
TAFRO Syndrome with Renal Thrombotic Microangiopathy: Insights into the Molecular Mechanism and Treatment Opportunities

 , ,
, ,
Abstract
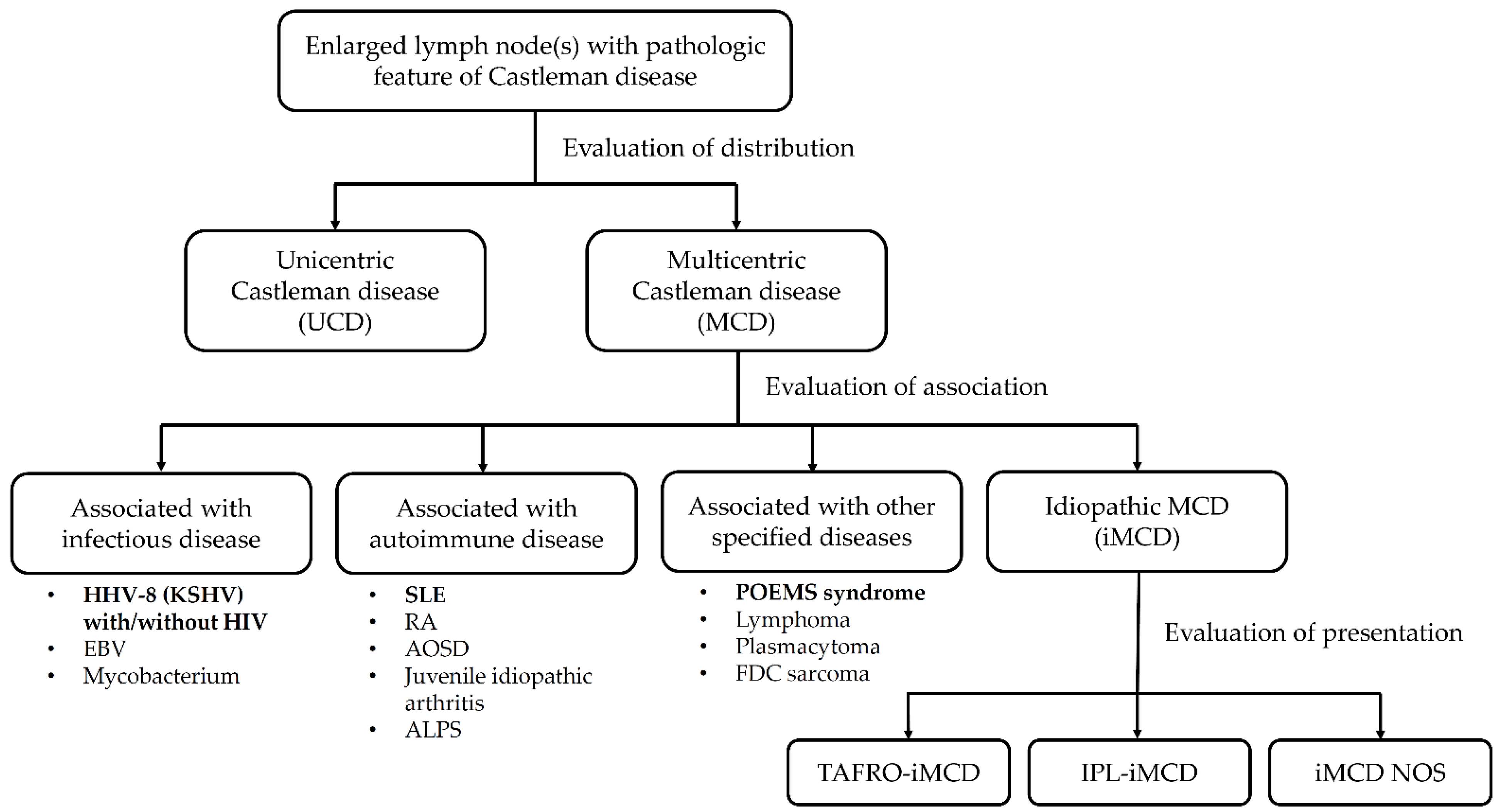
1. Introduction
2. Pathophysiologic Mechanisms of Castleman Disease and TAFRO Syndrome
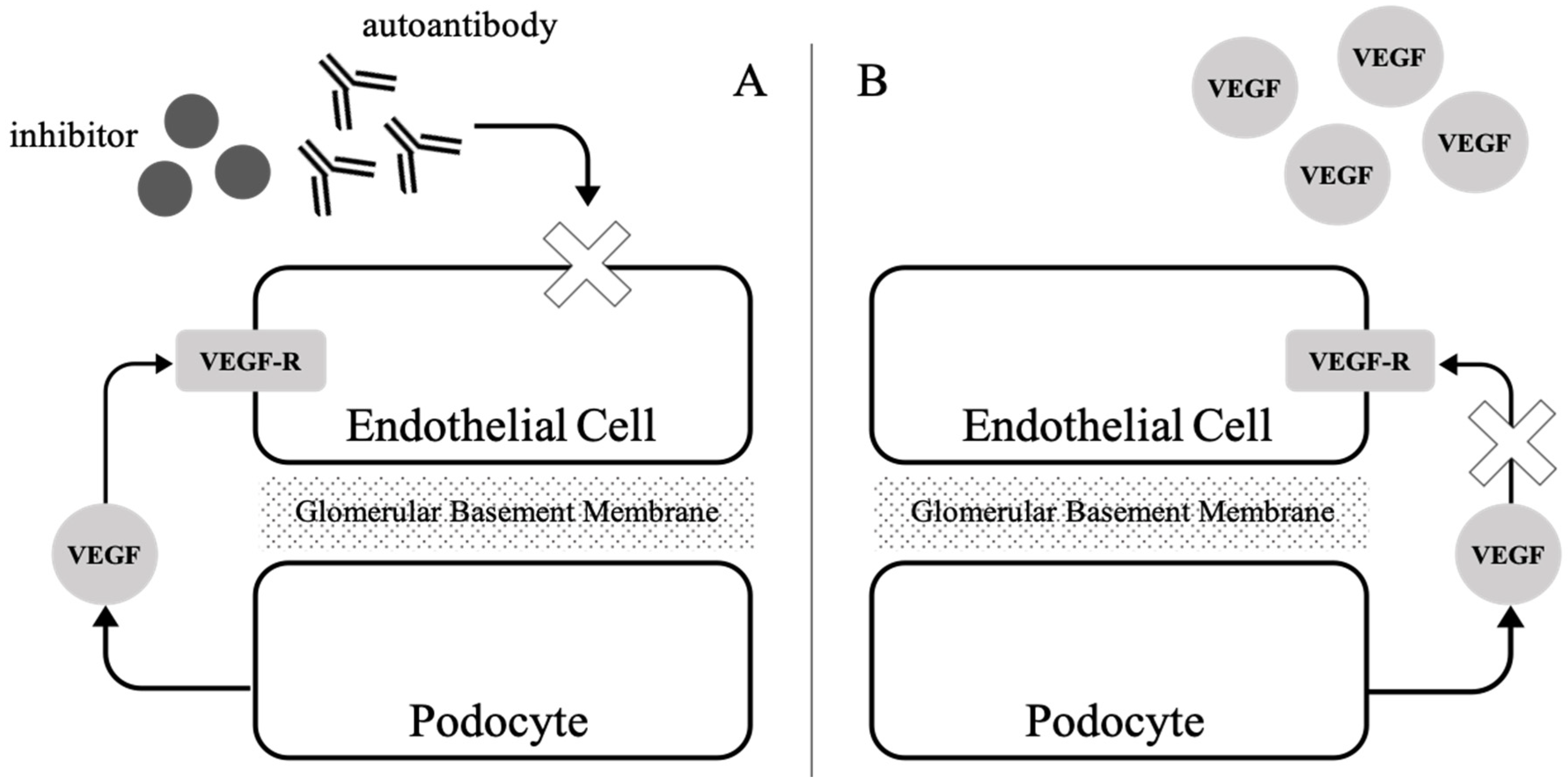
3. Renal Thrombotic Microangiopathy and Its Proposed Mechanism
4. From TAFRO Syndrome to Renal Thrombotic Microangiopathy
5. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Yu, L.; Tu, M.; Cortes, J.; Xu-Monette, Z.Y.; Miranda, R.N.; Zhang, J.; Orlowski, R.Z.; Neelapu, S.; Boddu, P.C.; Akosile, M.A.; et al. Clinical and pathological characteristics of HIV- and HHV-8-negative Castleman disease. Blood 2017, 129, 1658–1668. [Google Scholar] [CrossRef]
- Karoui, K.E.; Vuiblet, V.; Dion, D.; Izzedine, H.; Guitard, J.; Frimat, L.; Delahousse, M.; Remy, P.; Boffa, J.J.; Pillebout, E.; et al. Renal involvement in Castleman disease. Nephrol. Dial. Transplant. 2011, 26, 599–609. [Google Scholar] [CrossRef]
- Pan, Y.; Cui, Z.; Wang, S.; Zheng, D.; Deng, Z.; Tian, X.; Guo, H.; Bao, W.; Zhou, S.; Wang, Y. Idiopathic multicentric Castleman disease with Sjögren’s syndrome and secondary membranous nephropathy: A case report and review of the literature. BMC Nephrol. 2020, 21, 528. [Google Scholar] [CrossRef]
- Takai, K.; Nikkuni, K.; Shibuya, H.; Hashidate, H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki 2010, 51, 320–325. [Google Scholar] [PubMed]
- Sakashita, K.; Murata, K.; Takamori, M. TAFRO syndrome: Current perspectives. J. Blood Med. 2018, 9, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Srkalovic, G.; Marijanovic, I.; Srkalovic, M.B.; Fajgenbaum, D.C. TAFRO syndrome: New subtype of idiopathic multicentric Castleman disease. Bosn. J. Basic Med. Sci. 2017, 17, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Leurs, A.; Gnemmi, V.; Lionet, A.; Renaud, L.; Gibier, J.B.; Copin, M.C.; Hachulla, E.; Hatron, P.Y.; Launay, D.; Fajgenbaum, D.C.; et al. Renal Pathologic Findings in TAFRO syndrome: Is there a continuum between thrombotic microangiopathy and membranoproliferative glomerulonephritis? A case report and literature review. Front. Immunol. 2019, 10, 1489. [Google Scholar] [CrossRef]
- Kranich, J.; Krautler, N.J. How Follicular Dendritic Cells Shape the B-Cell Antigenome. Front. Immunol. 2016, 7, 225. [Google Scholar] [CrossRef]
- Wang, J.; Bu, D.F.; Li, T.; Zheng, R.; Zhang, B.X.; Chen, X.X.; Zhu, X.J. Autoantibody production from a thymoma and a follicular dendritic cell sarcoma associated with paraneoplastic pemphigus. Br. J. Dermatol. 2005, 153, 558–564. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 318–325. [Google Scholar] [CrossRef]
