et al. Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes. Proc. Natl. Acad. Sci. USA 2012, 109, 2978–2983. [Google Scholar] [CrossRef]
- Pober, B.R. Overview of epidemiology, genetics, birth defects, and chromosome abnormalities associated with CDH. Am. J. Med. Genet. Part. C: Semin. Med. Genet. 2007, 145C, 158–171. [Google Scholar] [CrossRef]
- Pober, B.R. Genetic aspects of human congenital diaphragmatic hernia. Clin. Genet. 2008, 74, 1–15. [Google Scholar] [CrossRef]
- Botha, G.S.M. The Anatomy of Phrenic Nerve Termination and the Motor Innervation of the Diaphragm. Thorax 1957, 12, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Shane Tubbs, R.; Rizk, E.; Shoja, M.M.; Loukas, M.; Barbaro, N.; Spinner, R.J. Volume 1: History, Embryology, Anatomy, Imaging and Diagnosis. In Nerves and Nerve Injuries; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- An, X.; Yue, B.; Lee, J.-H.; Lee, M.-S.; Lin, C.; Han, S.-H. Intramuscular distribution of the phrenic nerve in human diaphragm as shown by Sihler staining. Muscle Nerve 2012, 45, 522–526. [Google Scholar] [CrossRef]
- Bergman, R.A.; Thompson, S.A.; Saadeh, F.A. Compendium of Human Anatomic Variation; Urban Schwarz: Baltimore, MD, USA, 1988; Volume 1, pp. 138–139. [Google Scholar]
- Chandrasekharan, P.K.; Rawat, M.; Madappa, R.; Rothstein, D.H.; Lakshminrusimha, S. Congenital Diaphragmatic hernia—A review. Matern. Health Neonatol. Perinatol. 2017, 3, 1–16. [Google Scholar] [CrossRef]
- Veenma, D.; de Klein, A.; Tibboel, D. Developmental and genetic aspects of congenital diaphragmatic hernia. Pediatr. Pulmonol. 2012, 47, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Baglaj, M.; Spicer, R.; Ashworth, M. Unilateral agenesis of the diaphragm: A separate entity or an extremely large defect? Pediatr. Surg. Int. 1999, 15, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, K.G.; Herron, B.J.; Vargas, S.O.; Huang, H.; Tevosian, S.G.; Kochilas, L.; Rao, C.; Pober, B.R.; Babiuk, R.P.; Epstein, J.A.; et al. Fog2 Is Required for Normal Diaphragm and Lung Development in Mice and Humans. PLoS Genet. 2005, 1, e10–e65. [Google Scholar] [CrossRef] [PubMed]
- Jay, P.Y.; Bielinska, M.; Erlich, J.; Mannisto, S.; Pu, W.; Heikinheimo, M.; Wilson, D.B. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev. Biol. 2007, 301, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Sefton, E.M.; Gallardo, M.; Kardon, G. Developmental origin and morphogenesis of the diaphragm, an essential mammalian muscle. Dev. Biol. 2018, 440, 64–73. [Google Scholar] [CrossRef]
- Merrell, A.J.; Ellis, B.J.; Fox, Z.D.; Lawson, J.A.; Weiss, J.A.; Kardon, G. Muscle connective tissue controls development of the diaphragm and is a source of congenital diaphragmatic hernias. Nat. Genet. 2015, 47, 496–504. [Google Scholar] [CrossRef]
- Babiuk, R.P.; Zhang, W.; Clugston, R.; Allan, D.W.; Greer, J.J. Embryological origins and development of the rat diaphragm. J. Comp. Neurol. 2003, 455, 477–487. [Google Scholar] [CrossRef]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Merrell, A.J.; Kardon, G. Development of the diaphragm—A skeletal muscle essential for mammalian respiration. FEBS J. 2013, 280, 4026–4035. [Google Scholar] [CrossRef]
- Allan, D.W.; Greer, J.J. Embryogenesis of the phrenic nerve and diaphragm in the fetal rat. J. Comp. Neurol. 1997, 382, 459–468. [Google Scholar] [CrossRef]
- Uetani, N.; Chagnon, M.J.; Kennedy, T.E.; Iwakura, Y.; Tremblay, M.L. Mammalian Motoneuron Axon Targeting Requires Receptor Protein Tyrosine Phosphatases σ and δ. J. Neurosci. 2006, 26, 5872–5880. [Google Scholar] [CrossRef]
- Philippidou, P.; Walsh, C.M.; Aubin, J.; Jeannotte, L.; Dasen, J.S. Sustained Hox5 gene activity is required for respiratory motor neuron development. Nat. Neurosci. 2012, 15, 1636–1644. [Google Scholar] [CrossRef]
- Dietrich, S.; Abou-Rebyeh, F.; Brohmann, H.; Bladt, F.; Sonnenberg-Riethmacher, E.; Yamaai, T.; Lumsden, A.; Brand-Saberi, B.; Birchmeier, C. The role of SF/HGF and c-Met in the development of skeletal muscle. Development 1999, 126, 1621–1629. [Google Scholar] [CrossRef]
- Reiss, I.; Schaible, T.; Hout, L.V.D.; Capolupo, I.; Allegaert, K.; van Heijst, A.; Silva, M.G.; Greenough, A.; Tibboel, D. For the CDH EURO consortium Standardized Postnatal Management of Infants with Congenital Diaphragmatic Hernia in Europe: The CDH EURO Consortium Consensus. Neonatology 2010, 98, 354–364. [Google Scholar] [CrossRef]
- Sepulveda, W.; Wong, A.E.; Casasbuenas, A.; Solari, A.; Alcalde, J.L. Congenital diaphragmatic hernia in a first-trimester ultrasound aneuploidy screening program. Prenat. Diagn. 2008, 28, 531–534. [Google Scholar] [CrossRef]
- Tennant, P.W.; Pearce, M.S.; Bythell, M.; Rankin, J. 20-year survival of children born with congenital anomalies: A population-based study. Lancet 2010, 375, 649–656. [Google Scholar] [CrossRef]
- McGivern, M.R.; Best, K.; Rankin, J.; Wellesley, D.; Greenlees, R.; Addor, M.-C.; Arriola, L.; De Walle, H.; Barisic, I.; Beres, J.; et al. Epidemiology of congenital diaphragmatic hernia in Europe: A register-based study. Arch. Dis. Child. Fetal Neonatal Ed. 2014, 100, F137–F144. [Google Scholar] [CrossRef]
- Metkus, A.P.; Filly, R.A.; Stringer, M.D.; Harrison, M.R.; Adzick, N. Sonographic predictors of survival in fetal diaphragmatic hernia. J. Pediatr. Surg. 1996, 31, 148–152. [Google Scholar] [CrossRef]
- Mullassery, D.; Ba’Ath, M.E.; Jesudason, E.C.; Losty, P.D. Value of liver herniation in prediction of outcome in fetal congenital diaphragmatic hernia: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2010, 35, 609–614. [Google Scholar] [CrossRef]
- Adzick, N.S.; Vacanti, J.P.; Lillehei, C.W.; O’Rourke, P.P.; Crone, R.K.; Wilson, J.M. Fetal diaphragmatic hernia: Ultrasound diagnosis and clinical outcome in 38 cases. J. Pediatr. Surg. 1989, 24, 654–658. [Google Scholar] [CrossRef]
- Stringer, M.D.; Goldstein, R.B.; Filly, R.A.; Howell, L.J.; Sola, A.; Adzick, N.; Harrison, M.R. Fetal diaphragmatic hernia without visceral herniation. J. Pediatr. Surg. 1995, 30, 1264–1266. [Google Scholar] [CrossRef]
- Hatch, E.I.; Kendall, J.; Blumhagen, J. Stomach position as an in utero predictor of neonatal outcome in left-sided diaphragmatic hernia. J. Pediatr. Surg. 1992, 27, 778–779. [Google Scholar] [CrossRef]
- Basta, A.M.; Lusk, L.A.; Keller, R.L.; Filly, R.A. Fetal Stomach Position Predicts Neonatal Outcomes in Isolated Left-Sided Congenital Diaphragmatic Hernia. Fetal Diagn. Ther. 2016, 39, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Cordier, A.-G.; Jani, J.C.; Cannie, M.M.; Rodó, C.; Fabietti, I.; Persico, N.; Saada, J.; Carreras, E.; Senat, M.-V.; Benachi, A. Stomach position in prediction of survival in left-sided congenital diaphragmatic hernia with or without fetoscopic endoluminal tracheal occlusion. Ultrasound Obstet. Gynecol. 2014, 46, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Dommergues, M.; Louis-Sylvestre, C.; Mandelbrot, L.; Oury, J.F.; Herlicoviez, M.; Body, G.; Gamerre, M.; Dumez, Y. Congenital diaphragmatic hernia: Can prenatal ultrasonography predict outcome? Am. J. Obstet. Gynecol. 1996, 174, 1377–1381. [Google Scholar] [CrossRef]
- Lipshutz, G.S.; Albanese, C.T.; Feldstein, V.A.; Jennings, R.W.; Housley, H.T.; Beech, R.; Farrell, J.A.; Harrison, M.R. Prospective analysis of lung-to-head ratio predicts survival for patients with prenatally diagnosed congenital diaphragmatic hernia. J. Pediatr. Surg. 1997, 32, 1634–1636. [Google Scholar] [CrossRef]
