1. Introduction
Chaperone proteins have developed in evolution for the protection of cells against a variety of abiotic stresses, including drought, high or low temperature, osmotic stress, different salts, or heavy metals. These protective proteins come in a variety of shapes and forms as they need to be able to function under unfavorable conditions and on a plethora of different clients. One of the largest groups of chaperones are heat shock proteins (HSPs), originally identified as being expressed under conditions of elevated temperatures. These are complex globular proteins that come in a variety of sizes and utilize intricate molecular structures and interaction networks to fulfil their functions [
1]. Nevertheless, not only globular proteins can serve as chaperones, as a growing body of information underlines the importance and efficiency of a chaperone class encompassing disordered proteins [
2,
3].
Dehydrins, which are categorized as Group 2 LEA (Late Embryogenesis Abundant) proteins, are such disordered chaperones that function in an ATP-independent manner [
2]. Proteins in this family have multifunctional roles in stress response in many taxa [
4]. They are generally characterized by high net charge and the presence of polar amino acids; they are enriched in glycine and lysine residues but are depleted in cysteine and tryptophan [
5], suggesting a disordered character. Their categorization is based on the highly conserved segments that can be found in their sequences [
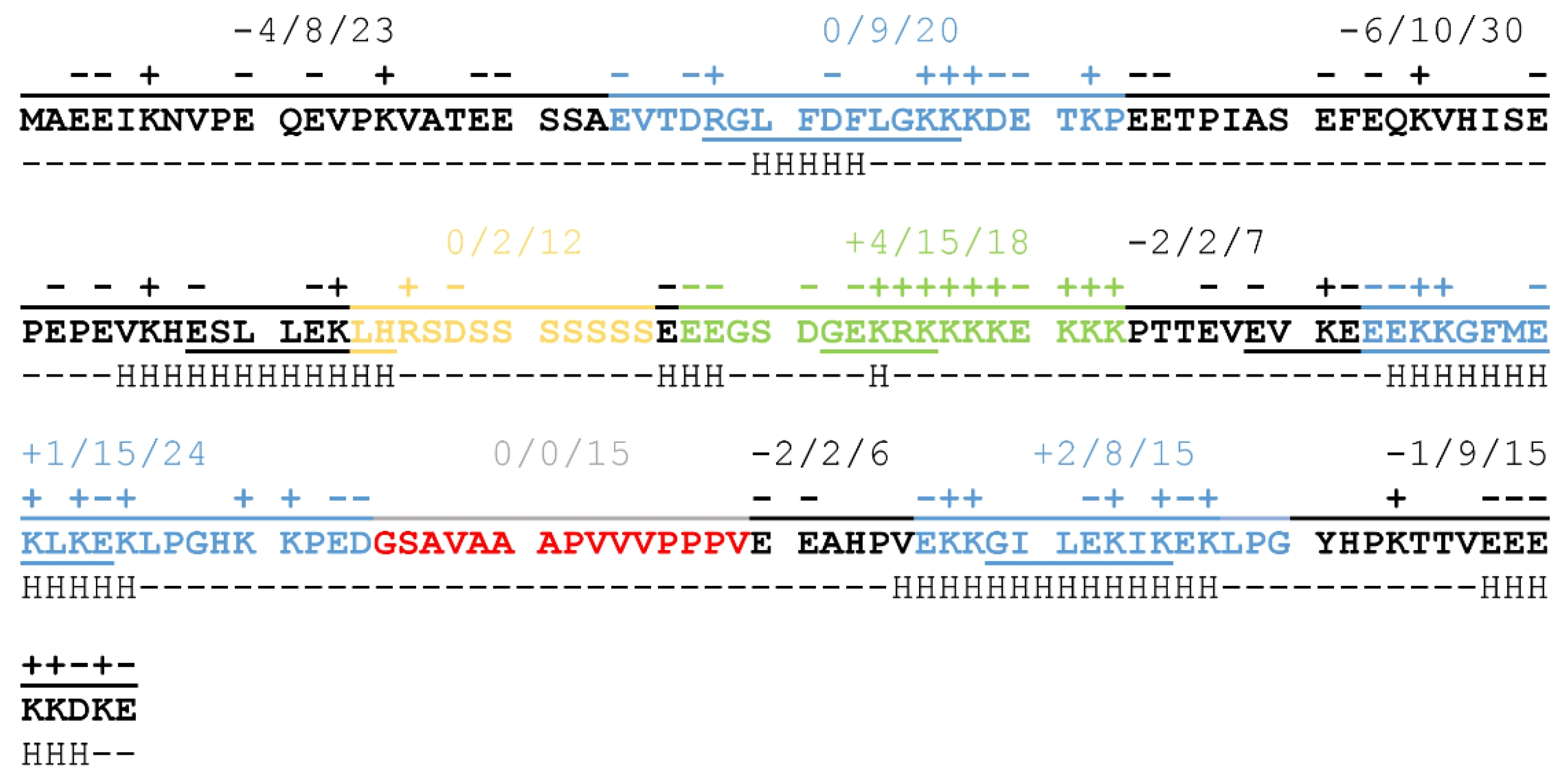
6] in different combinations: (i) The K-segment is an approximately 15 amino acid long conserved lysine-rich motif present in all cold-expressed dehydrins. It can be found in multiple copies (1–12) within a dehydrin and, according to in silico predictions, it tends to adopt an amphipathic α-helix structure [
7,
8]. Presumably, K-segments can play a role in shaping the interaction with partner molecules, which is supported by our earlier observations [
9]. (ii) The S-segment is a serine-rich region that can be phosphorylated, and, in most examples, it is wedged between the K-segments. The exact function of its phosphorylation is not yet known, but it may play a role in the protein’s delivery to the nucleus or in membrane stabilization. (iii) The Y-segment is a conserved region at the N-terminus of dehydrins that shows homology to the nucleotide binding regions of plant proteins and bacterial chaperone proteins. (iv) The ChP-segment is a highly charged, lysine-rich region that bears a strong resemblance to the charged linker region of the Hsp-90 chaperone protein found in animals. (v) The q-segment is a less conserved, apolar region composed of glycine or proline and alanine amino acids, usually located between two K-segments [
6,
10].