- El-Osta, H.E.; Kurzrock, R. Castleman’s disease: From basic mechanisms to molecular therapeutics. Oncologist 2011, 16, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Katsume, A.; Saito, H.; Yamada, Y.; Yorozu, K.; Ueda, O.; Akamatsu, K.; Nishimoto, N.; Kishimoto, T.; Yoshizaki, K.; Ohsugi, Y. Anti-interleukin 6 (IL-6) receptor antibody suppresses Castleman’s disease like symptoms emerged in IL-6 transgenic mice. Cytokine 2002, 20, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Kellum, J.A. Interleukin-6. Crit. Care Med. 2005, 33, S463–S465. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef]
- Huang, S.P.; Wu, M.S.; Shun, C.T.; Wang, H.P.; Lin, M.T.; Kuo, M.L.; Lin, J.T. Interleukin-6 increases vascular endothelial growth factor and angiogenesis in gastric carcinoma. J. Biomed. Sci. 2004, 11, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Aoki, C.; Yoshio-Hoshino, N.; Takayama, K.; Curiel, D.T.; Nishimoto, N. Interleukin-6 induces both cell growth and VEGF production in malignant mesotheliomas. Int. J. Cancer 2006, 119, 1303–1311. [Google Scholar] [CrossRef]
- Tanaka, T.; Kishimoto, T. Targeting interleukin-6: All the way to treat autoimmune and inflammatory diseases. Int. J. Biol. Sci. 2012, 8, 1227–1236. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; van Rhee, F.; Nabel, C.S. HHV-8-negative, idiopathic multicentric Castleman disease: Novel insights into biology, pathogenesis, and therapy. Blood 2014, 123, 2924–2933. [Google Scholar] [CrossRef]
- Leger-Ravet, M.B.; Peuchmaur, M.; Devergne, O.; Audouin, J.; Raphael, M.; van Damme, J.; Galanaud, P.; Diebold, J.; Emiliem, D. Interleukin-6 gene expression in Castleman’s disease. Blood 1991, 78, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, K.; Matsuda, T.; Nishimoto, N. Pathogenic significance of interleukin-6 (IL-6/ BSF-2) in Castleman’s disease. Blood 1989, 74, 1360–1367. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Uldrick, T.S.; Bagg, A.; Frank, D.; Wu, D.; Srkalovic, G.; Simpson, D.; Liu, A.Y.; Menke, D.; Chandrakasan, S.; et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017, 129, 1646–1657. [Google Scholar] [CrossRef]
- Lee, J.P.; Kim, D.K.; Oh, D.Y. Successfully treated multicentric Castleman’s disease with renal thrombotic microangiopathy using rituximab and corticosteroid. Clin. Nephrol. 2011, 75, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Kanakura, Y.; Aozasa, K.; Johkoh, T.; Nakamura, M.; Nakano, S.; Nakano, N.; Ikeda, Y.; Sasaki, T.; Nishioka, K.; et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005, 106, 2627–2632. [Google Scholar] [CrossRef]
- Masaki, Y.; Kawabata, H.; Takai, K.; Kojima, M.; Tsukamoto, N.; Ishigaki, Y.; Kurose, N.; Ide, M.; Murakami, J.; Nara, K.; et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int. J. Hematol. 2016, 103, 686–692. [Google Scholar] [CrossRef]