- Jani, J.; Keller, R.L.; Benachi, A.; Nicolaides, K.H.; Favre, R.; Gratacos, E.; Laudy, J.; Eisenberg, V.; Eggink, A.; Vaast, P.; et al. Prenatal prediction of survival in isolated left-sided diaphragmatic hernia. Ultrasound Obstet. Gynecol. 2005, 27, 18–22. [Google Scholar] [CrossRef]
- Gentili, A.; Pasini, L.; Iannella, E.; Landuzzi, V.; Lima, M.; Reggiani, M.L.B.; Baroncini, S. Predictive outcome indexes in neonatal Congenital Diaphragmatic Hernia. J. Matern. Neonatal Med. 2014, 28, 1–6. [Google Scholar] [CrossRef]
- Ba’Ath, M.E.; Jesudason, E.C.; Losty, P.D. How useful is the lung-to-head ratio in predicting outcome in the fetus with congenital diaphragmatic hernia? A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2007, 30, 897–906. [Google Scholar] [CrossRef]
- Jani, J.; Nicolaides, K.H.; Keller, R.L.; Benachi, A.; Peralta, C.F.A.; Favre, R.; Moreno, O.; Tibboel, D.; Lipitz, S.; Eggink, A.; et al. Observed to expected lung area to head circumference ratio in the prediction of survival in fetuses with isolated diaphragmatic hernia. Ultrasound Obstet. Gynecol. 2007, 30, 67–71. [Google Scholar] [CrossRef]
- Deprest, J.; Flemmer, A.W.; Gratacos, E.; Nicolaides, K. Antenatal prediction of lung volume and in-utero treatment by fetal endoscopic tracheal occlusion in severe isolated congenital diaphragmatic hernia. Semin. Fetal Neonatal. Med. 2009, 14, 8–13. [Google Scholar] [CrossRef]
- Snoek, K.G.; Peters, N.C.J.; Van Rosmalen, J.; van Heijst, A.; Eggink, A.J.; Sikkel, E.; Wijnen, R.M.; Ijsselstijn, H.; Cohen-Overbeek, T.; Titia, E.; et al. The validity of the observed-to-expected lung-to-head ratio in congenital diaphragmatic hernia in an era of standardized neonatal treatment, a multicenter study. Prenat. Diagn. 2017, 37, 658–665. [Google Scholar] [CrossRef]
- Weidner, M.; Hagelstein, C.; Debus, A.; Walleyo, A.; Weiss, C.; Schoenberg, S.O.; Schaible, T.; Büsing, K.A.; Kehl, S.; Neff, K.W. MRI-Based Ratio of Fetal Lung Volume to Fetal Body Volume as a New Prognostic Marker in Congenital Diaphragmatic Hernia. Am. J. Roentgenol. 2014, 202, 1330–1336. [Google Scholar] [CrossRef]
- Ruano, R.; Aubry, M.-C.; Barthe, B.; Mitanchez, D.; Dumez, Y.; Benachi, A. Quantitative analysis of fetal pulmonary vasculature by 3-dimensional power Doppler ultrasonography in isolated congenital diaphragmatic hernia. Am. J. Obstet. Gynecol. 2006, 195, 1720–1728. [Google Scholar] [CrossRef]
- Lupo, E.; Castoldi, F.M.L.R.; Maestri, L.; Rustico, M.; Dani, C.; Lista, G. Outcome of congenital diaphragmatic hernia: Analysis of implicated factors. Minerva Pediatr. 2013, 65. [Google Scholar]
- Werner, N.L.; Coughlin, M.; Kunisaki, S.M.; Hirschl, R.; Ladino-Torres, M.; Berman, D.; Kreutzman, J.; Mychaliska, G.B. Prenatal and postnatal markers of severity in congenital diaphragmatic hernia have similar prognostic ability. Prenat. Diagn. 2015, 36, 107–111. [Google Scholar] [CrossRef]
- The Congenital Diaphragmatic Hernia Study Group*. Defect Size Determines Survival in Infants with Congenital Diaphragmatic Hernia. Pediatrics 2007, 120, e651–e657. [Google Scholar] [CrossRef]
- Brindle, M.E.; Cook, E.F.; Tibboel, D.; Lally, P.A.; Lally, K.P.; on behalf of the Congenital Diaphragmatic Hernia Study Group. A Clinical Prediction Rule for the Severity of Congenital Diaphragmatic Hernias in Newborns. Pediatry 2014, 134, e413–e419. [Google Scholar] [CrossRef]
- Baird, R.; Macnab, Y.C.; Skarsgard, E. Mortality prediction in congenital diaphragmatic hernia. J. Pediatr. Surg. 2008, 43, 783–787. [Google Scholar] [CrossRef]
- Langham, M.R., Jr.; Kays, D.W.; Ledbetter, D.J.; Frentzen, B.; Sanford, L.L.; Richards, D.S. Congenital diaphragmatic hernia. Epidemi-ology and outcome. Clin. Perinatol. 1996, 23, 671. [Google Scholar] [CrossRef] [PubMed]
- Downard, C.D.; Jaksic, T.; Garza, J.J.; Dzakovic, A.; Nemes, L.; Jennings, R.W.; Wilson, J.M. Analysis of an improved survival rate for congenital diaphragmatic hernia. J. Pediatr. Surg. 2003, 38, 729–732. [Google Scholar] [CrossRef]
- Chin, K.M.; Kim, N.H.S.; Rubin, L.J. The right ventricle in pulmonary hypertension. Coron. Artery Dis. 2005, 16, 13–18. [Google Scholar] [CrossRef]
- Aggarwal, S.; Stockmann, P.; Klein, M.D.; Natarajan, G. Echocardiographic measures of ventricular function and pulmonary artery size: Prognostic markers of congenital diaphragmatic hernia? J. Perinatol. 2011, 31, 561–566. [Google Scholar] [CrossRef]
- Byrne, F.A.; Keller, R.L.; Meadows, J.; Miniati, D.; Brook, M.M.; Silverman, N.H.; Moon-Grady, A.J. Severe left diaphragmatic hernia limits size of fetal left heart more than does right diaphragmatic hernia. Ultrasound Obstet. Gynecol. 2015, 46, 688–694. [Google Scholar] [CrossRef]
- Kinsella, J.P.; Ivy, D.D.; Abman, S.H. Pulmonary Vasodilator Therapy in Congenital Diaphragmatic Hernia: Acute, Late, and Chronic Pulmonary Hypertension. Semin. Perinatol. 2005, 29, 123–128. [Google Scholar] [CrossRef]
- Yamoto, M.; Inamura, N.; Terui, K.; Nagata, K.; Kanamori, Y.; Hayakawa, M.; Tazuke, Y.; Yokoi, A.; Takayasu, H.; Okuyama, H.; et al. Echocardiographic predictors of poor prognosis in congenital diaphragmatic hernia. J. Pediatr. Surg. 2016, 51, 1926–1930. [Google Scholar] [CrossRef]
- Stoll, C.; Alembik, Y.; Dott, B.; Roth, M.-P. Associated malformations in cases with congenital diaphragmatic hernia. Genet. Couns. 2008, 19, 331–339. [Google Scholar]
- Yu, L.; Hernan, R.R.; Wynn, J.; Chung, W.K. The influence of genetics in congenital diaphragmatic hernia. Semin. Perinatol. 2020, 44, 151169. [Google Scholar] [CrossRef]
- Tibboel, D.; Gaag, A.V. Etiologic and Genetic Factors in Congenital Diaphragmatic Hernia. Clin. Perinatol. 1996, 23, 689–699. [Google Scholar] [CrossRef]
- Holder, A.; Klaassens, M.; Tibboel, D.; de Klein, A.; Lee, B.; Scott, D. Genetic Factors in Congenital Diaphragmatic Hernia. Am. J. Hum. Genet. 2007, 80, 825–845. [Google Scholar] [CrossRef] [PubMed]
- Garne, E.; Haeusler, M.; Barisic, I.; Gjergja, R.; Stoll, C.; Clementi, M. Congenital diaphragmatic hernia: Evaluation of prenatal diagnosis in 20 European regions. Ultrasound Obstet. Gynecol. 2002, 19, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Lurie, I.W. Where to look for the genes related to diaphragmatic hernia? Genet. Couns. 2003, 14, 12725592. [Google Scholar]
- Honoré, L.H.; Torfs, C.P.; Curry, C.J.R. Possible association between the hernia of Morgagni and trisomy 21. Am. J. Med. Genet. 1993, 47, 255–256. [Google Scholar] [CrossRef]
- Pokorny, W.J.; McGill, C.W.; Harberg, F.J. Morgagni hernias during infancy: Presentation and associated anomalies. J. Pediatr. Surg. 1984, 19, 394–397. [Google Scholar] [CrossRef]
- Jetley, N.K.; Al-Assiri, A.H.; Al-Helal, A.S.; Ali, A.M.A.-B. Down’s syndrome as a factor in the diagnosis, management, and outcome in patients of Morgagni hernia. J. Pediatr. Surg. 2011, 46, 636–639. [Google Scholar] [CrossRef]
- Cigdem, M.K.; Onen, A.; Okur, H.; Otcu, S. Associated malformations in Morgagni hernia. Pediatr. Surg. Int. 2007, 23, 1101–1103. [Google Scholar] [CrossRef]
- Harris, G.J.; Soper, R.T.; Kimura, K.K. Foramen of morgagni hernia in identical twins: Is this an inheritable defect? J. Pediatr. Surg. 1993, 28, 177–178. [Google Scholar] [CrossRef]
