
Tau Seeding Mouse Models with Patient Brain-Derived Aggregates

Abstract
1. Introduction
2. Tau Protein Properties in Health and Disease
2.1. Tau Physiology
2.2. Tau Pathophysiology
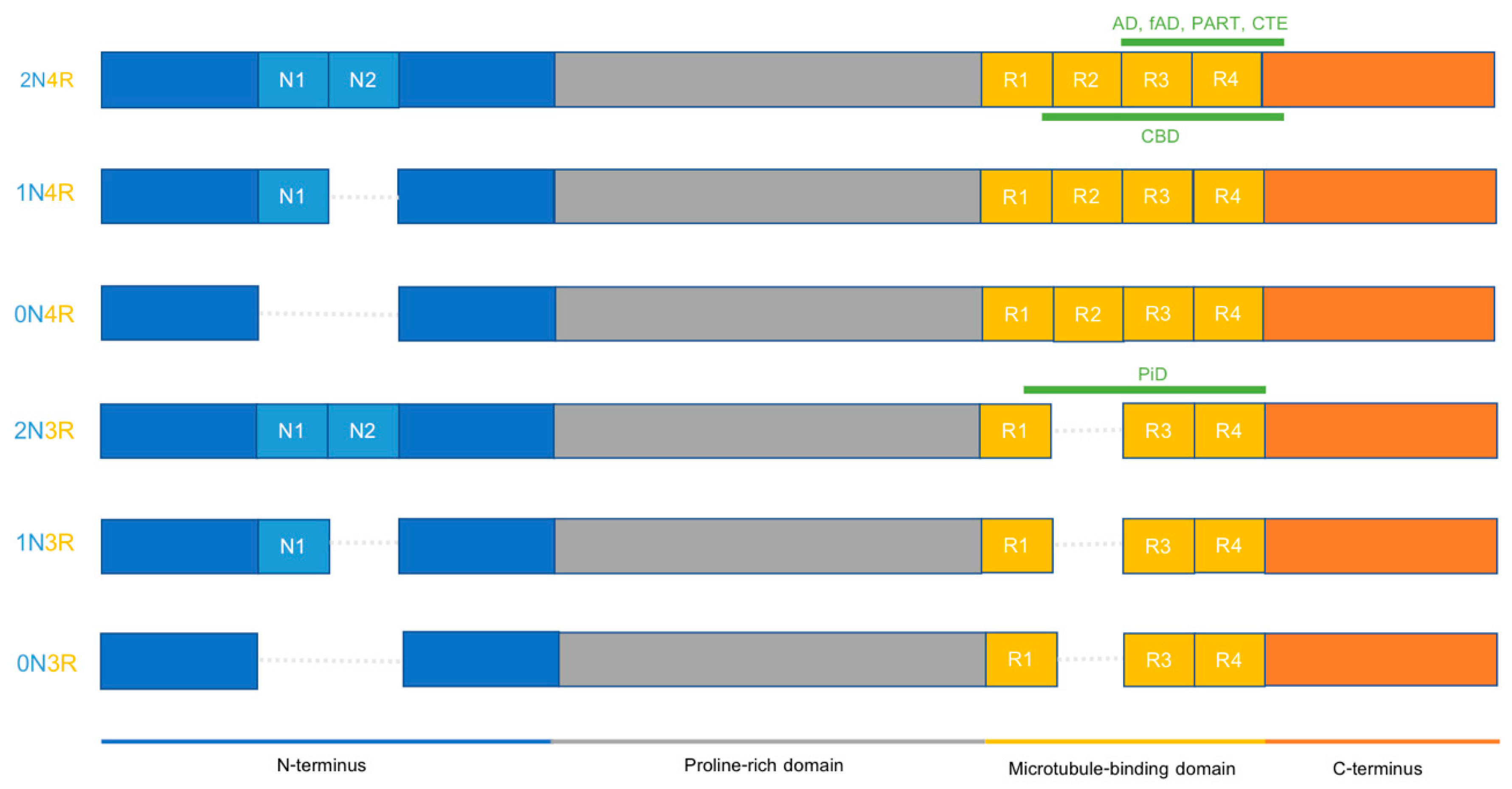
2.3. Different Tauopathies Exhibit Different Pathological Features
3. Animal Models of Tau Propagation
3.1. Transgenic Mouse Models
3.2. Transgenic Mouse Models with Localized Expression
3.3. Viral Expression of Human Tau in Wild-Type Mouse Models
3.4. Seeding with Tg Mouse Brain Lysates
3.5. Seeding with Recombinant Tau Fibrils
4. Patient-Derived Tau Seeding Models
4.1. Inoculation of Tg Mice Overexpressing Human Tau with FTD Mutation
4.2. Injection in Tg Mice Overexpressing A Single WT Human Tau Isoform
4.3. Injection into WT Mouse Models
4.4. Inoculation into Mice with a Humanized MAPT Sequence
4.5. Towards More Integrative AD Models
5. Applications of Patient-Derived Tau Seeding Models
5.1. Identification of Seed-Competent Tau Species
5.2. Intracellular Mechanisms and Tau Aggregate Degradation
5.3. Role of Astrocytes in Tau Spreading
5.4. Role of Microglia in Tau Spreading
5.5. Tau Pathology in Lewy Body Dementia
5.6. Tau-Targeted AD Treatments
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| AD | Alzheimer’s disease |
| AGD | argyrophilic grain disease |
| APP | amyloid precursor protein |
| ARTAG | aging-related tau astrogliopathy |
| ASO | antisense oligonucleotide |
| Aβ | amyloid-β |
| CBD | corticobasal degeneration |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| Cryo-EM | cryogenic electron microscopy |
| CSF | cerebrospinal fluid |
| CTE | chronic traumatic encephalopathy |
| DLB | dementia with Lewy bodies |
| EC | entorhinal cortex |
| fAD | familial Alzheimer’s disease |
| FTD | frontotemporal dementia |
| GFP | green fluorescent protein |
| GGT | globular glial tauopathy |
| GVB | granulovacuolar degeneration body |
| iPSC | induced pluripotent stem cell |
| KI | knock-in |
| KO | knock-out |
| LBD | Lewy body dementia |
| LRP1 | lipoprotein receptor-related protein 1 |
| LRRK2 | leucine-rich repeat kinase |
| mAB | monoclonal antibody |
| MAPT | microtubule-associated protein tau |
| MTBR | microtubule-binding region |
| NFT | neurofibrillary tangle |
| PSEN | presenilin |
| P-tau | phosphorylated tau |
| PART | primary age-related tauopathy |
| PD | Parkinson’s disease |
| PDD | Parkinson’s disease dementia |
| PET | positron emission tomography |
| PFF | preformed fibril |
| PiD | Pick’s disease |
| PP2A | protein phosphatase 2A |
| PSP | progressive supranuclear palsy |
| PTM | post-translational modification |
| R | microtubule-binding repeat |
| Tg | transgenic |
| tTA | tetracycline transactivator |
| VCP | valosin-containing protein |
| WT | wild-type |
| α-syn | α-synuclein |
References
- Scheltens, P.; de Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021. [Google Scholar] [CrossRef]
- Vogels, T.; Leuzy, A.; Cicognola, C.; Ashton, N.J.; Smolek, T.; Novak, M.; Blennow, K.; Zetterberg, H.; Hromadka, T.; Zilka, N.; et al. Propagation of Tau Pathology: Integrating Insights from Postmortem and In Vivo Studies. Biol. Psychiatry 2020, 87, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Bejanin, A.; Schonhaut, D.R.; la Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Smith, R.; Ohlsson, T.; Strandberg, O.; Mattsson, N.; Insel, P.S.; Palmqvist, S.; Hansson, O. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology 2019, 92, e601–e612. [Google Scholar] [CrossRef] [PubMed]