The
Arabidopsis thaliana genome encodes for 10 different dehydrins that probably have partially overlapping functions [
11,
12]. ERD14 is one of the
A. thaliana dehydrins that belongs to the SK2 dehydrins subgroup, and it contains an additional hydrophobic region (H region) in its sequence. Its expression is induced under different stress conditions, such as heat and osmotic stress, and it can also be found in high quantities in rapidly dividing tissues, such as meristems [
13]. In vitro and in vivo studies have indicated that ERD14 is a potent chaperone for a number of substrates [
14,
15]. Experiments with lysozyme, alcohol dehydrogenase, luciferase, and citrate synthase model substrates have shown that this protein is capable of preventing heat-induced loss of activity and subsequent aggregation of the enzymes [
3]. They did not show, however, a chaperone activity in reactivating denatured proteins, and the exact mechanism of its chaperone function is yet unknown.
Conserved segments in ERD14 create a specific pattern of charge- and structure-distribution that may be crucial for the proper chaperone function of the protein.
The balanced distribution of charged amino acids throughout the full length of the protein results in a sequence that has a low overall negative charge (−9 at neutral pH resulting from 46 negatively and 37 positively charged sidechains) carrying a significant portion of charged regions (
Figure 1). The N-terminal half of the protein is negatively charged, with two non-conserved, disordered regions of strong negative charge, while the C-terminal region partly compensates for this, mainly through the region of the Chp-segment consisting of 15 charged residues out of 18 (in green on
Figure 1). K-segments (blue on
Figure 1) achieve neutral net charges by alternating positively and negatively charged residues, a pattern that might be important in partner binding [
9]. The S-segment (yellow on
Figure 1), on the other hand, consists mostly of polar, non-charged amino acids, whose exact relevance is not yet clear, although phosphorylation of the serine sidechains may have a regulatory function. The H-segment (red on
Figure 1) also lacks charged residues, but it is highly hydrophobic and contains several proline residues. This may be important for the recognition of exposed hydrophobic patches on partner proteins while also helping to keep ERD14 in a disordered structural state.
In our previous work, we identified several different
E. coli proteins bound by ERD14, supporting the idea of its promiscuous protein binding and protection. We provided evidence that ERD14 is localized in the cytoplasm during heat stress, arguing against its functioning through membrane protection [
9]. Our in-cell NMR measurements also confirmed that the conserved K-segments that sample helical structural states in vitro [
16] are the ones that participate mainly in molecular interactions, while most of the other regions remain disordered. Nevertheless, being able to bind proteins is not the only prerequisite of chaperones, highlighting the importance of the regions outside of K-segments in the function of ERD14.
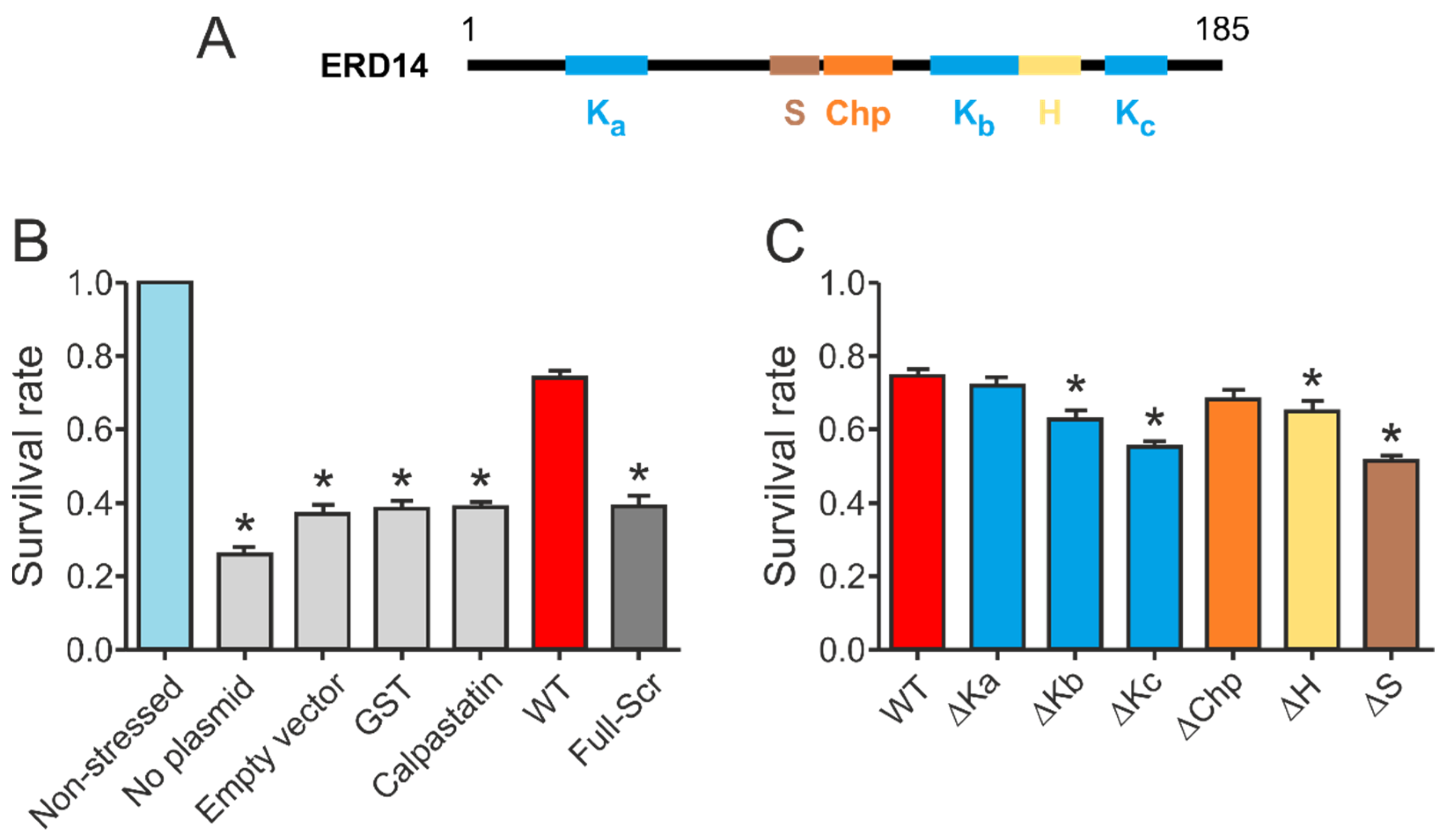
In order to gain deeper insight into the molecular architecture needed for the protective function of ERD14, we designed several mutants by deleting one or more of the conserved regions (1x: ∆Ka, ∆Kb, ∆Kc, ∆Chp, ∆H, ∆S; 2x: ∆Kab, ∆Kbc, ∆Kac; 3x: ∆Kabc) or prepared scrambled mutant sequences (Full-Scr, Scr-KabcS, Scr-Kabc, ScrKcS, Scr-Kc, Scr-S) and determined the heat-resistance of E. coli cells, expressing the different mutants and studying the structural consequences of the deletions or sequence modifications.