- Snyder, W.H., Jr.; Greaney, E.M., Jr. Congenital diaphragmatic hernia; 77 consecutive cases. Surgery 1965, 57, 576. [Google Scholar] [PubMed]
- Quah, B.S.; Menon, B.S. Down syndrome associated with a retroperitoneal teratoma and Morgagni hernia. Clin. Genet. 2008, 50, 232–234. [Google Scholar] [CrossRef]
- Kubiak, R.; Platen, C.; Schmid, E.; Gruber, R.; Ludwig, K.-H.; Rauh, W. Delayed appearance of bilateral morgagni herniae in a child with Down’s syndrome. Pediatr. Surg. Int. 1998, 13, 600–601. [Google Scholar] [CrossRef]
- Chen, C.-P. Omphalocele and congenital diaphragmatic hernia associated with fetal trisomy 18. Prenat. Diagn. 2005, 25, 421–423. [Google Scholar] [CrossRef]
- PK, A.J.; Yk, P.K.S. Congenital diaphragmatic hernia in a case of patau syndrome: A rare association. J. Neonatal. Surg. 2015, 1, 4. [Google Scholar]
- Sahin, S.; Kutman, K.H.; Bozkurt, O.; Canpolat, F.E.; Uras, N.; Oguz, S.S.; Topcu, V.; Ozdemir, O.; Dilmen, U. A trisomy 13 case prsenting with congenita diaphragmatic hernia and microphthalmia. Genet. Couns. 2015, 26, 263. [Google Scholar] [PubMed]
- Ruano, R.; Bunduki, V.; Silva, M.M.; Yoshizaki, C.T.; Tanuri, U.; Macksoud, J.G.; Zugaib, M. Prenatal diagnosis and perinatal outcome of 38 cases with congenital diaphragmatic hernia: 8-year experience of a tertiary Brazilian center. Clinics 2006, 61, 197–202. [Google Scholar] [CrossRef]
- Zaiss, I.; Kehl, S.; Link, K.; Neff, W.; Schaible, T.; Sütterlin, M.; Siemer, J. Associated Malformations in Congenital Diaphragmatic Hernia. Am. J. Perinatol. 2010, 28, 211–218. [Google Scholar] [CrossRef]
- Pallister, P.D.; Meisner, L.F.; Elejalde, B.R.; Francke, U.; Herrmann, J.; Spranger, J.; Tiddy, W.; Inhorn, S.L.; Opitz, J.M. The pallister mosaic syndrome. Birth Defect. Orig. Artic. Ser. 1977, 13, 103. [Google Scholar] [PubMed]
- Peltomäki, P.; Knuutila, S.; Ritvanen, A.; Kaitila, I.; De La Chapelle, A. Pallister-Killian syndrome: Cytogenetic and molecular studies. Clin. Genet. 2008, 31, 399–405. [Google Scholar] [CrossRef]
- Warburton, D.; Anyane-Yeboa, K.; Francke, U.; Reynolds, J.F. Mosaic tetrasomy 12p: Four new cases, and confirmation of the chromosomal origin of the supernumerary chromosome in one of the original Pallister-Mosaic syndrome cases. Am. J. Med. Genet. 1987, 27, 275–283. [Google Scholar] [CrossRef]
- Schinzel, A. Tetrasomy 12p (Pallister-Killian syndrome). J. Med. Genet. 1991, 28, 122–125. [Google Scholar] [CrossRef]
- Blyth, M.; Maloney, V.; Beal, S.; Collinson, M.; Huang, S.; Crolla, J.; Temple, I.K.; Baralle, D. Pallister-Killian syndrome: A study of 22 British patients. J. Med. Genet. 2015, 52, 454–464. [Google Scholar] [CrossRef]
- Thakur, S.; Gupta, R.; Tiwari, B.; Singh, N.; Saxena, K. Pallister-Killian syndrome: Review of fetal phenotype. Clin. Genet. 2018, 95, 79–84. [Google Scholar] [CrossRef]
- Salzano, E.; Raible, S.; Kaur, M.; Wilkens, A.; Sperti, G.; Tilton, R.; Bettini, L.; Rocca, A.; Cocchi, G.; Selicorni, A.; et al. Prenatal profile of Pallister-Killian syndrome: Retrospective analysis of 114 pregnancies, literature review and approach to prenatal diagnosis. Am. J. Med. Genet. Part. A 2018, 176, 2575–2586. [Google Scholar] [CrossRef]
- Wilkens, A.; Liu, H.; Park, K.; Campbell, L.B.; Jackson, M.; Kostanecka, A.; Pipan, M.; Izumi, K.; Pallister, P.; Krantz, I.D. Novel clinical manifestations in Pallister-Killian syndrome: Comprehensive evaluation of 59 affected individuals and review of previously reported cases. Am. J. Med. Genet. Part. A 2012, 158A, 3002–3017. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Theisen, A.; Bejjani, B.A.; Ballif, B.C.; Aylsworth, A.S.; Lim, C.; McDonald, M.; Ellison, J.W.; Kostiner, D.; Saitta, S.; et al. The discovery of microdeletion syndromes in the post-genomic era: Review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet. Med. 2007, 9, 607–616. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Lacassie, Y.; El-Khechen, D.; Escobar, L.F.; Reggin, J.; Heuer, C.; Chen, E.; Jenkins, L.S.; Collins, A.T.; Zinner, S.; et al. New cases and refinement of the critical region in the 1q41q42 microdeletion syndrome. Eur. J. Med. Genet. 2011, 54, 42–49. [Google Scholar] [CrossRef]
- Slavotinek, A.M.; Moshrefi, A.; Jiminez, N.L.; Chao, R.; Mendell, A.; Shaw, G.M.; Pennacchio, L.A.; Bates, M.D. Sequence variants in theHLXgene at chromosome 1q41-1q42 in patients with diaphragmatic hernia. Clin. Genet. 2009, 75, 429–439. [Google Scholar] [CrossRef]
- Hentsch, B.; Lyons, I.; Li, R.; Hartley, L.; Lints, T.J.; Adams, J.M.; Harvey, R.P. Hlx homeo box gene is essential for an inductive tissue interaction that drives expansion of embryonic liver and gut. Genes Dev. 1996, 10, 70–79. [Google Scholar] [CrossRef]
- Farrell, S.A.; Sodhi, S.; Marshall, C.R.; Guerin, A.; Slavotinek, A.; Paton, T.; Chong, K.; Sirkin, W.L.; Scherer, S.W.; Bérubé-Simard, F.-A.; et al. HLX is a candidate gene for a pattern of anomalies associated with congenital diaphragmatic hernia, short bowel, and asplenia. Am. J. Med. Genet. Part. A 2017, 173, 3070–3074. [Google Scholar] [CrossRef]
- Kantarci, S.; Ackerman, K.G.; Russell, M.K.; Longoni, M.; Sougnez, C.; Noonan, K.M.; Hatchwell, E.; Zhang, X.; Vanmarcke, R.P.; Anyane-Yeboa, K.; et al. Characterization of the chromosome 1q41q42.12 region, and the candidate gene DISP1, in patients with CDH. Am. J. Med. Genet. Part. A 2010, 152A, 2493–2504. [Google Scholar] [CrossRef]
- Clark, R.D.; Fenner-Gonzales, M. Apparent Fryns syndrome in a boy with a tandem duplication of 1q24-31. Am. J. Med. Genet. 1989, 34, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.E.X.; Lu, J.; Tso, P.; Blaner, W.S.; Levin, M.S.; Li, E. Increased Neonatal Mortality in Mice Lacking Cellular Retinol-binding Protein II. J. Biol. Chem. 2002, 277, 36617–36623. [Google Scholar] [CrossRef]
- Greer, J.J.; Babiuk, R.P.; Thébaud, B. Etiology of Congenital Diaphragmatic Hernia: The Retinoid Hypothesis. Pediatr. Res. 2003, 53, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, J.; Brown, J.; Masters, K.G.; Wright, C.; English, C.J. Blepharophimosis sequence and diaphragmatic hernia associated with interstitial deletion of chromosome 3 (46,XY,del(3)(q21q23)). J. Med. Genet. 1994, 31, 647–648. [Google Scholar] [CrossRef][Green Version]
- Dillon, E.; Renwick, M.; Wright, C. Congenital diaphragmatic herniation: Antenatal detection and outcome. Br. J. Radiol. 2000, 73, 360–365. [Google Scholar] [CrossRef]
- Hirschhorn, K.; Cooper, H.L.; Firschein, I.L. Deletion of short arms of chromosome 4–5 in a child with defects of midline fusion. Qual. Life Res. 1965, 1, 479–482. [Google Scholar] [CrossRef]
- Wolf, U.; Reinwein, H.; Porsch, R.; Schröter, R.; Baitsch, H. Deficiency on the short arms of a chromosome No. 4. Humangenetik 1965, 1, 397–413. [Google Scholar]
- Battaglia, A.; Filippi, T.; Carey, J.C. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: Experience with 87 patients and recommendations for routine health supervision. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2008, 148C, 246–251. [Google Scholar] [CrossRef]
- Casaccia, G.; Mobili, L.; Braguglia, A.; Santoro, F.; Bagolan, P. Distal 4p microdeletion in a case of Wolf-Hirschhorn syndrome with congenital diaphragmatic hernia. Birth Defects Res. Part. A Clin. Mol. Teratol. 2006, 76, 210–213. [Google Scholar] [CrossRef]