- Digma, L.A.; Madsen, J.R.; Reas, E.T.; Dale, A.M.; Brewer, J.B.; Banks, S.J. Tau and atrophy: Domain-specific relationships with cognition. Alzheimer Res. Ther. 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D.; et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef]
- Hansson, O.; Grothe, M.J.; Strandberg, T.O.; Ohlsson, T.; Hägerström, D.; Jögi, J.; Smith, R.; Schöll, M. Tau pathology distribution in Alzheimer’s disease corresponds differentially to cognition-relevant functional brain networks. Front. Neurosci. 2017, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Clavaguera, F.; Tolnay, M. Tauopathy models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Respondek, G.; Kovacs, G.G. New classification of tauopathies. Rev. Neurol. 2018, 174, 664–668. [Google Scholar] [CrossRef]
- Narasimhan, S.; Lee, V.M.Y. The use of mouse models to study cell-to-cell transmission of pathological tau. Methods Cell Biol. 2017, 141, 287–305. [Google Scholar] [PubMed]
- He, Z.; McBride, J.D.; Xu, H.; Changolkar, L.; Kim, S.-J.; Zhang, B.; Narasimhan, S.; Gibbons, G.S.; Guo, J.L.; Kozak, M.; et al. Transmission of tauopathy strains is independent of their isoform composition. Nat. Commun. 2020, 11, 7. [Google Scholar] [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Boluda, S.; Iba, M.; Zhang, B.; Raible, K.M.; Lee, V.M.Y.; Trojanowski, J.Q. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015, 129, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2021. Alzheimer Dement. Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef]
- Lee, G.; Cowan, N.; Kirschner, M. The Primary Structure and Heterogeneity of Tau Protein from Mouse Brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich1, J.; Crowther, R.A. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and Novel Exons of the Human tau Gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Combs, B.; Gamblin, T.C. FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry 2012, 51, 8597–8607. [Google Scholar] [CrossRef]
- Falcon, B.; Cavallini, A.; Angers, R.; Glover, S.; Murray, T.K.; Barnham, L.; Jackson, S.; O’Neill, M.J.; Isaacs, A.M.; Hutton, M.L.; et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J. Biol. Chem. 2015, 290, 1049–1065. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Pittman, A.M.; Myers, A.J.; Duckworth, J.; Bryden, L.; Hanson, M.; Abou-Sleiman, P.; Wood, N.W.; Hardy, J.; Lees, A.; de Silva, R. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum. Mol. Genet. 2004, 13, 1267–1274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Myers, A.J.; Pittman, A.M.; Zhao, A.S.; Rohrer, K.; Kaleem, M.; Marlowe, L.; Lees, A.; Leung, D.; McKeith, I.G.; Perry, R.H.; et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis. 2007, 25, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Caillet-Boudin, M.L.; Buée, L.; Sergeant, N.; Lefebvre, B. Regulation of human MAPT gene expression. Mol. Neurodegener. 2015, 10, 28. [Google Scholar] [CrossRef]
- Kouri, N.; Ross, O.A.; Dombroski, B.; Younkin, C.S.; Serie, D.J.; Soto-Ortolaza, A.; Baker, M.; Finch, N.C.A.; Yoon, H.; Kim, J.; et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 2015, 6, 7247. [Google Scholar] [CrossRef]