2. Results
ERD14 is a typical representative of the SK2 group that harbors several potentially important sequential features. In addition to its unique charge-distribution, secondary structural elements are also dispersed along the sequence of ERD14. As indicated on
Figure 1, short helical regions can be found mostly in the K-segments, but a non-conserved region at the border of the S-segment, as well as the ChP-segment, samples helical conformations in vitro, as suggested by NMR spectroscopy [
16]. The observed patterning of charges and secondary structural elements suggest that the different regions have specific functions and possibly cooperate with each other to achieve biological function.
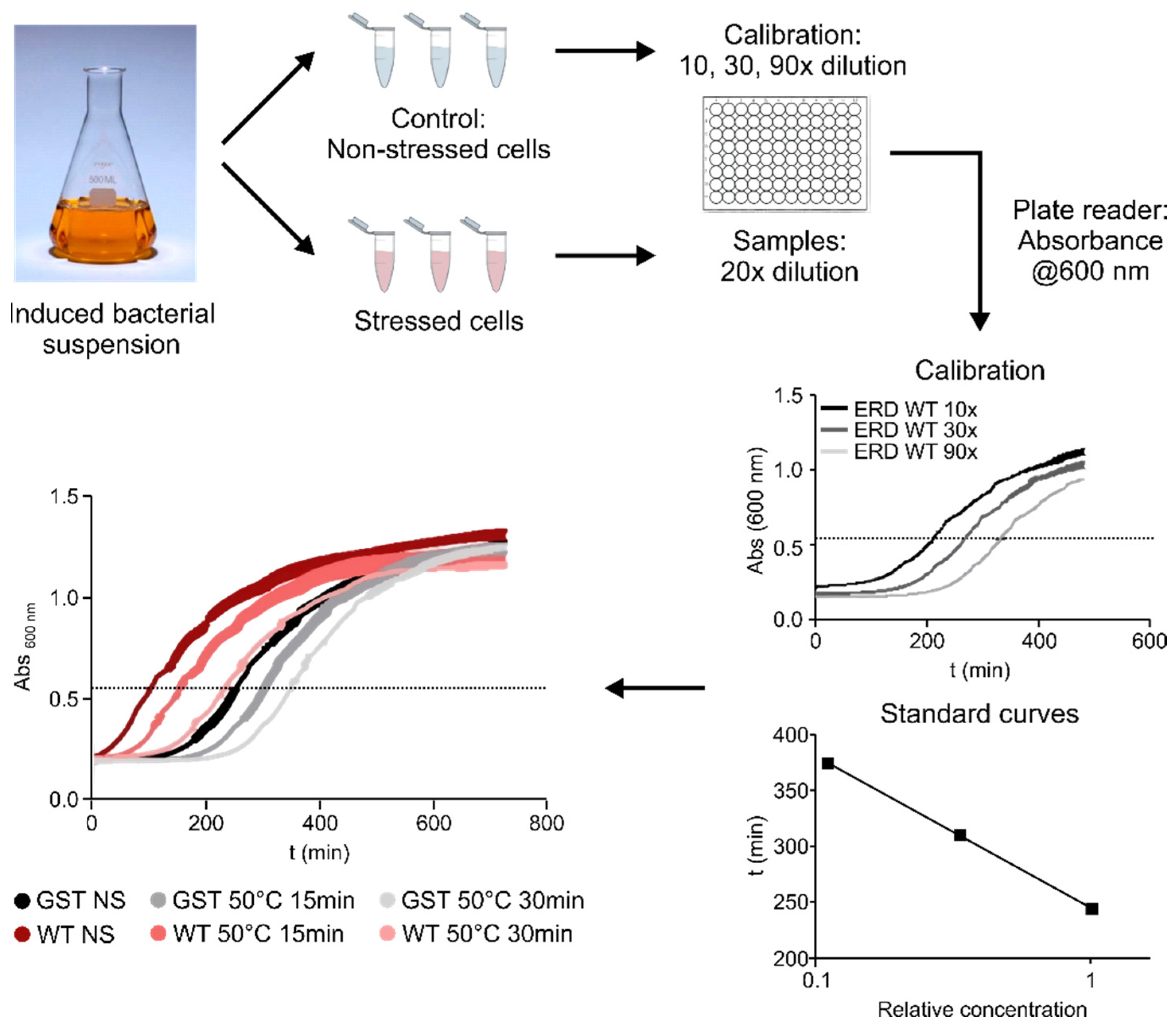
To assess the contribution of different regions of ERD14 to the protective effect of the protein, we developed a system using a heat-treatment that reduces the viability of
E. coli cells to around 25% of the original value (
Figure 2 and
Figure 3B). After testing various conditions (
Figure 2), we chose a treatment of 50 °C for 15 min, as this gave the optimal difference between the stressed and non-stressed cells, allowing a wide dynamic range to detect differences in survival rates. The protective effect of each ERD14 variant was assessed as the difference of the survival rate (DSR) of the cells compared to the ones expressing the wild-type protein. This system has already proved to be useful for the determination of the in vivo structural state and the molecular partners of ERD14 [
9], and it has several beneficial aspects for our purposes. Most importantly, it enabled us to investigate several different protein constructs in a timely manner and, by eliminating the redundancy of plant chaperones [
12], it largely simplified the interpretation of the results. Because dehydrins are known to have broad substrate specificity [