- Van Dooren, M.; Brooks, A.; Hoogeboom, A.; Hoonaard, T.V.D.; De Klein, J.; Wouters, C.; Tibboel, D. Early diagnosis of Wolf-Hirschhorn syndrome triggered by a life-threatening event: Congenital diaphragmatic hernia. Am. J. Med. Genet. 2004, 127A, 194–196. [Google Scholar] [CrossRef]
- Tachdjian, G.; Fondacci, G.; TAPlA, S.; Huten, Y.; Blot, P.; Nessmann, C. The Wolf-Hirschhorn syndrome in fetuses. Clin. Genet. 2008, 42, 281–287. [Google Scholar] [CrossRef]
- Sergi, C.; Schulze, B.R.; Hager, H.D.; Beedgen, B.; Zilow, E.; Linderkamp, O.; Otto, H.F.; Tariverdian, G. Wolf-Hirschhorn syndrome: Case report and review of the chromosomal aberrations associated with diaphragmatic defects. Pathology 1998, 90, 285–293. [Google Scholar]
- Batanian, J.R.; Grange, D.K.; Fleming, R.; Gadre, B.; Wetzel, J. Two unbalanced translocations involving a common 6p25 region in two XY female patients. Clin. Genet. 2001, 59, 52–57. [Google Scholar] [CrossRef]
- Temple, I.K.; Barber, J.C.; James, R.S.; Burge, D. Diaphragmatic herniae and translocations involving 8q22 in two patients. J. Med. Genet. 1994, 31, 735–737. [Google Scholar] [CrossRef][Green Version]
- Longoni, M.; Russell, M.; High, F.; Darvishi, K.; Maalouf, F.; Kashani, A.; Tracy, A.; Coletti, C.; Loscertales, M.; Lage, K.; et al. Prevalence and penetrance ofZFPM2mutations and deletions causing congenital diaphragmatic hernia. Clin. Genet. 2015, 87, 362–367. [Google Scholar] [CrossRef]
- Longoni, M.; Lage, K.; Russell, M.K.; Loscertales, M.; Abdul-Rahman, O.A.; Baynam, G.; Bleyl, S.B.; Brady, P.D.; Breckpot, J.; Chen, C.P.; et al. Congenital diaphragmatic hernia interval on chromosome 8p23.1 characterized by genetics and protein interaction networks. Am. J. Med. Genet. Part. A 2012, 158A, 3148–3158. [Google Scholar] [CrossRef]
- Wat, M.J.; Shchelochkov, O.A.; Holder, A.M.; Breman, A.M.; Dagli, A.; Bacino, C.; Scaglia, F.; Zori, R.T.; Cheung, S.W.; Scott, D.A.; et al. Chromosome 8p23.1 deletions as a cause of complex congenital heart defects and diaphragmatic hernia. Am. J. Med. Genet. Part. A 2009, 149A, 1661–1677. [Google Scholar] [CrossRef]
- Shimokawa, O.; Miyake, N.; Yoshimura, T.; Sosonkina, N.; Harada, N.; Mizuguchi, T.; Kondoh, S.; Kishino, T.; Ohta, T.; Remco, V.; et al. Molecular characterization of del(8)(p23.1p23.1) in a case of congenital diaphragmatic hernia. Am. J. Med. Genet. Part. A 2005, 136A, 49–51. [Google Scholar] [CrossRef]
- Pecile, V.; Petroni, M.G.; Fertz, M.C.; Filippi, G. Deficiency of distal 8p -: Report of two cases and review of the literature. Clin. Genet. 2008, 37, 271–278. [Google Scholar] [CrossRef]
- Faivre, L.; Morichon-Delvallez, N.; Viot, G.; Narcy, F.; Loison, S.; Mandelbrot, L.; Aubry, M.C.; Raclin, V.; Edery, P.; Munnich, A.; et al. Prenatal diagnosis of an 8p23.1 deletion in a fetus with a diaphragmatic hernia and review of the literature. Prenat. Diagn. 1998, 18, 1055–1060. [Google Scholar] [CrossRef]
- Borys, D.; Taxy, J.B. Congenital Diaphragmatic Hernia and Chromosomal Anomalies: Autopsy Study. Pediatr. Dev. Pathol. 2004, 7, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, K.G.; Wang, J.; Luo, L.; Fujiwara, Y.; Orkin, S.H.; Beier, D.R. Gata4Is Necessary for Normal Pulmonary Lobar Development. Am. J. Respir. Cell Mol. Biol. 2007, 36, 391–397. [Google Scholar] [CrossRef] [PubMed]
- MorenoFuenmayor, H.; Meilinger, K.L.; Rucknagel, D.L.; Mohrenweiser, H.L.; Chu, E.H.Y.; Optiz, J.M. Duplication 8p syndrome: Studies in a family with a reciprocal translocation between chromosomes 8 and 12. Am. J. Med. Genet. 1980, 7, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Moog, U.; Engelen, J.J.M.; Albrechts, J.C.M.; Baars, L.G.M.; de Die-Smulders, C.E.M. Familial dup(8)(p12p21.1): Mild phenotypic effect and review of partial 8p duplications. Am. J. Med. Genet. 2000, 94, 306–310. [Google Scholar] [CrossRef]
- Ringer, K.; Rogers, J.; Pasztor, L.M. Inversion duplication of chromosome 8 with diaphragmatic hernia. Am. J. Human Genet. Suppl. 1995, 57, A124107. [Google Scholar]
- Liberfarb, R.M.; Atkins, L.; Holmes, L.B. A clinical syndrome associated with 5p duplication and 9p deletion. Ann. Génét. 1980, 23, 26–30. [Google Scholar]
- Donnenfeld, A.E.; Campbell, T.J.; Byers, J.; Librizzi, R.J.; Weiner, S. Tissue-specific mosaicism among fetuses with prenatally diagnosed diaphragmatic hernia. Am. J. Obstet. Gynecol. 1993, 169, 1017–1021. [Google Scholar] [CrossRef]
- Scott, D.; Cooper, M.; Stankiewicz, P.; Patel, A.; Potocki, L.; Cheung, S. Congenital diaphragmatic hernia in WAGR syndrome. Am. J. Med. Genet. Part. A 2005, 134A, 430–433. [Google Scholar] [CrossRef]
- Gustavson, K.-H.; Anneren, G.; Wranne, L. Two cases of 11p13 interstitial deletion and unusual clinical features. Clin. Genet. 2008, 26, 247–249. [Google Scholar] [CrossRef]
- Carmona, R.; Cañete, A.; Cano, E.; Ariza, L.; Rojas, A.; Muñoz-Chápuli, R. Conditional deletion of WT1 in the septum transversum mesenchyme causes congenital diaphragmatic hernia in mice. eLife 2016, 5, e16009. [Google Scholar] [CrossRef]
- Klaassens, M.; Scott, D.; van Dooren, M.; Hochstenbach, R.; Eussen, H.; Cai, W.; Galjaard, R.; Wouters, C.; Poot, M.; Laudy, J.; et al. Congenital diaphragmatic hernia associated with duplication of 11q23-qter. Am. J. Med. Genet. Part. A 2006, 140A, 1580–1586. [Google Scholar] [CrossRef]
- Park, J.P.; McDermet, M.K.; Doody, A.M.; Moeschler, J.B.; Marin-Padilla, J.M.; Wurster-Hill, D.H. Familial t(11;13)(q21;q14) and the duplication 11q, 13q phenotype. Am. J. Med. Genet. 1993, 45, 46–48. [Google Scholar] [CrossRef]
- Klaassens, M.; Van Dooren, M.; Eussen, H.J.; Douben, H.; Dekker, A.T.D.; Lee, C.; Donahoe, P.K.; Galjaard, R.J.; Goemaere, N.; De Krijger, R.R.; et al. Congenital diaphragmatic hernia and chromosome 15q26: Determination of a Candidate Region by Use of Fluorescent In Situ Hybridization and Array-Based COmparative Genomic Hybridization. Am. J. Hum. Genet. 2005, 76, 877–882. [Google Scholar] [CrossRef]
- Marguet, F.; Vezain, M.; Marcorelles, P.; Audebert-Bellanger, S.; Cassinari, K.; Drouot, N.; Chambon, P.; Gonzalez, B.J.; Horowitz, A.; Laquerriere, A.; et al. Neuropathological hallmarks of fetal hydrocephalus linked to CCDC88C pathogenic variants. Acta Neuropathol. Commun. 2021, 9, 104. [Google Scholar] [CrossRef]
- Enns, G.M.; Cox, V.A.; Goldstein, R.B.; Gibbs, D.L.; Harrison, M.R.; Golabi, M. Congenital diaphragmatic defects and associated syn-dromes, malformations, and chromosome anomalies: A retrospective study of 60 patients and literature review. Am. J. Med. Genet. 1998, 79, 215. [Google Scholar] [CrossRef] [PubMed]
- Kardon, G.; Ackerman, K.G.; McCulley, D.J.; Shen, Y.; Wynn, J.; Shang, L.; Bogenschutz, E.; Sun, X.; Chung, W.K. Congenital diaphragmatic hernias: From genes to mechanisms to therapies. Dis. Model. Mech. 2017, 10, 955–970. [Google Scholar] [CrossRef]
- Pilia, G.; Hughes-Benzie, R.M.; MacKenzie, A.E.; Baybayan, P.; Chen, E.Y.; Huber, R.; Neri, G.; Cao, A.; Forabosco, A.; Schlessinger, D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet. 1996, 12, 241–247. [Google Scholar] [CrossRef]
- Cottereau, E.; Mortemousque, I.; Moizard, M.-P.; Bürglen, L.; Lacombe, D.; Gilbert-Dussardier, B.; Sigaudy, S.; Boute, O.; David, A.; Faivre, L.; et al. Phenotypic Spectrum of Simpson-Golabi-Behmel Syndrome in a Series of 42 Cases with a Mutation in GPC 3 and Review of the Literature. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2013, 163, 92–105. [Google Scholar] [CrossRef]