- Heckman, M.G.; Brennan, R.R.; Labbé, C.; Soto, A.I.; Koga, S.; Deture, M.A.; Murray, M.E.; Petersen, R.C.; Boeve, B.F.; Van Gerpen, J.A.; et al. Association of mapt subhaplotypes with risk of progressive supranuclear palsy and severity of tau pathology. JAMA Neurol. 2019, 76, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; McCarty-Wood, E.; van Deerlin, V.M.; Lee, V.M.-Y.; Trojanowski, J.Q. Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am. J. Pathol. 2013, 183, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.; Defensor, E.; Mok, S.A.; et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.; Stieler, J.T.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, Y.; Chung D eun, C.; Yue, M.; Castanedes-Casey, M.; Madden, B.J.; Dunmore, J.; Tong, J.; DeTure, M.; Dickson, D.W.; Petrucelli, L.; et al. An Acetylation–phosphorylation switch that regulates tau aggregation propensity and function. J. Biol. Chem. 2017, 292, 15277–15286. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V.W. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Lebbadi, M.; Salem, N.; Bennett, D.A.; Calon, F. Sirtuin 1 Reduction Parallels the Accumulation of Tau in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef]
- Min, S.W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.-A.; Gestwicki, J.E.; Gan, L. iSIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef] [PubMed]
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, M.; Rolkova, G.; Kontsekova, E.; Novak, M. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588. [Google Scholar] [CrossRef]
- McMillan, P.J.; Kraemer, B.C.; Robinson, L.; Leverenz, J.B.; Raskind, M.; Schellenberg, G. Truncation of tau at E391 Promotes Early Pathologic Changes in Transgenic Mice. J. Neuropathol. Exp. Neurol. 2011, 70, 1006–1019. [Google Scholar] [CrossRef]
- Zimova, I.; Brezovakova, V.; Hromadka, T.; Weisova, P.; Cubinkova, V.; Valachova, B.; Filipcik, P.; Jadhav, S.; Smolek, T.; Novak, M.; et al. Human Truncated Tau Induces Mature Neurofibrillary Pathology in a Mouse Model of Human Tauopathy. J. Alzheimer Dis. 2016, 54, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, S.; Sprenger, F.; Skachokova, Z.; Fraser, G.; Abramowski, D.; Clavaguera, F.; Probst, A.; Frank, S.; Müller, M.; Staufenbiel, M.; et al. Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol. Psychiatry 2016, 21, 1790–1798. [Google Scholar] [CrossRef]
- Kim, Y.D.; Choi, H.; Lee, W.J.; Park, H.; Kam, T.I.; Hong, S.-H.; Nah, J.; Jung, S.; Shin, B.; Lee, H.; et al. Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol. Dis. 2016, 87, 19–28. [Google Scholar] [CrossRef]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.-M.; Mandelkow, E. Assembly of protein into Alzheimer paired helical filaments depends on a local sequence motif (306 VQIVYK 311) forming structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local β-Structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Wegmann, S.; Cho, H.; Devos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef] [PubMed]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. eLife 2018, 7, e36584. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Schöll, M.; Ossenkoppele, R.; Strandberg, O.; Palmqvist, S.; Jögi, J.; Ohlsson, T.; Smith, R.; Hansson, O. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 2017, 140, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Sarazin, M.; Lagarde, J.; Bottlaender, M. Distinct tau PET imaging patterns in typical and atypical Alzheimer’s disease. Brain 2016, 139, 1321–1324. [Google Scholar] [CrossRef]