21,
22,
23], and we were able to identify several partner proteins of ERD14 in
E. coli cells, we found this system to be an appropriate model system for our study.
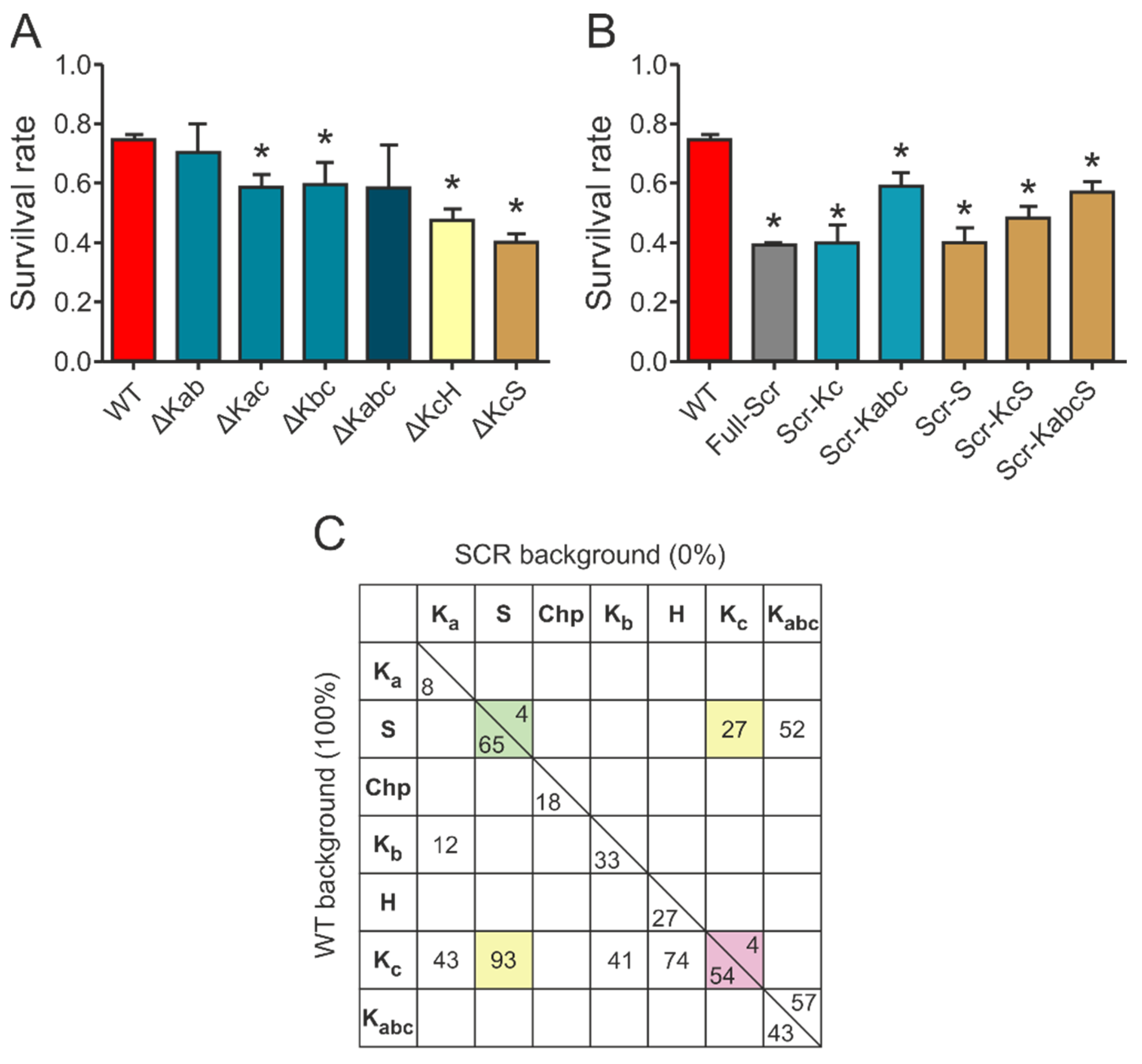
Our initial results confirmed that wild type ERD14 is capable of protecting cells against the applied heat stress, increasing their survival rate to 70% (
Figure 3B). None of the control proteins were capable of such protection, and induction of an empty expression vector did not affect stress resistance either. The importance of specific sequential components (
Figure 3A) in the function of the protein was suggested by the fact that a variant with similar amino acid composition but randomized sequence (Full-Scr) did not protect the
E. coli cells against heat stress (
Figure 3B).
Our structural studies indicated that the conserved K-segments were primarily involved in direct partner binding within the cells [
9], prompting us to design a series of deletion mutants targeting these regions (for constructs, cf.
Supplementary Table S1). Even though the other conserved ERD14 regions, the S-, Chp-, and H-segments, did not appear to play major roles in partner recognition, we have also created deletions of these as well to determine if they contributed to the chaperone effect. The constructs were expressed in
E. coli (at levels comparable to that of WT ERD14, cf.
Supplementary Figure S1), and their protective effects in the cell viability assay were measured (
Figure 2).