- Chen, E.; Johnson, J.P.; Cox, V.A.; Golabi, M. Simpson-Golabi-Behmel syndrome: Congenital diaphragmatic hernia and radiologic findings in two patients and follow-up of a previously reported case. Am. J. Med. Genet. 1993, 46, 574–578. [Google Scholar] [CrossRef]
- Twigg, S.R.F.; Kan, R.; Babbs, C.; Bochukova, E.G.; Robertson, S.P.; Wall, S.A.; Morriss-Kay, G.M.; Wilkie, A.O.M. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8652–8657. [Google Scholar] [CrossRef]
- Vasudevan, P.C.; Twigg, S.R.F.; Mulliken, J.B.; Cook, J.A.; Quarrell, O.W.J.; Wilkie, A.O.M. Expanding the phenotype of craniofrontonasal syndrome: Two unrelated boys with EFNB1 mutations and congenital diaphragmatic hernia. Eur. J. Hum. Genet. 2006, 14, 884–887. [Google Scholar] [CrossRef]
- Brooks, A.S.; Van Dooren, M.; Hoogeboom, J.; Gischler, S.; Willems, P.J.; Tibboel, D. Congenital diaphragmatic hernia in a female patient with craniofrontonasal syndrome. Clin. Dysmorphol. 2002, 11, 151–153. [Google Scholar] [CrossRef]
- Laporte, J.; Hu, L.J.; Kretz, C.; Mandel, J.-L.; Kioschis, P.; Coy, J.F.; Klauck, S.M.; Poustka, A.; Dahl, N. A gene mutated in X–linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996, 13, 175–182. [Google Scholar] [CrossRef]
- Grogan, P.M.; Tanner, S.M.; Orstavik, K.H.; Knudsen, G.; Saperstein, D.S.; Vogel, H.; Barohn, R.J.; Herbelin, L.L.; McVey, A.L.; Katz, J.S. Myopathy with skeletal asymmetry and hemidiaphragm elevation is caused by myotubularin mutations. Neurology 2005, 64, 1638–1640. [Google Scholar] [CrossRef]
- Quaderi, N.A.; Schweiger, S.; Gaudenz, K.; Franco, B.; Rugarli, E.; Berger, W.; Feldman, G.J.; Volta, M.; Andolfi, G.; Gilgenkrantz, S.; et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp. Nat. Genet. 1997, 17, 285–291. [Google Scholar] [CrossRef]
- Taylor, J.; Aftimos, S. Congenital diaphragmatic hernia is a feature of Opitz G/BBB syndrome. Clin. Dysmorphol. 2010, 19, 225–226. [Google Scholar] [CrossRef]
- Lowe, C.; Terrey, M.; MacLachlan, E.A. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardationa clinical entity. Arch. Pediatr. Adolesc. Med. 1952, 83, 164. [Google Scholar] [CrossRef]
- Loi, M. Lowe syndrome. Orphanet. J. Rare Dis. 2006, 1, 16. [Google Scholar] [CrossRef]
- Race, H.J.; Elhadi, N.; Holder, S.E. Congenital diaphragmatic hernia in Lowe syndrome: A rare association? Clin. Dysmorphol. 2010, 19, 226. [Google Scholar] [CrossRef]
- Wang, X.; Sutton, V.R.; Peraza-Llanes, J.O.; Yu, Z.; Rosetta, R.; Kou, Y.-C.; Eble, T.N.; Patel, A.; Thaller, C.; Fang, P.; et al. Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat. Genet. 2007, 39, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Goltz, R.W.; Peterson, W.C.; Gorlin, R.J.; Ravits, H.G. Focal Dermal Hypoplasia. Arch. Dermatol. 1962, 86, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, X.; Zhao, X.; Liu, D.; Hu, T.; Li, W.; Jiang, L.; Dan, H.; Zeng, X.; Chen, Q. Focal dermal hypoplasia: Updates. Oral Dis. 2013, 20, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, R.; Jakubiak, A.; Lombardi, M.P.; Jaworski, W.; Slezak, R.; Patkowski, D.; Hennekam, R.C. Co-occurrence of severe Goltz-Gorlin syndrome and pentalogy of Cantrell—Case report and review of the literature. Am. J. Med. Genet. Part. A 2011, 155, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.M.; Lombardi, M.P.; Van Essen, A.J.; Wakeling, E.; Castle, B.; Temple, I.K.; Kumar, V.K.A.; Writzl, K.; Hennekam, R.C.M. Phenotype and genotype in 17 patients with Goltz-Gorlin syndrome. J. Med. Genet. 2009, 46, 716–720. [Google Scholar] [CrossRef]
- Wimplinger, I.; Morleo, M.; Rosenberger, G.; Iaconis, D.; Orth, U.; Meinecke, P.; Lerer, I.; Ballabio, A.; Gal, A.; Franco, B.; et al. Mutations of the Mitochondrial Holocytochrome c–Type Synthase in X-Linked Dominant Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2006, 79, 878–889. [Google Scholar] [CrossRef]
- Veyver, I.V.D. Microphthalmia with linear skin defects (MLS), Aicardi, and Goltz syndromes: Are they related X-linked dominant male-lethal disorders? Cytogenet. Genome Res. 2002, 99, 289–296. [Google Scholar] [CrossRef]
- Krantz, I.D.; McCallum, J.; De Scipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef]
- Ireland, M.; Donnai, D.; Burn, J. Brachmann-de Lange syndrome. Delineation of the clinical phenotype. Am. J. Med. Genet. 1993, 47, 959–964. [Google Scholar] [CrossRef]
- Martínez-Frías, M.L.; Bermejo, E.; Félix, V.; Jiménez, N.; Gómez-Ullate, J.; López, J.A.; Aparicio, P.; Ayala, A.; Gairi, J.M.; Galán, E.; et al. Síndrome de Brachmann de Lange en nuestro medio: Características clínicas y epidemiológicas [Brachmann-de-Lange syndrome in our population: Clinical and epidemiological characteristics]. An. Esp. Pediatr. 1998, 48, 293–298. (In Spanish) [Google Scholar]
- Marino, T.; Wheeler, P.G.; Simpson, L.L.; Craigo, S.D.; Bianchi, D.W. Fetal diaphragmatic hernia and upper limb anomalies suggest Brachmann-de Lange syndrome. Prenat. Diagn. 2002, 22, 144–147. [Google Scholar] [CrossRef]
- Jelsema, R.D.; Isada, N.B.; Kazzi, N.J.; Sargent, K.; Harrison, M.R.; Johnson, M.P.; Evans, M.I. Prenatal diagnosis of congenital diaphragmatic hernia not amenable to prenatal or neonatal repair: Brachmann-de Lange syndrome. Am. J. Med. Genet. 1993, 47, 1022–1023. [Google Scholar] [CrossRef]
- Schrier, S.A.; Sherer, I.; Deardorff, M.A.; Clark, D.; Audette, L.; Gillis, L.; Kline, A.D.; Ernst, L.; Loomes, K.; Krantz, I.D.; et al. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am. J. Med. Genet. Part. A 2011, 155, 3007–3024. [Google Scholar] [CrossRef]
- Pelletier, J.; Bruening, W.; Kashtan, C.E.; Mauer, S.M.; Manivel, J.C.; Striegel, J.E.; Houghton, D.C.; Junien, C.; Habib, R.; Fouser, L. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 1991, 67, 437–447. [Google Scholar] [CrossRef]
- Devriendt, K.; DeLoof, E.; Moerman, P.; Legius, E.; Vanhole, C.; De Zegher, F.; Proesmans, W.; Devlieger, H. Diaphragmatic hernia in Denys-Drash syndrome. Am. J. Med. Genet. 1995, 57, 97–101. [Google Scholar] [CrossRef]
- Cho, H.Y.; Lee, B.S.; Kang, C.H.; Kim, W.-H.; Ha, I.S.; Cheong, H.I.; Choi, Y. Hydrothorax in a patient with Denys-Drash syndrome associated with a diaphragmatic defect. Pediatr. Nephrol. 2006, 21, 1909–1912. [Google Scholar] [CrossRef]
- Antonius, T.; van Bon, B.; Eggink, A.; van der Burgt, I.; Noordam, K.; van Heijst, A. Denys–Drash syndrome and congenital diaphragmatic hernia: Another case with the 1097G > A(Arg366His) mutation. Am. J. Med. Genet. Part. A 2008, 146A, 496–499. [Google Scholar] [CrossRef]
- Faivre, L.; Collod-Beroud, G.; Loeys, B.; Child, A.; Binquet, C.; Gautier, E.; Callewaert, B.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Effect of Mutation Type and Location on Clinical Outcome in 1,013 Probands with Marfan Syndrome or Related Phenotypes and FBN1 Mutations: An International Study. Am. J. Hum. Genet. 2007, 81, 454–466. [Google Scholar] [CrossRef]
- Aubart, M.; Gazal, S.; Arnaud, P.; Benarroch, L.; Gross, M.-S.; Buratti, J.; Boland, A.; Meyer, V.; Zouali, H.; Hanna, N.; et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur. J. Hum. Genet. 2018, 26, 1759–1772. [Google Scholar] [CrossRef]
- Universal Mutation Database (UMD) FBN. Available online: http://www.umd.be/FBN1/;http://www.umd.be/FBN1/4DACTION/w_mutations (accessed on 27 November 2020).