- Day, G.S.; Gordon, B.A.; Jackson, K.; Christensen, J.J.; Rosana Ponisio, M.; Su, Y.; Ances, B.M.; Benzinger, T.L.S.; Morris, J.C. Tau-PET Binding Distinguishes Patients With Early-stage Posterior Cortical Atrophy From Amnestic Alzheimer Disease Dementia. Alzheimer Dis. Assoc. Disord. 2017, 31, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 871–881. [Google Scholar] [CrossRef]
- Franzmeier, N.; Dewenter, A.; Frontzkowski, L.; Dichgans, M.; Rubinski, A.; Neitzel, J.; Smith, R.; Strandberg, O.; Ossenkoppele, R.; Buerger, K.; et al. Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer’s disease. Sci. Adv. 2020, 6, eabd1327. [Google Scholar] [CrossRef]
- Li, L.; Shi, R.; Gu, J.; Tung, Y.C.; Zhou, Y.; Zhou, D.; Wu, R.; Chu, D.; Jin, N.; Deng, K.; et al. Alzheimer’s disease brain contains tau fractions with differential prion-like activities. Acta Neuropathol. Commun. 2021, 9, 28. [Google Scholar] [CrossRef]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M.; et al. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Müller, H.M.; Nachman, E.; Steringer, J.P.; de Almodovar, C.M.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef]
- Merezhko, M.; Brunello, C.A.; Yan, X.; Vihinen, H.; Jokitalo, E.; Uronen, R.L.; Huttunen, H.J. Secretion of Tau via an Unconventional Non-vesicular Mechanism. Cell Rep. 2018, 25, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Rauch, J.N.; Chen, J.J.; Sorum, A.W.; Miller, G.M.; Sharf, T.; See, S.K.; Hsieh-Wilson, L.C.; Kampmann, M.; Kosik, K.S. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 2018, 8, 6382. [Google Scholar] [CrossRef]
- Evans, L.D.; Strano, A.; Campbell, A.; Karakoc, E.; Iorio, F.; Bassett, A.R.; Livesey, F.J. Whole genome CRISPR screens identify LRRK2-regulated endocytosis as a major mechanism for extracellular tau uptake by human neurons. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cooper, J.M.; Lathuiliere, A.; Migliorini, M.; Arai, A.L.; Wani, M.M.; Dujardin, S.; Muratoglu, S.C.; Hyman, B.T.; Strickland, D.K. Regulation of tau internalization, degradation, and seeding by LRP1 reveals multiple pathways for tau catabolism. J. Biol. Chem. 2021, 296, 100715. [Google Scholar] [CrossRef]
- Morozova, V.; Cohen, L.S.; Makki, A.E.H.; Shur, A.; Pilar, G.; el Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell. Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713. [Google Scholar] [CrossRef]
- Moloney, C.M.; Lowe, V.J.; Murray, M.E. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: A clinicopathologic perspective for biomarker research. Alzheimer Dement. 2021. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Shi, Y.; Murzin, A.G.; Falcon, B.; Epstein, A.; Machin, J.; Tempest, P.; Newell, K.L.; Vidal, R.; Garringer, H.J.; Sahara, N.; et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol. 2021, 141, 697–708. [Google Scholar] [CrossRef]
- Scheres, S.H.; Zhang, W.; Falcon, B.; Goedert, M. Cryo-EM structures of tau filaments. Curr. Opin. Struct. Biol. 2020, 64, 17–25. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Götz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C.; Kress, Y.; Espinoza, M.; de Silva, R.; Tucker, K.L.; Barde, Y.A.; Duff, K.; Davies, P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef]
- Götz, J.J.; Götz, J. Experimental Models of Tauopathy–From Mechanisms to Therapies. Adv. Exp. Med. Biol. 2019, 1184, 381–391. [Google Scholar]
- Eckermann, K.; Mocanu, M.M.; Khlistunova, I.; Biernat, J.; Nissen, A.; Hofmann, A.; Schönig, K.; Bujard, H.; Haemisch, A.; Mandelkow, E.; et al. The β-propensity of tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J. Biol. Chem. 2007, 282, 31755–31765. [Google Scholar] [CrossRef]