- Fujimoto, S.; Sakai, T.; Kawabata, H.; Kurose, N.; Yamada, S.; Takai, K.; Aoki, S.; Kuroda, J.; Ide, M.; Setoguchi, K.; et al. Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am. J. Hematol. 2019, 94, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, N.; Fajgenbaum, D.C.; Nabel, C.S.; Gion, Y.; Kondo, E.; Kawano, M.; Masunari, T.; Yoshida, I.; Moro, H.; Nikkuni, K.; et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am. J. Hematol. 2016, 91, 220–226. [Google Scholar] [CrossRef]
- Fujiwara, S.; Mochinaga, H.; Nakata, H. Successful treatment of TAFRO syndrome, a variant type of multicentric Castleman disease with thrombotic microangiopathy, with anti-IL-6 receptor antibody and steroids. Int. J. Hematol. 2016, 103, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef]
- Wang, L.; Chen, H.; Shi, J.; Tang, H.; Li, H.; Zheng, W.; Zhang, F. Castleman disease mimicking systemic lupus erythematosus: A case report. Medicine 2018, 97, e12291. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Udvardi, A.; Hertl, M.; Eming, R.; Schmidt, E.; Zillikens, D.; Volc-Platzer, B. Paraneoplastic pemphigus with anti-BP180 autoantibodies and Castleman disease. Br. J. Dermatol. 2017, 176, 824–826. [Google Scholar] [CrossRef]
- Marietta, M.; Pozzi, S.; Luppi, M.; Bertesi, M.; Cappi, C.; Morselli, M.; Torelli, G. Acquired haemophilia in HIV negative, HHV-8 positive multicentric Castleman’s disease: A case report. Eur. J. Haematol. 2003, 70, 181–182. [Google Scholar] [CrossRef]
- Liu, A.Y.; Nabel, C.S.; Finkelman, B.S.; Ruth, J.R.; Kurzrock, R.; van Rhee, F.; Krymskaya, V.P.; Kelleher, D.; Rubenstein, A.H.; Fajgenbaum, D.C. Idiopathic multicentric Castleman’s disease: A systematic literature review. Lancet Haematol. 2016, 3, e163–e175. [Google Scholar] [CrossRef]
- Morita, K.; Fujiwara, S.I.; Ikeda, T.; Kawaguchi, S.I.; Toda, Y.; Ito, S.; Ochi, S.I.; Nagayama, T.; Mashima, K.; Umino, K.; et al. TAFRO Syndrome with an Anterior Mediastinal Mass and Lethal Autoantibody-Mediated Thrombocytopenia: An Autopsy Case Report. Acta. Haematol. 2019, 141, 158–163. [Google Scholar] [CrossRef] [PubMed]
- George, J.N. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010, 116, 4060–4069. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.M. Atypical Hemolytic Uremic Syndrome: Beyond Hemolysis and Uremia. Am. J. Med. 2019, 132, 161–167. [Google Scholar] [CrossRef]
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic Microangiopathy and the Kidney. Clin. J. Am. Soc. Nephrol. 2018, 13, 300–317. [Google Scholar] [CrossRef] [PubMed]
- Chiasakul, T.; Cuker, A. Clinical and laboratory diagnosis of TTP: An integrated approach. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 530–538. [Google Scholar] [CrossRef]