- Jacobs, A.M.; Toudjarska, I.; Racine, A.; Tsipouras, P.; Kilpatrick, M.W.; Shanske, A. A Recurring FBN1 Gene Mutation in Neonatal Marfan Syndrome. Arch. Pediatr. Adolesc. Med. 2002, 156, 1081–1085. [Google Scholar] [CrossRef]
- Revencu, N.; Quenum, G.; Detaille, T.; Verellen, G.; De Paepe, A.; Verellen-Dumoulin, C. Congenital diaphragmatic eventration and bilateral uretero-hydronephrosis in a patient with neonatal Marfan syndrome caused by a mutation in exon 25 of the FBN1 gene and review of the literature. Eur. J. Nucl. Med. Mol. Imaging 2003, 163, 33–37. [Google Scholar] [CrossRef]
- Veiga-Fernández, A.; Prieto, L.J.; Álvarez, T.; Ruiz, Y.; Pérez, R.; Gámez, F.; Abad, V.O.; Yllana, F.; De León-Luis, J. Perinatal diagnosis and management of early-onset Marfan syndrome: Case report and systematic review. J. Matern. Neonatal Med. 2019, 33, 2493–2504. [Google Scholar] [CrossRef]
- Bergman, J.E.; Janssen, N.; Hoefsloot, L.H.; Jongmans, M.C.; Hofstra, R.M.; Van Ravenswaaij-Arts, C.M.A. CHD7 mutations and CHARGE syndrome: The clinical implications of an expanding phenotype. J. Med. Genet. 2011, 48, 334–342. [Google Scholar] [CrossRef]
- Janssen, N.; Bergman, J.E.H.; Swertz, M.; Tranebjaerg, L.; Lodahl, M.; Schoots, J.; Hofstra, R.M.W.; van Ravenswaaij-Arts, C.M.A.; Hoefsloot, L.H. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum. Mutat. 2012, 33, 1149–1160. [Google Scholar] [CrossRef]
- Pagon, R.A.; Graham, J.M.; Zonana, J.; Yong, S.-L. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr. 1981, 99, 223–227. [Google Scholar] [CrossRef]
- Hall, B.D. Choanal atresia and associated multiple anomalies. J. Pediatr. 1979, 95, 395–398. [Google Scholar] [CrossRef]
- Blake, K.D.; Davenport, S.L.H.; Hall, B.D.; Hefner, M.A.; Pagon, R.A.; Williams, M.S.; Lin, A.E.; Graham, J.J.M. CHARGE Association: An Update and Review for the Primary Pediatrician. Clin. Pediatr. 1998, 37, 159–173. [Google Scholar] [CrossRef]
- Verloes, A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am. J. Med. Genet. Part. A 2005, 133A, 306–308. [Google Scholar] [CrossRef]
- Casaccia, G.; Digilio, M.C.; Seymandi, P.L.; Bagolan, P. Congenital diaphragmatic hernia in CHARGE syndrome. Pediatr. Surg. Int. 2007, 24, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.J.; Admiraal, R.J.; Van Der Donk, K.P.; Vissers, L.E.L.M.; Baas, A.F.; Kapusta, L.; Van Hagen, J.M.; Donnai, D.; De Ravel, T.J.; Veltman, J.; et al. CHARGE syndrome: The phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2005, 43, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Philip, N.; Gambarelli, D.; Guys, J.M.; Camboulives, J.; Ayme, S. Epidemiological study of congenital diaphragmatic defects with special reference to aetiology. Eur. J. Nucl. Med. Mol. Imaging 1991, 150, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Dillon, E.; Renwick, M. Antenatal detection of congenital diaphragmatic hernias: The northern region experience. Clin. Radiol. 1993, 48, 264–267. [Google Scholar] [CrossRef]
- Neville, H.L.; Jaksic, T.; Wilson, J.M.; Lally, P.A.; Hardin, W.D.; Hirschl, R.B.; Langham, M.R.; Lally, K.P. Fryns syndrome in children with congenital diaphragmatic hernia. J. Pediatr. Surg. 2002, 37, 1685–1687. [Google Scholar] [CrossRef]
- Brady, P.; Moerman, P.; De Catte, L.; Deprest, J.; Devriendt, K.; Vermeesch, J. Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic Congenital Diaphragmatic Hernia. Eur. J. Med. Genet. 2014, 57, 487–493. [Google Scholar] [CrossRef]
- McInerney-Leo, A.M.; Harris, J.E.; Gattas, M.; Peach, E.E.; Sinnott, S.; Dudding-Byth, T.; Rajagopalan, S.; Barnett, C.P.; Anderson, L.K.; Wheeler, L.; et al. Fryns Syndrome Associated with Recessive Mutations in PIGN in two Separate Families. Hum. Mutat. 2016, 37, 695–702. [Google Scholar] [CrossRef]
- Lin, A.E.; Pober, B.R.; Mullen, M.P.; Slavotinek, A.M. Cardiovascular malformations in Fryns syndrome: Is there a pathogenic role for neural crest cells? Am. J. Med. Genet. Part. A 2005, 139, 186–193. [Google Scholar] [CrossRef]
- Fryns, J.P.; Moerman, F.; Goddeeris, P.; Bossuyt, C.; Berghe, H.V.D. A new lethal syndrome with cloudy corneae, diaphragmatic defects and distal limb deformities. Qual. Life Res. 1979, 50, 65–70. [Google Scholar] [CrossRef]
- Goddeeris, P.; Fryns, J.P.; van den Berghe, H. Diaphragmatic defects, craniofacial dysmorphism, cleft palate and distal limb de-formities—A new lethal syndrome. J. Genet. Hum. 1980, 28, 57. [Google Scholar] [PubMed]
- Donnai, D.; Barrow, M. Diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness: A newly recognized autosomal recessive disorder? Am. J. Med. Genet. 1993, 47, 679–682. [Google Scholar] [CrossRef]
- Chassaing, N.; Lacombe, D.; Carles, D.; Calvas, P.; Saura, R.; Bieth, E. Donnai-Barrow syndrome: Four additional patients. Am. J. Med. Genet. 2003, 121A, 258–262. [Google Scholar] [CrossRef]
- Kantarci, S.; Al-Gazali, L.; Hill, R.S.; Donnai, D.; Black, G.C.M.; Bieth, E.; Chassaing, N.; Lacombe, D.; Devriendt, K.; Teebi, A.; et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat. Genet. 2007, 39, 957–959. [Google Scholar] [CrossRef]
- Raila, J.; Willnow, T.E.; Schweigert, F. Megalin-Mediated Reuptake of Retinol in the Kidneys of Mice Is Essential for Vitamin A Homeostasis. J. Nutr. 2005, 135, 2512–2516. [Google Scholar] [CrossRef]
- Kling, D.E.; Schnitzer, J.J. Vitamin A deficiency (VAD), teratogenic, and surgical models of congenital diaphragmatic hernia (CDH). Am. J. Med. Genet. Part. C Semin. Med. Genet. 2007, 145C, 139–157. [Google Scholar] [CrossRef]
- Khalifa, O.; Al-Sahlawi, Z.; Imtiaz, F.; Ramzan, K.; Allam, R.; Al-Mostafa, A.; Abdel-Fattah, M.; Abuharb, G.; Nester, M.; Verloes, A.; et al. Variable expression pattern in Donnai-Barrow syndrome: Report of two novel LRP2 mutations and review of the literature. Eur. J. Med. Genet. 2015, 58, 293–299. [Google Scholar] [CrossRef]
- Kantarci, S.; Donahoe, P.K. Congenital diaphragmatic hernia (CDH) etiology as revealed by pathway genetics. Am. J. Med. Genet. Part. C: Semin. Med. Genet. 2007, 145C, 217–226. [Google Scholar] [CrossRef]
- Goodman, D.S. The Retinoids; Sporn, M.B., Boberts, A.B., Goodman, D.S., Eds.; Academic Press: New York, NY, USA, 1984. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A Membrane Receptor for Retinol Binding Protein Mediates Cellular Uptake of Vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; FitzPatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 Cause a Broad Spectrum of Malformations Including Anophthalmia, Congenital Heart Defects, Diaphragmatic Hernia, Alveolar Capillary Dysplasia, Lung Hypoplasia, and Mental Retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef]