Deletion of different conserved regions affected the chaperone activity of ERD14 to different extents (
Figure 3C). Of the three K-segments, removal of Kc (∆Kc) impaired activity the most, reducing the remaining cell viability from 74.5% (wild type) to 54.9% (DSR: 19.6%). The observed level of protection was always normalized to the actual expression level of the given construct, assuming linearity of response (cf. Experimental Procedures and
Supplementary Figure S2). Survival rates changed linearly with increasing ERD14 concentrations for the wild type and the ∆Kc mutant ERD14 when we applied an IPTG-inducible system, as well as in an arabinose-inducible set-up. The latter enabled us to avoid the side effects of changing cellular density during elongated expression times. The reduction in protection by deleting the S-segment was the most pronounced (DSR 23.4%), and it is commensurable with that of Kc. The deletion of Kb (DSR 11.9%), H (DSR 9.8%), and Chp (DSR 6.6%) have intermediate effects, whereas deletion of Ka (DSR 3%) causes almost no change in activity. These results suggest that Kc and S are the primary sites of activity, together with varying contributions of other segments. These notions were further confirmed by the in vitro chaperone activities of these mutants (
Supplementary Figure S3 and Table S4). In these experiments, the ∆Kc and ∆S variants showed similarly reduced chaperone activities as the FS ERD14 variant. This is an interesting observation, especially in light of the fact that all K-segments participate in partner binding within a cell, whereas the S-segment does not [
9]. Although partner binding in itself is a property of large amounts of proteins, only a small portion of them exhibit chaperone activity. Beyond the necessary partner binding regions, a chaperone is expected to have segments that contribute to or are responsible for chaperone function, such as the S-segment.
Because the ChP-segment and the three K-segments tend to adopt α-helical conformations, as detected by NMR [
9,
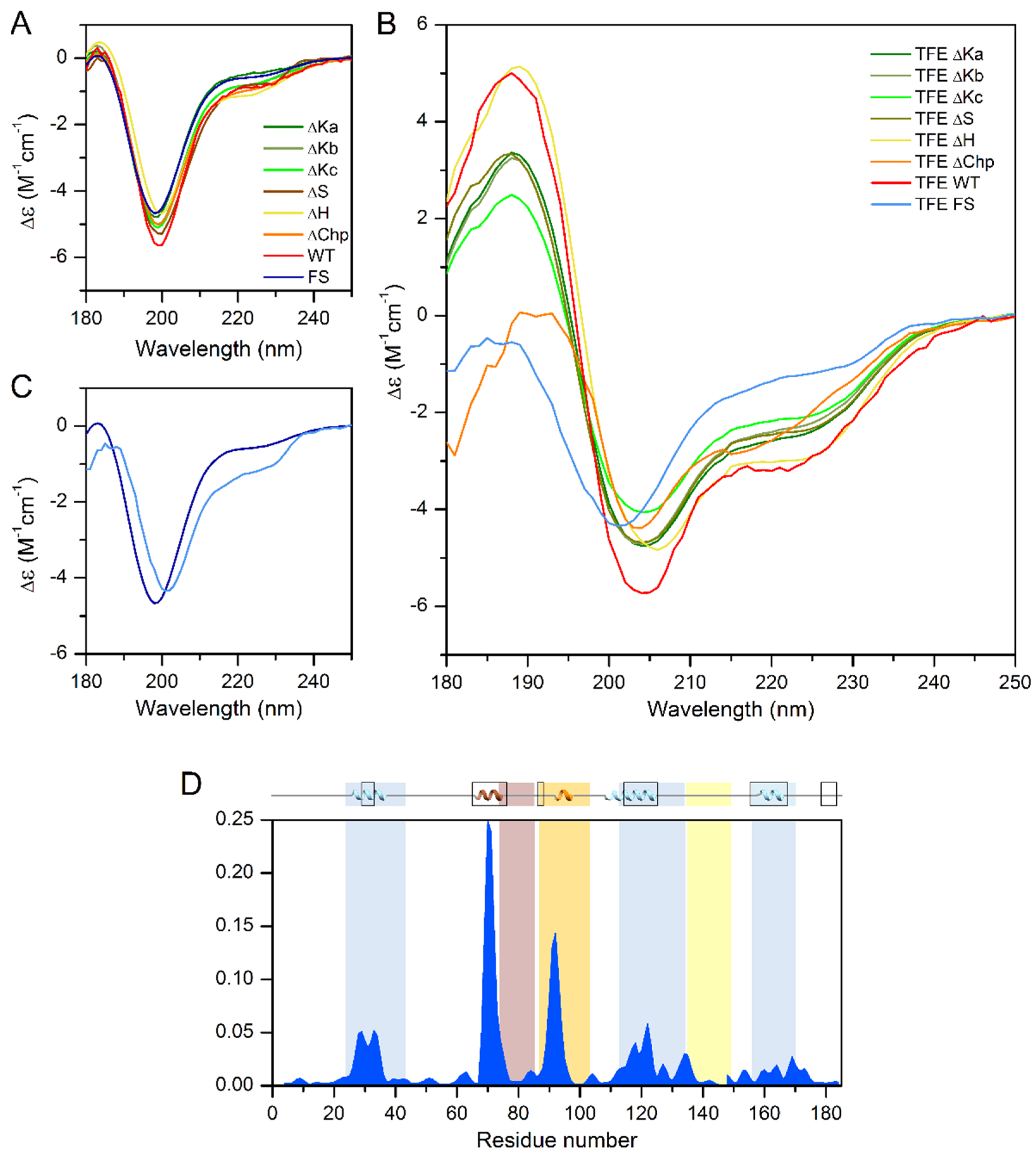
16], we compared the helix content of the different single deletion mutants by CD spectroscopy. In phosphate buffer, all tested proteins showed a CD spectral shape characteristic of a disordered protein (
Figure 4A), very similar to that of the wild-type ERD14. Because CD spectroscopy reflects the average secondary structure content, this indicates that the free ERD14 molecules are essentially disordered, and the helices shown by NMR [
9] are truly only transient and might be stabilized by interacting with partner molecules. The inherent tendency of the wild-type protein and the deletion mutants to form α-helical structures was confirmed by testing their structure in the presence of the helix-inducing trifluoro-ethanol (TFE). The addition of 30% TFE increased the helicity of the wild-type protein from 5% to 25% (
Figure 4B). Deletion of the K segments reduced the helicity in TFE to 16–21% (21%, 20%, and 16% for ∆Ka, b, and c, respectively), indicating that the overall structure of the protein was probably not perturbed in these mutants. Similar observations were made for the S-segment, which had a residual helicity of 20% in TFE. Removal of the H-segment resulted in a minor change in helix-forming tendency (23% helix content in TFE), and the ∆Chp variant also showed a somewhat reduced helix-forming tendency with 22% helix content. In contrast to the single deletion mutants, the Full-Scr ERD14 variant remained almost completely disordered, with only 9% helix content even in the presence of TFE (
Figure 4C,D). Intriguingly, the loss of helicity by deletion of the individual segments sums up significantly more than the overall helix content of WT ERD14 (67 residues vs. 46, respectively), which indicates that the structure-forming tendencies of the segments are not independent, nor additive, and there might be an interplay between ERD segments.