- Maeda, S.; Djukic, B.; Taneja, P.; Yu, G.; Lo, I.; Davis, A.; Craft, R.; Guo, W.; Wang, X.; Kim, D.; et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016, 17, 530–551. [Google Scholar] [CrossRef] [PubMed]
- Bondulich, M.K.; Guo, T.; Meehan, C.; Manion, J.; Rodriguez Martin, T.; Mitchell, J.C.; Hortobagyi, T.; Yankova, N.; Stygelbout, V.; Brion, J.-P.; et al. Tauopathy induced by low level expression of a human brain-derived tau fragment in mice is rescued by phenylbutyrate. Brain 2016, 139, 2290–2306. [Google Scholar] [CrossRef]
- Lewis, J.; Mcgowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; van Slegtenhorst, M.; Gwinn-Hardy, K.; Murphy, M.P.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Forman, M.S.; Lal, D.; Zhang, B.; Dabir, D.V.; Swanson, E.; Lee, V.M.-Y.; Trojanowski, J.Q. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J. Neurosci. 2005, 25, 3539–3550. [Google Scholar] [CrossRef]
- Higuchi, M.; Zhang, B.; Forman, M.S.; Yoshiyama, Y.; Trojanowski, J.Q.; Lee, V.M.Y. Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J. Neurosci. 2005, 25, 9434–9443. [Google Scholar] [CrossRef]
- Forman, M.S.; Lal, D.; Zhang, B.; Dabir, D.V.; Swanson, E.; Lee, V.M.Y.; Trojanowski, J.Q. Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J. Neurosci. 2006, 26, 644–654. [Google Scholar]
- Gamache, J.; Benzow, K.; Forster, C.; Kemper, L.; Hlynialuk, C.; Furrow, E.; Ashe, K.H.; Koob, M.D. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat. Commun. 2019, 10, 2479. [Google Scholar] [CrossRef]
- Kodama, L.; Guzman, E.; Etchegaray, J.I.; Li, Y.; Sayed, F.A.; Zhou, L.; Zhan, L.; Le, D.; Udeochu, J.C.; Clelland, C.D.; et al. Microglial microRNAs mediate sex-specific responses to tau pathology. Nat. Neurosci. 2020, 23, 167–171. [Google Scholar] [CrossRef]
- Harris, J.A.; Koyama, A.; Maeda, S.; Ho, K.; Devidze, N.; Dubal, D.B.; Yu, G.-Q.; Masliah, E.; Mucke, L. Human P301L-Mutant Tau Expression in Mouse Entorhinal-Hippocampal Network Causes Tau Aggregation and Presynaptic Pathology but No Cognitive Deficits. PLoS ONE 2012, 7, e45881. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.; DeVos, S.L.; Nicholls, S.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O.; et al. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015, 34, 3028–3041. [Google Scholar] [CrossRef]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Bégard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- de Gérando, A.M.; d’Orange, M.; Augustin, E.; Joséphine, C.; Aurégan, G.; Gaudin-Guérif, M.; Guillermier, M.; Hérard, A.-S.; Stimmer, L.; Petit, F.; et al. Neuronal tau species transfer to astrocytes and induce their loss according to tau aggregation state. Brain 2021, 144, 1167–1182. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; van Ingelgom, A.J.; Linares, C.; Kurti, A.; Knight, J.A.; Heckman, M.G.; Diehl, N.N.; Shinohara, M.; Martens, Y.A.; et al. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat. Commun. 2018, 9, 4388. [Google Scholar] [CrossRef]
- Rodrigues, S.; Anglada-Huguet, M.; Hochgräfe, K.; Kaniyappan, S.; Wegmann, S.; Mandelkow, E.-M. Spreading of Tau Protein Does Not Depend on Aggregation Propensity. 2021. Available online: https://assets.researchsquare.com/files/rs-356350/v1/36b64d6c-8143-4d1b-8afe-282465fea9e3.pdf (accessed on 5 May 2021).