- Andijani, A.A.; Shajira, E.S.; Abushaheen, A.; Al-Matary, A. Microphthalmia Syndrome 9: Case Report of a Newborn Baby with Pulmonary Hypoplasia, Diaphragmatic Eventration, Microphthalmia, Cardiac Defect and Severe Primary Pulmonary Hypertension. Am. J. Case Rep. 2019, 20, 354–360. [Google Scholar] [CrossRef]
- Srour, M.; Chitayat, D.; Caron, V.; Chassaing, N.; Bitoun, P.; Patry, L.; Cordier, M.-P.; Capo-Chichi, J.-M.; Francannet, C.; Calvas, P.; et al. Recessive and Dominant Mutations in Retinoic Acid Receptor Beta in Cases with Microphthalmia and Diaphragmatic Hernia. Am. J. Hum. Genet. 2013, 93, 765–772. [Google Scholar] [CrossRef]
- Park, Y.; Gong, G.; Choe, G.; Yu, E.; Kim, K.S.; Lee, I. Jarcho-Levin syndrome: A report of an autopsy case with cytogenetic analysis. J. Korean Med. Sci. 1993, 8, 471–475. [Google Scholar] [CrossRef]
- Lam, Y.H.; Eik-Nes, S.H.; Tang, M.H.Y.; Lee, C.P.; Nicholls, J.M. Prenatal sonographic features of spondylocostal dysostosis and diaphragmatic hernia in the first trimester. Ultrasound Obstet. Gynecol. 1999, 13, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Day, R.; Fryer, A. Diaphragmatic hernia and preaxial polydactyly in spondylothoracic dysplasia. Clin. Dysmorphol. 2003, 12, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, L.M.; García-García, I.; Correa-Rivas, M.S.; García-Fragoso, L. Pulmonary hypoplasia in Jarcho-Levin syndrome. Puerto Rico Health Sci. J. 2004, 23, 65–67. [Google Scholar]
- Cornier, A.; Ramírez, N.; Arroyo, S.; Acevedo, J.; García, L.; Carlo, S.; Korf, B. Phenotype characterization and natural history of spondylothoracic dysplasia syndrome: A series of 27 new cases. Am. J. Med. Genet. Part. A 2004, 128A, 120–126. [Google Scholar] [CrossRef]
- Turnpenny, P.D.; Alman, B.; Cornier, A.S.; Giampietro, P.F.; Offiah, A.; Tassy, O.; Pourquie, O.; Kusumi, K.; Dunwoodie, S. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev. Dyn. 2007, 236, 1456–1474. [Google Scholar] [CrossRef]
- Bulman, M.P.; Kusumi, K.; Frayling, T.M.; McKeown, C.; Garrett, C.; Lander, E.S.; Krumlauf, R.; Hattersley, A.T.; Ellard, S.; Turnpenny, P.D. Mutations in the human Delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 2000, 24, 438–441. [Google Scholar] [CrossRef]
- Kuo, C.T.; Morrisey, E.E.; Anandappa, R.; Sigrist, K.; Lu, M.M.; Parmacek, M.S.; Soudais, C.; Leiden, J.M. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997, 11, 1048–1060. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Lin, Q.; Duncan, S.A.; Olson, E.N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997, 11, 1061–1072. [Google Scholar] [CrossRef]
- Yu, L.; Wynn, J.; Cheung, Y.H.; Shen, Y.; Mychaliska, G.B.; Crombleholme, T.M.; Azarow, K.S.; Lim, F.Y.; Chung, D.H.; Potoka, D.; et al. Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Qual. Life Res. 2012, 132, 285–292. [Google Scholar] [CrossRef]
- Cai, K.Q.; Capo-Chichi, D.C.; Rula, M.E.; Yang, D.-H.; Xu, X.-X. Dynamic GATA6 expression in primitive endoderm formation and maturation in early mouse embryogenesis. Dev. Dyn. 2008, 237, 2820–2829. [Google Scholar] [CrossRef]
- Allen, H.L.; The International Pancreatic Agenesis Consortium; Flanagan, S.; Shaw-Smith, C.; De Franco, E.; Akerman, I.; Caswell, R.; Ferrer, J.; Hattersley, A.T.; Ellard, S. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 2011, 44, 20–22. [Google Scholar] [CrossRef]
- Keijzer, R.; van Tuyl, M.; Meijers, C.; Post, M.; Tibboel, D.; Grosveld, F.; Koutsourakis, M. The transcription factor GATA6 is essential for branching morphogenesis and epithelial cell differentiation during fetal pulmonary development. Development 2001, 128, 503–511. [Google Scholar] [CrossRef]
- Zhang, Y.; Goss, A.M.; Cohen, E.D.; Kadzik, R.; Lepore, J.J.; Muthukumaraswamy, K.; Yang, J.; DeMayo, F.; Whitsett, J.A.; Parmacek, M.S.; et al. A Gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat. Genet. 2008, 40, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Kodo, K.; Nishizawa, T.; Furutani, M.; Arai, S.; Yamamura, E.; Joo, K.; Takahashi, T.; Matsuoka, R.; Yamagishi, H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13933–13938. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, M.; Shield, J.; Acerini, C.; Walker, J.; Ellard, S.; Marchand, M.; Polak, M.; Vaxillaire, M.; Crolla, J.; Bunyan, D.; et al. Pancreatic hypoplasia presenting with neonatal diabetes mellitus in association with congenital heart defect and developmental delay. Am. J. Med. Genet. Part. A 2010, 152A, 340–346. [Google Scholar] [CrossRef]
- Lin, X.; Huo, Z.; Liu, X.; Zhang, Y.; Li, L.; Zhao, H.; Yan, B.; Liu, Y.; Yang, Y.; Chen, Y.-H. A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. J. Hum. Genet. 2010, 55, 662–667. [Google Scholar] [CrossRef]
- Maitra, M.; Koenig, S.N.; Srivastava, D.; Garg, V. Identification of GATA6 Sequence Variants in Patients with Congenital Heart Defects. Pediatr. Res. 2010, 68, 281–285. [Google Scholar] [CrossRef]
- Zhao, R.; Watt, A.J.; Li, J.; Luebke-Wheeler, J.; Morrisey, E.E.; Duncan, S.A. GATA6 Is Essential for Embryonic Development of the Liver but Dispensable for Early Heart Formation. Mol. Cell. Biol. 2005, 25, 2622–2631. [Google Scholar] [CrossRef]
- Morrisey, E.E.; Tang, Z.; Sigrist, K.; Lu, M.M.; Jiang, F.; Ip, H.S.; Parmacek, M.S. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998, 12, 3579–3590. [Google Scholar] [CrossRef]
- Yu, L.; Bennett, J.T.; Wynn, J.; Carvill, G.L.; Cheung, Y.H.; Shen, Y.; Mychaliska, G.B.; Azarow, K.S.; Crombleholme, T.M.; Chung, D.H.; et al. Whole exome sequencing identifies de novo mutations inGATA6associated with congenital diaphragmatic hernia. J. Med. Genet. 2014, 51, 197–202. [Google Scholar] [CrossRef]
- Brady, P.D.; Van Houdt, J.; Callewaert, B.; Deprest, J.; Devriendt, K.; Vermeesch, J.R. Exome sequencing identifies ZFPM2 as a cause of familial isolated congenital diaphragmatic hernia and possibly cardiovascular malformations. Eur. J. Med. Genet. 2014, 57, 247–252. [Google Scholar] [CrossRef]
- Qiu, Y.; Krishnan, V.; Pereira, F.A.; Tsai, S.Y.; Tsai, M.J. Chicken ovalbumin upstream promoter-transcription factors and their reg-ulation. J. Steroid. Biochem. Mol. Biol. 1996, 56, 81–85. [Google Scholar] [CrossRef]
- You, L.-R.; Takamoto, N.; Yu, C.-T.; Tanaka, T.; Kodama, T.; DeMayo, F.; Tsai, S.Y.; Tsai, M.-J. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc. Natl. Acad. Sci. USA 2005, 102, 16351–16356. [Google Scholar] [CrossRef] [PubMed]
- High, F.A.; Bhayani, P.; Wilson, J.M.J.M.; Bult, C.J.; Donahoe, P.K.P.K.; Longoni, M. De Novo Frameshift Mutation in COUP-TFII (NR2F2) in Human Congenital Diaphragmatic Hernia. Am. J. Med. Genet. Part. A 2016, 170, 2457–2461. [Google Scholar] [CrossRef] [PubMed]
- Clugston, R.D.; Zhang, W.; Greer, J.J. Gene expression in the developing diaphragm: Significance for congenital diaphragmatic hernia. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L665–L675. [Google Scholar] [CrossRef]