Table 1 shows the changes in the number of residues that are in α-helical conformation in the presence of TFE in the single deletion mutants, compared to the wild-type protein, thus we can gain valuable insight into the helix-forming tendencies of each conserved segment.
Intriguingly, the PredictProtein online bioinformatics structure analysis tool ([
20]) predicts α-helix for 50 residues in the ERD14 sequence, which is far more than experimentally observed by CD under native conditions (9 residues); however, the prediction fits well to the CD analysis of the TFE-induced structure (46 residues in α-helix). NMR measurements are also in good correlation with this, as 44 residues show transient helical conformations in vitro [
16]. These are not stable enough for the CD to pick up in phosphate buffer, but as we can see, they can be stabilized and measured by the addition of TFE. PredictProtein assigns all three K-segments as helical; however, similarly to CD (
Table 1), it predicts the lowest helicity for Ka. It also indicates some helicity for Chp and S and for regions that are located outside of the conserved segments. Apart from three residues located sporadically in β-sheet, the rest of ERD14 is predicted to be disordered. These bioinformatics results are partly fit to the experimental ones of CD and the earlier NMR, and while being useful, should be handled with caution.
If we compare the structural effects observed by CD spectroscopy with the changes in the survival rates of bacteria expressing different constructs (
Table 1 and
Table S3), we can draw interesting conclusions regarding the structure-function relations of ERD14. While the structural changes induced by removing each of the K-segments were substantial, Kc causing the highest, their effect on the activity of the protein differed more significantly. While the deletion of Kc caused the highest activity decrease, Ka, despite its helix contribution, had almost no effect on activity. Conversely, the ∆H mutant was a significantly less effective chaperone than the WT protein, even though the change in α-helical content was the lowest (
Figure 3B and
Table 1). The deletion of the ChP-segment resulted in a moderate reduction of helicity, while it did not significantly affect the protective activity. Finally, removing all structural tendencies and sequence motifs by randomizing the full sequence of the protein results in a complete loss of activity (
Figure 3A and
Table 1).
Taken together, these observations suggest that secondary structure formation is a necessary, but not sufficient, prerequisite of the protective effect of ERD14, as partner binding and a detectable redundancy of the regions also play important roles.
Based on the results of the survival assays and the CD measurements, we concluded that there is a significant redundancy of the motifs that manifests at several levels in the apparent exchangeability of individual motifs. This is shown by deleting multiple motifs (
Figure 5A). For example, the chaperone activity of the deletion mutant ∆Kc is not further reduced by the deletion of the Ka or Kb segment (cf. ∆Kac: DSR 15.7%, ∆Kbc: DSR 14.7%, and ∆Kabc: DSR 15.6%), i.e., the Ka and Kb segments are rather redundant and, without the more effective Kc or S-segment, are of little additional effect. Furthermore, ∆Kab (DSR 4.2%) is almost as effective a chaperone as WT ERD14, i.e., even Ka and Kb combined have little protective function. Surprisingly, the single deletion of Kb caused more significant reduction (DSR 11.9%), suggesting that there might be a complex interplay between the segments. Combined deletion of the Kc and S-segments renders the protein almost completely ineffective (∆KcS: DSR 34.1% vs. Full-Scr: DSR 35.6%), while deletion of the H-segment together with the Kc increases the activity loss of either of the segments deleted alone (∆KcH: DSR 26.8% vs. ∆Kc: DSR 19.5% and ∆H: DSR 9.8%).
In an attempt to gain a deeper insight into the functional interplay of the different conserved segments, we created a further variety of mutants in which different elements of ERD14 were modified, either individually or in various combinations (cf.
Supplementary Table S1). First, we have investigated if any of the effective motifs, combined with the rest of the proteins, scrambled (kept disordered but without any recognizable sequence motifs), has a protective effect (
Figure 5B). Keeping any of the most effective segments (Scr-Kc or Scr-S) results in a protein with negligible chaperone activity, similar to the entirely scrambled protein (Full-Scr). Keeping two segments (Scr-KcS) results in a more active protein, whereas keeping three (Scr-Kabc) or four (Scr-KabcS) makes it an even more potent chaperone. The importance of the other sequential elements is underlined by the fact that keeping even four segments intact cannot completely rescue the chaperone activity of the protein.
These results support a mechanism where binding at multiple points is complemented by the functional role of a disordered linker and/or terminal regions of ERD14.