- John, P.G.; George, M.R.; Bertil, G.; Daniel, A.A.; Robert, T.M.; Alan, F.L.; Frederick, R.A.; Angela, D.; Todd, A.F. Wintrobe’s Clinical Hematology, 14th ed.; LWW: Philadelphia, CA, USA, 2018. [Google Scholar]
- Nester, C.M.; Barbour, T.; de Cordoba, S.R.; Dragon-Durey, M.A.; Fremeaux-Bacchi, V.; Goodship, T.H.J.; Kavanagh, D.; Noris, M.; Pickering, M.; Sanchez-Corral, P.; et al. Atypical aHUS: State of the art. Mol. Immunol. 2015, 67, 31–42. [Google Scholar] [CrossRef]
- George, J.N.; Nester, C.M. Syndromes of thrombotic microangiopathy. N. Engl. J. Med. 2014, 371, 654–666. [Google Scholar] [CrossRef]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef]
- Schrijvers, B.F.; Flyvbjerg, A.; de Vriese, A.S. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int. 2004, 65, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Cui, S.; Gerber, H.; Ferrara, N.; Haigh, J.; Nagy, A.; Ema, M.; Rossant, J.; Jothy, S.; Miner, J.H.; et al. Vascular endothelial growth factor: A signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J. Am. Soc. Nephrol. 2006, 17, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Keir, L.S.; Firth, R.; Aponik, L.; Feitelberg, D.; Sakimoto, S.; Aguilar, E.; Welsh, G.I.; Richards, A.; Usui, Y.; Satchell, S.C.; et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J. Clin. Investig. 2017, 127, 199–214. [Google Scholar] [CrossRef]
- Beck, B.B.; van Spronsen, F.; Diepstra, A.; Berger, R.M.F.; Kömhoff, M. Renal Thrombotic Microangiopathy in Patients with cblC Defect: Review of An under-Recognized Entity. Pediatr. Nephrol. 2017, 32, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Delvaeye, M.; Noris, M.; de Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar] [CrossRef]
- Quaggin, S.E. DGKE and atypical HUS. Nat. Genet. 2013, 45, 475–476. [Google Scholar] [CrossRef]
- Tarr, P.I. Shiga toxin-associated hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: Distinct mechanisms of pathogenesis. Kidney Int. Suppl. 2009, 112, S29–S32. [Google Scholar] [CrossRef]
- Song, D.; Wu, L.H.; Wang, F.M.; Yang, X.W.; Zhu, D.; Chen, M.; Yu, F.; Liu, G.; Zhao, M.H. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res. Ther. 2013, 15, R12. [Google Scholar] [CrossRef]
- Johnsen, S.J.A.; Valborgland, T.; Gudlaugsson, E.; Bostad, L.; Omdal, R. Thrombotic microangiopathy and the antiphospholipid syndrome. Lupus 2010, 19, 1569–1572. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Chang, A. Complement and renal thrombotic microangiopathy associated with hypertension and scleroderma. Adv. Chronic Kidney Dis. 2020, 27, 149–154. [Google Scholar] [CrossRef]