- Hong, S.-H.; David, G.; Wong, C.-W.; Dejean, A.; Privalsky, M.L. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor (RAR) and PLZF-RAR oncoproteins associated with acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 9028–9033. [Google Scholar] [CrossRef]
- Aljohani, O.; Haldeman, S.; Rao, R. Abstracts from the 11th International Conference on Neonatal and Childhood Pulmonary Vascular Disease. Pulm. Circ. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Rossetti, L.Z.; Glinton, K.; Yuan, B.; Liu, P.; Pillai, N.; Mizerik, E.; Magoulas, P.; Rosenfeld, J.A.; Karaviti, L.; Sutton, V.R.; et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am. J. Med. Genet. Part. A 2019, 179, 1376–1382. [Google Scholar] [CrossRef]
- Qi, H.; Yu, L.; Zhou, X.; Wynn, J.; Zhao, H.; Guo, Y.; Zhu, N.; Kitaygorodsky, A.; Hernan, R.; Aspelund, G.; et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018, 14, e1007822. [Google Scholar] [CrossRef]
- Slavotinek, A.M. Single gene disorders associated with congenital diaphragmatic hernia. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2007, 145C, 172–183. [Google Scholar] [CrossRef]
- Brady, P.D.; Dekoninck, P.; Fryns, J.P.; Devriendt, K.; Deprest, J.; Vermeesch, J.R. Identification of dosage-sensitive genes in fetuses referred with severe isolated congenital diaphragmatic hernia. Prenat. Diagn. 2013, 33, 1283–1292. [Google Scholar] [CrossRef]
- Yu, L.; Wynn, J.; Ma, L.; Guha, S.; Mychaliska, G.B.; Crombleholme, T.M.; Azarow, K.S.; Lim, F.Y.; Chung, D.H.; Potoka, D.; et al. De novo copy number variants are associated with congenital diaphragmatic hernia. J. Med. Genet. 2012, 49, 650–659. [Google Scholar] [CrossRef]
- Beurskens, N.; Klaassens, M.; Rottier, R.; de Klein, A.; Tibboel, D. Linking animal models to human congenital diaphragmatic hernia. Birth Defects Res. Part. A: Clin. Mol. Teratol. 2007, 79, 565–572. [Google Scholar] [CrossRef]
- Longoni, M.; High, F.A.; Qi, H.; Joy, M.P.; Hila, R.; Coletti, C.M.; Wynn, J.; Loscertales, M.; Shan, L.; Bult, C.J.; et al. Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Qual. Life Res. 2017, 136, 679–691. [Google Scholar] [CrossRef]
- The Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nat. Cell Biol. 2015, 519, 223–228. [Google Scholar] [CrossRef]
- Yu, L.; Sawle, A.; Wynn, J.; Aspelund, G.; Stolar, C.J.; Arkovitz, M.S.; Potoka, D.; Azarow, K.S.; Mychaliska, G.B.; Shen, Y.; et al. Increased burden ofde novopredicted deleterious variants in complex congenital diaphragmatic hernia. Hum. Mol. Genet. 2015, 24, 4764–4773. [Google Scholar] [CrossRef]
- Ackerman, K.G.; Greer, J.J. Development of the diaphragm and genetic mouse models of diaphragmatic defects. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2007, 145C, 109–116. [Google Scholar] [CrossRef]
- Best, S.; Wou, K.; Vora, N.; Van Der Veyver, I.B.; Wapner, R.; Chitty, L.S. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat. Diagn. 2018, 38, 10–19. [Google Scholar] [CrossRef]
- Meier, N.; Bruder, E.; Lapaire, O.; Hoesli, I.; Kang, A.; Hench, J.; Hoeller, S.; De Geyter, J.; Miny, P.; Heinimann, K.; et al. Exome sequencing of fetal anomaly syndromes: Novel phenotype–genotype discoveries. Eur. J. Hum. Genet. 2019, 27, 730–737. [Google Scholar] [CrossRef]
- Qiao, L.; Wynn, J.; Yu, L.; Ms, R.H.; Zhou, X.; Duron, V.; Aspelund, G.; Farkouh-Karoleski, C.; Zygumunt, A.; Krishnan, U.S.; et al. Likely damaging de novo variants in congenital diaphragmatic hernia patients are associated with worse clinical outcomes. Genet. Med. 2020, 22, 2020–2028. [Google Scholar] [CrossRef]
- Scott, T.M.; Campbell, I.M.; Hernandez-Garcia, A.; Lalani, S.R.; Liu, P.; Shaw, C.A.; Rosenfeld, J.A.; Scott, D.A. Clinical exome sequencing data reveal high diagnostic yields for congenital diaphragmatic hernia plus (CDH+) and new phenotypic expansions involving CDH. J. Med. Genet. 2021. [Google Scholar] [CrossRef]
- Nakamura, H.; Doi, T.; Puri, P.; Friedmacher, F. Transgenic animal models of congenital diaphragmatic hernia: A comprehensive overview of candidate genes and signaling pathways. Pediatr. Surg. Int. 2020, 36, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Russo, F.M.; Debeer, A.; De Coppi, P.; Devriendt, K.; Crombag, N.; Hubble, T.; Power, B.; Benachi, A.; Deprest, J. What should we tell parents? Congenital diaphragmatic hernia. Prenat. Diagn. 2020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Locus | Protein Activity | Expression | Function | Anomalies Associated |
|---|---|---|---|---|---|
| COUP-TFII | 15q26.2 | Nuclear receptor subfamily 2 group F: steroid/thyroid hormone receptor superfamily | Lung mesenchyme | Transcription factor | CHD, pancreatic agenesis |
| SIN3A | 15q24.2 | Corepressor that interacts with the retinoic acid receptor (RARs) | Transcription factor | Microcephaly, intellectual disabilities | |
| FOG2 | 8q23.1 | Zinc finger protein that modulates the transcriptional activity of GATA proteins | Lung mesenchyme and diaphragm | Transcription cofactor | Cardiac defects, lung hypoplasia, eventration |
| GATA4 | 8q23.1 | Zinc finger transcription factor; retinoic signaling pathway | Liver, lung mesenchyme, diaphragm | Transcription factor | Cardiac defects, eventration of the diaphragm |
| GATA6 | 18q11.1-q11.2 | Zinc finger containing transcription factors | Lung epithelium and diaphragm | Transcription factor | Organogenesis defects of the pancreas and heart and defect in the development of the diaphragm and pericardium |
| MYRF | 11q12.2 | Membrane-associated transcription factor | Diaphragm, lung, heart, genitourinary system | Transcription factor | CHD, genitourinary anomalies, and pulmonary hypoplasia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannata, G.; Caporilli, C.; Grassi, F.; Perrone, S.; Esposito, S. Management of Congenital Diaphragmatic Hernia (CDH): Role of Molecular Genetics. Int. J. Mol. Sci. 2021, 22, 6353. https://doi.org/10.3390/ijms22126353
Cannata G, Caporilli C, Grassi F, Perrone S, Esposito S. Management of Congenital Diaphragmatic Hernia (CDH): Role of Molecular Genetics. International Journal of Molecular Sciences. 2021; 22(12):6353. https://doi.org/10.3390/ijms22126353
Chicago/Turabian StyleCannata, Giulia, Chiara Caporilli, Federica Grassi, Serafina Perrone, and Susanna Esposito. 2021. "Management of Congenital Diaphragmatic Hernia (CDH): Role of Molecular Genetics" International Journal of Molecular Sciences 22, no. 12: 6353. https://doi.org/10.3390/ijms22126353
APA StyleCannata, G., Caporilli, C., Grassi, F., Perrone, S., & Esposito, S. (2021). Management of Congenital Diaphragmatic Hernia (CDH): Role of Molecular Genetics. International Journal of Molecular Sciences, 22(12), 6353. https://doi.org/10.3390/ijms22126353