Our previous observations [
9,
14,
16], combined with the results detailed above, suggest that ERD14 functions by an interplay of transient partner binding via short binding motifs and space filling by significant residual disorder in the linker and flanking regions. To gain a deeper insight into the structure-function relationship of the protective effect of ERD14, we devised a strategy to quantitate and compare the contribution of different segments of the protein to its in vivo activity. We found that scrambled ERD14 has no activity (0%) and have taken the activity of WT ERD14 (under the given conditions) as 100%. We then argued that, for a deletion construct in the WT background, the addition of the given motif would restore activity to 100%, i.e., its contribution corresponds to the reduction of activity upon its deletion. For scrambled mutants, we argue that the addition of the given region to the inactive scrambled construct (Full-Scr) raises activity from 0% to the level measured, i.e., the contribution of the segment in the Scr background corresponds to the activity measured for the given construct. The numbers thus calculated (
Supplementary Table S2 and
Figure 5C) show that the two numbers for the same region (i.e., for WT and Scr background) are different, which points to a strong synergy between different, conserved, and linker regions of the protein and allows several mechanistic conclusions.
4. Materials and Methods
4.1. Cell Viability Assays
Cell viability was measured in the LB medium, by observing the time it takes for cells to reach an optical density of 0.6 at 600 nm of appropriately diluted samples. To this end, BL21 Star (DE3)
E. coli cells, overexpressing one of various ERD14 constructs or control proteins (GST, calpastatin (CAST)), were grown in 5 mL LB medium containing 50 µg/mL carbenicillin overnight at 37 °C under continuous shaking at 200 rpm. The next day, 50 µL aliquots were transferred into 5 mL LB medium containing 50 µg/mL carbenicillin and were grown for 2 h. Protein expression was induced with 0.5 mM IPTG for 3 h; 0.5 mL aliquots were taken and either stressed or not stressed before appropriate dilution into LB medium into the wells of a 96-well plate. Measurements of absorbance at 600 nm were taken every 2 min in a BioTek Synergy Mx microplate reader for 12 h at 37 °C by 200 rpm shaking; viability was associated with the time taken for the cells to reach an optical density of 0.6, roughly the half-maximal value they attain after long incubation periods (cf.
Figure 2). From a number of different conditions, we selected 50 °C × 15 min for an optimal stress effect—as defined by a survival rate of the untransformed cells between 20% and 30%. This provides a wide dynamic range to assess the protective effects of the different overexpressed protein constructs. The observed protection effect was normalized to the actual expression level of the given construct in the given experiment, enabled by the linearity of dependence (cf.
Supplementary Figure S2). In short, the actual concentration was determined, and the expected protection at a concentration matching that of WT ERD14 was calculated by linear intra- or extrapolation. Further details, such as optimization of the assay, and varying concentration of constructs, are given in
Supplementary Information.
4.2. Constructs, Recombinant Proteins, Mutagenesis
Various deletion constructs of ERD14 were generated from the wild-type sequence by PCR. Scrambled sequences (Full-Scr, Scr-Kc, Scr-S, Scr-SKc, Scr-Kabc, and Scr-KabcS), designed in silico, were generated by total cDNA synthesis. For in-cell stability determination, ERD14 was FLAG-tagged by inserting it into the expression vector pT7-FLAG 2. Sequences of all constructs and the control proteins are delineated in
Supplementary Table S1, whereas construction details can be found in
Supplementary Information.
Differential survival rate (DSR) was calculated as follows: percentage of cells expressing specific mutant constructs that survived the applied heat stress was extracted from the percentage of survival when the WT ERD14 was expressed, i.e., if the survival rate was 80% for the WT and 50% for a mutant, then the DSR of the specific mutant is 30%. This means that the larger the DSR, the less efficient a chaperone the mutant is.
4.3. Scrambled ERD Mutants
We used a multistep approach during the design of the scrambled ERD14 mutants: (i) we generated 10,000 random sequences by using the amino acid composition of ERD14, keeping the sequence set non redundant during the process; (ii) we used IUPred [
33] (long disorder prediction) to predict disorder propensity on all sequences and filtered out those that were significantly different from the disorder characteristics of the original ERD14 (different average disorder score or different disorder score variance); (iii) as the main reason for working with scrambled mutants was to test the effect of the amino acid composition only, we used the ANCHOR algorithm [
34] to predict potential interaction regions and exclude sequences with high confidence interaction regions; (iv) as the randomized sequences turned out to be highly prone to generating regions with high coiled-coil propensity, we introduced a further filtering step: we used the COILS server [
35] to predict coiled-coil regions and exclude sequences with high coiled-coil propensity; (v) as a final step, we used a charge distribution plot on the selected candidates (
https://www.bioinformatics.nl/cgi-bin/emboss/charge (accessed on various dates in October 2016 and February 2017) to visually filter out sequences with uneven charge distribution. A more detailed description of the selection and post-filtering can be found in the
Supplementary Methods.
4.4. Expression and Purification of Recombinant Proteins
The pET-22b (+) vector containing the appropriate construct was transformed into competent E. coli BL21 * (DE3) pLysS cells and grown in LB medium containing 0.05 mg/mL carbenicillin overnight at 37 °C with shaking at 200 rpm. After inoculation with the starter cell culture into fresh LB medium containing 0.05 mg/mL carbenicillin, the cells were grown to OD600 = 0.6–0.8. Protein expression was induced with the addition 0.5 mM IPTG, at 37 °C.
After 3 h of expression, cells were harvested by centrifugation at 4 °C at 4000 rpm for 20 min. The pellet was resuspended in lysis buffer (50 mM Tris, 150 mM NaCl, 0.4 mM DTE, pH 7.5) supplemented with a protease inhibitor tablet (Roche, Basel, Switzerland) prior to usage. Sonication was performed for 6 × 15 s with 30 s pauses, on ice. Samples were centrifuged at 4 °C 5100 rpm for 20 min to remove cell debris. Using the high temperature resistance of ERD14, boiling the supernatant was performed for 10–15 min to precipitate globular proteins. The denatured proteins were removed by centrifugation at 4 °C, 16,000 rpm for 30 min. The supernatant was filtered through a 0.2 µm filter. The protein extract was purified with ÄKTAexplorer™ (GE Healthcare, Chicago, IL, USA) LC purification system. The first purification step was via buffer exchange on a HiPrep 26/10 Desalting column (GE Healthcare, Chicago, IL, USA). The sample was transferred to a 50 mM Tris buffer (pH 7.5).