- Robson, M.; Côte, I.; Abbs, I.; Koffman, G.; Goldsmith, D. Thrombotic micro-angiopathy with sirolimus-based immunosuppression: Potentiation of calcineurin-inhibitor-induced endothelial damage? Am. J. Transplant. 2003, 3, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Gavriilaki, E.; Sakellari, I.; Anagnostopoulos, A.; Brodsky, R.A. Transplant-associated thrombotic microangiopathy: Opening Pandora’s box. Bone Marrow. Transplant. 2017, 52, 1355–1360. [Google Scholar] [CrossRef]
- Cohen, E.P.; Robbins, M.E. Radiation nephropathy. Semin. Nephrol. 2003, 23, 486–499. [Google Scholar] [CrossRef]
- Pène, F.; Papo, T.; Brudy-Gulphe, L.; Cariou, A.; Piette, J.C.; Vinsonneau, C. Septic shock and thrombotic microangiopathy due to mycobacterium tuberculosis in a non-immunocompromised patient. Arch. Intern. Med. 2001, 161, 1347–1348. [Google Scholar] [CrossRef]
- Weitz, I.C. Thrombotic microangiopathy in cancer. Thromb. Res. 2018, 164 (Suppl. 1), S103–S105. [Google Scholar] [CrossRef]
- Pourrat, O.; Coudroy, R.; Pierre, F. Differentiation between severe HELLP syndrome and thrombotic microangiopathy, thrombotic thrombocytopenic purpura and other imitators. Eur. J. Obstet. Gynecol. Reprod. Biol. 2015, 189, 68–72. [Google Scholar] [CrossRef]
- Timmermans, S.A.M.E.G.; Wérion, A.; Damoiseaux, J.G.M.C.; Morelle, J.; Reutelingsperger, C.P.; van Paassen, P. Diagnostic and risk factors for complement defects in hypertensive emergency and thrombotic microangiopathy. Hypertension 2020, 75, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. 3), 4–10. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Fajenbaum, D.C. Overviews of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| 2015 Iwaki Criteria for TAFRO-iMCD | 2015 Masaki Criteria for TAFRO Syndrome | 2017 International Consensus Criteria for iMCD |
|---|---|---|
| Histopathologic criteria (prerequisite) • Compatible with pathological findings of lymph nodes as TAFRO-iMCD • Negative LANA-1 for HHV-8 Major criteria (all required) • Presents 3 of 5 TAFRO symptoms • - Thrombocytopenia - Anasarca - Fever - Reticulin fibrosis - Organomegaly • Absence of hypergammaglobulinemia • Small volume lymphadenopathy Minor criteria (1 or more required) • Hyper/normoplasia of megakaryocytes in the bone marrow • High levels of serum ALP without markedly elevated serum transaminase | Major categories (all required) • Anasarca, including pleural effusion, ascites, and general edema • Thrombocytopenia defined as a pre-treatment platelet count ≤ 100,000/μL • Systemic inflammation defined as fever of unknown etiology above 37.5 °C and/or serum CRP ≥ 2 mg/dL Minor categories (need 2 or more) • Castleman’s disease-like features on lymph node biopsy • Reticulin myelofibrosis and/or increased number of megakaryocytes in the bone marrow • Mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy • Progressive renal insufficiency Diseases to be excluded: • Malignancy, autoimmune disorders, infectious