Fractions containing ERD14 were purified on a Resource Q 6 mL (GE Healthcare, Chicago, IL, USA) anion exchange column. Elution was performed in 50 mM Tris buffer (pH 7.5) with a linear salt gradient up to 500 mM NaCl. Fractions selected from the chromatogram were checked by SDS PAGE on a 15% gel. Based on the gel image, the fractions containing pure protein were dialyzed into MQ water. Dialysis was performed at 4 °C O/N. The samples were lyophilized and stored at −20 °C until usage.
4.5. In Vitro Chaperone Assay
Thermal denaturation of citrate synthase (CS) was monitored in the presence of WT, FS, ∆Kc, and ∆S ERD variants, using CD spectroscopy and an aggregation assay.
Thermal induced transitions were monitored in CD experiments by recording the ellipticity at 220 nm, which is characteristic for the α-helix structural component of citrate synthase. Changes in ellipticity induced by the increasing temperature were correlated with the thermal unfolding of the enzyme.
In the aggregation assay, citrate synthase was incubated at elevated temperature in itself or in the presence of different ERD14 constructs (WT, FS, ∆Kc, or ∆S) The amount of denatured CS was determined by measuring the protein concentration before and after heat stress, after removing the aggregated proteins by centrifugation. Detailed description of the methods can be found in the
Supplementary Methods.
4.6. Statistical Analysis
To assess the statistical significance of the observed differences, a large number of intra- and inter-experiment replicates were done. The survival rates showed minimal standard deviation within the same experiment, and only slightly larger deviation between experiments with a Gaussian distribution. Unpaired t-tests were used to quantify the significance of the differences between survival rates of the differently transformed bacteria.
4.7. CD Spectroscopy Measurements
Circular dichroism (CD) spectroscopy experiments were carried out on a Jasco J-810 instrument (Japan Spectroscopic Co., Tokyo, Japan) equipped with a Peltier-controlled thermostat. For secondary structure determination in 10 mM Na-phosphate, pH 7.4 buffer, and in the presence of 30% trifluoro-ethanol (TFE), a quartz cell of 10 μM pathlength was used with a protein concentration of ~5 mg/mL. Four scans were accumulated by using a scanning rate of 20 nm/min, a bandwidth of 1 nm, and a data integration time of 4 s.
SRCD spectra were recorded at the DISCO SRCD beamline of a SOLEIL Synchrotron (Gif-sur-Yvette, France). Experimental parameters were set by following the guidelines of Micsonai et al. [
36]. Protein concentration was ~5 mg/mL in 10 mM Na-phosphate, pH 7.4, using a 12 µm CaF
2 cell (Hellma GmbH, Müllcheim, Germany). At least 12 scans were accumulated in the 175–270 nm wavelength range at 1 nm steps with a lock-in time constant of 300 msec and integration time of 1200 msec. The spectra were treated with the CDTool package [
37] (averaging, baseline subtraction, correction with camphorsulphonic acid (CSA), normalization). Secondary structure composition was analyzed by the BeStSel algorithm [
38].
For the CD measurements, the protein concentrations were determined directly in the CD cuvettes by the absorbances at 205 and 214 nm. Measurements with the dialysis buffers were used as reference. The advantage of this method is that the extinction coefficients are large, even in the absence of aromatic residues, which is often the case for IDPs. Moreover, the actual CD samples are measured and thus any dilution errors or pathlength variations are taken into account. The extinction coefficients were calculated from the amino acid sequences on the BeStSel webserver (
https://bestsel.elte.hu (accessed on various dates in April, 2021) following the protocol of Antis and Clore [
39] and Kuipers and Gruppen [
40] for 205 and 214 nm, respectively.
4.8. Quantification of the Contribution of ERD14 Motifs to Cell Protection
The protection of ERD14 and its various deletion and scrambled constructs of cells is primarily visualized through cell viability values measured after the applied stress (
Supplementary Table S2). As viability before stress is taken as 100%, we observe that the viability of control cells (cells with an empty vector, or cells expressing GST, calpastatin, or scrambled ERD14) drops to 38.2% upon stress, whereas cells expressing ERD14 retain a viability of 74.5%. To derive the contributions of individual motifs or their combinations to this protective activity, we developed a % metric, which is 0% if addition of the motif keeps the activity of a given construct unchanged, whereas it is 100% if it raises its activity to the level of WT ERD14. As the background activity of a construct without the motif differs for the deletion and scrambling experiments, we have worked out a way to derive these two different values for the deletion and scrambled variants. For deletion constructs (e.g., ∆Kc in WT background), we took the viability (V) of the deletion construct after stress and assumed that adding back the deleted segment(s) would increase viability back to 100% (as it reconstitutes WT ERD14), thus the contribution of the segment is:
For example, viability of cells expressing ∆Kc is 0.549, which, upon “adding” Kc back, increases to 0.745. That is, the Kc segment increases the activity by 0.745–0.549, 53.99% of the potential maximum (cf.
Table S2 and
Figure 5C).
For scrambled mutants (e.g., Scr-Kc), we assumed that activity without the motif at 38.2% chaperone activity is 0% (because Full-Scr ERD14 exerts no protection), thus the measured actual V value represents the activity associated with the motif:
For example, viability of Scr-Kc is 0.397, i.e., Kc alone can add to the activity of Full-Scr ERD14 0.397–0.382, about 4.13% of the possible maximum (cf.
Supplementary Table S2 and
Figure 5C).


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}