disorders, POEMS syndrome, IgG4-related disease, cirrhosis, and TTP/HUS | Major criteria (both required) • Histopathologic lymph node features consistent with the iMCD spectrum • Enlarged lymph nodes (≥1 cm in short-axis diameter) in ≥2 lymph node stations Minor criteria (2 or more required with at least 1 laboratory criterion) Laboratory criteria • Elevated CRP (>10 mg/L) or ESR (>15 mm/h) • Anemia (hemoglobin < 12.5 g/dL for males, hemoglobin <11.5 g/dL for females) • Thrombocytopenia (platelet count <150 k/mL) or thrombocytosis (platelet count >400 k/mL) • Hypoalbuminemia (albumin <3.5 g/dL) • Renal dysfunction (eGFR <60 mL/min/1.73 m2) or proteinuria (total protein 150 mg/24 h or 10 mg/100 mL) • Polyclonal hypergammaglobulinemia (total γ-globulin or IgG > 1700 mg/dL) Clinical criteria • Constitutional symptoms: night sweats, fever (>38 °C), weight loss, or fatigue (≥2 CTCAE lymphoma score for B-symptoms) • Large spleen and/or liver • Fluid accumulation: edema, anasarca, ascites, or pleural effusion • Eruptive cherry hemangiomatosis or violaceous papules • Lymphocytic interstitial pneumonitis Exclusion criteria • Infection-related disorder, autoimmune/autoinflammatory disease, malignant/lymphoproliferative disorders |
| Disease Category | Major Mechanism of Renal Injury |
|---|---|
| Activation of coagulation system with secondary endothelial injury | |
| ∙ Genetic or acquired TTP | ADAMTS13 deficiency from gene mutation or autoantibody causes large vWF multimers formation and platelet-rich thrombosis |
| ∙ HUS with a defect in the cobalamin and coagulation pathway | Genetic mutation of MMACHC, THBD, DKGE affect the regulation of the coagulation system |
| ∙ Drugs: clopidogrel, ticlopidine | Associated with the presence of anti-ADAMTS13 autoantibodies or inhibitors |
| Direct endothelial injury with prothrombotic state | |
| ∙ Toxin-related HUS: Shiga-like toxin, neuraminidases | Shiga-like toxins bind to Gb3 receptors of endothelial cells and directly cause damage; neuraminidases expose cryptic antigens of endothelial cells and elicit immunologic damage |
| ∙ Autoantibody: SLE, APS, systemic sclerosis | Antiphospholipid antibody or anti-endothelial antibody directly cause endothelial injury |
| ∙ Drug: calcineurin inhibitor (CNI) | CNI reduce the prostacyclin synthesis and formation of activated protein C, which causes direct endothelial damage |
| Radiation | Direct endothelial injury from radiation effect |
| Indirect endothelial injury with prothrombotic state | |
| ∙ HUS with a defect in complement regulation | Gene mutation or autoantibody-inhibition of complement regulatory factors, e.g., CFH, CFI, MCP, CFB, result in uncontrolled activation of the alternative complement pathway, followed by endothelial injury and prothrombotic state |
| ∙ VEGF blockade: preeclampsia and eclampsia | Overexpression of sFlt1 from the placenta acts as an antagonist of VEGF, causing endothelial injury |
| ∙ VEGF blockade: anti-VEGF, tyrosine kinase inhibitor | Inhibition of the VEGF pathway directly causes endothelial swelling and disruption of cell integrity |
| ∙ VEGF blockade: mTOR inhibitor | mTOR regulates the intracellular pathway of VEGF production. mTOR inhibitor results in a decrease in VEGF production, followed by endothelial injury |
| Miscellaneous: DIC, neoplasm, HELLP syndrome, malignant hypertension, idiopathic multicentric Castleman disease, TAFRO syndrome. | |
| Disease Category | Possible Treatment |
|---|---|
| iMCD or TAFRO-iMCD | Anti-IL6 (siltuximab, tocilizumab) Corticosteroid Anti-CD20 (rituximab) Proteasome inhibitors (Bortezomib) IVIG Calcineurin inhibitors (cyclosporin) mTOR inhibitors (sirolimus) Immunomodulatory drugs (Thalidomide, Lenalidomide) Chemotherapy (R-CHOP, R-CVP) Autologous stem cell transplantation |
| HHV-8 associated MCD | If HIV exists, provide antiviral agents. Anti-CD20 (rituximab) Interferon Chemotherapy: Etoposide, Doxorubicin. |
| POEMS associated MCD | If no bone lesion, treated as iMCD. If bone lesion exists, treated as plasmacytoma or multiple myeloma. |
| Other association | Treat as associated disease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, K.-H.; Fan, P.-Y.; Chen, T.-D.; Chuang, W.-Y.; Wu, C.-Y.; Ku, C.-L.; Tian, Y.-C.; Yang, C.-W.; Fang, J.-T.; Yang, H.-Y. TAFRO Syndrome with Renal Thrombotic Microangiopathy: Insights into the Molecular Mechanism and Treatment Opportunities. Int. J. Mol. Sci. 2021, 22, 6286. https://doi.org/10.3390/ijms22126286
Tu K-H, Fan P-Y, Chen T-D, Chuang W-Y, Wu C-Y, Ku C-L, Tian Y-C, Yang C-W, Fang J-T, Yang H-Y. TAFRO Syndrome with Renal Thrombotic Microangiopathy: Insights into the Molecular Mechanism and Treatment Opportunities. International Journal of Molecular Sciences. 2021; 22(12):6286. https://doi.org/10.3390/ijms22126286
Chicago/Turabian StyleTu, Kun-Hua, Pei-Yi Fan, Tai-Di Chen, Wen-Yu Chuang, Chao-Yi Wu, Cheng-Lung Ku, Ya-Chung Tian, Chih-Wei Yang, Ji-Tseng Fang, and Huang-Yu Yang. 2021. "TAFRO Syndrome with Renal Thrombotic Microangiopathy: Insights into the Molecular Mechanism and Treatment Opportunities" International Journal of Molecular Sciences 22, no. 12: 6286. https://doi.org/10.3390/ijms22126286
APA StyleTu, K.-H., Fan, P.-Y., Chen, T.-D., Chuang, W.-Y., Wu, C.-Y., Ku, C.-L., Tian, Y.-C., Yang, C.-W., Fang, J.-T., & Yang, H.-Y. (2021). TAFRO Syndrome with Renal Thrombotic Microangiopathy: Insights into the Molecular Mechanism and Treatment Opportunities. International Journal of Molecular Sciences, 22(12), 6286. https://doi.org/10.3390/ijms22126286