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef]
- Wegmann, S.; Bennett, R.E.; Delorme, L.; Robbins, A.B.; Hu, M.; Mackenzie, D.; Kirk, M.J.; Schiantarelli, J.; Tunio, N.; Amaral, A.C.; et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019, 5, eaaw6404. [Google Scholar] [CrossRef]
- Vogels, T.; Vargova, G.; Brezovakova, V.; Quint, W.H.; Hromadka, T. Viral Delivery of Non-Mutated Human Truncated Tau to Neurons Recapitulates Key Features of Human Tauopathy in Wild-Type Mice. J. Alzheimer Dis. 2020, 77, 551–568. [Google Scholar] [CrossRef]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014, 127, 667–683. [Google Scholar] [CrossRef]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.Y. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef]
- Peeraer, E.; Bottelbergs, A.; van Kolen, K.; Stancu, I.C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Donck, L.V.; Torremans, A.; Sluydts, E.; et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Banks, R.A.; Kim, B.; Xu, H.; Changolkar, L.; Leight, S.N.; Riddle, D.M.; Li, C.; Gathagan, R.J.; Brown, H.J.; et al. GFP-mutant human tau transgenic mice develop tauopathy following CNS injections of Alzheimer’s brain-derived pathological tau or synthetic mutant human tau fibrils. J. Neurosci. 2017, 37, 11485–11494. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Narasimhan, S.; Changolkar, L.; He, Z.; Stieber, A.; Zhang, B.; Gathagan, R.J.; Iba, M.; McBride, J.D.; Trokanowski, J.Q.; et al. Unique pathological tau conformers from alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 2016, 213, 2635–2654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Anthony Crowther, R.; Goedert, M.; Scheres, S.H.W. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 2019, 8, e43584. [Google Scholar] [CrossRef]
- Saito, T.; Mihira, N.; Matsuba, Y.; Sasaguri, H.; Hashimoto, S.; Narasimhan, S.; Zhang, B.; Murayama, S.; Higuchi, M.; Lee, V.M.Y.; et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem. 2019, 294, 12754–12765. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; de Decker, R.; vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef]
- Skachokova, Z.; Martinisi, A.; Flach, M.; Sprenger, F.; Naegelin, Y.; Steiner-Monard, V.; Sollberger, M.; Monsch, A.U.; Goedert, M.; Tolnay, M.; et al. Cerebrospinal fluid from Alzheimer’s disease patients promotes tau aggregation in transgenic mice. Acta Neuropathol. Commun. 2019, 7, 72. [Google Scholar] [CrossRef]
- Hayashi, T.; Shimonaka, S.; Elahi, M.; Matsumoto, S.-E.; Ishiguro, K.; Takanashi, M.; Hattori, N.; Motoi, Y. Learning Deficits Accompanied by Microglial Proliferation After the Long-Term Post-Injection of Alzheimer’s Disease Brain Extract in Mouse Brains. J. Alzheimer Dis. 2021, 79, 1701–1711. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Yamazaki, N.; Hashimoto, S.; Saido, T.C. Calpain activation in Alzheimer’s model mice is an artifact of APP and presenilin overexpression. J. Neurosci. 2016, 36, 9933–9936. [Google Scholar] [CrossRef] [PubMed]
- Croft, C.L.; Goodwin, M.S.; Ryu, D.H.; Lessard, C.B.; Tejeda, G.; Marrero, M.; Vause, A.R.; Paterno, G.; Cruz, P.E.; Lewis, J.; et al. Photodynamic studies reveal rapid formation and appreciable turnover of tau inclusions. Acta Neuropathol. 2021, 141, 359–381. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef]
- Audouard, E.; Houben, S.; Masaracchia, C.; Yilmaz, Z.; Suain, V.; Authelet, M.; De Decker, R.; Buée, L.; Boom, A.; Leroy, K.; et al. High–Molecular-Weight Paired Helical Filaments from Alzheimer Brain Induces Seeding of Wild-Type Mouse Tau into an Argyrophilic 4R Tau Pathology in Vivo. Am. J. Pathol. 2016, 186, 2709–2722. [Google Scholar] [CrossRef]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef]
- Ferrer, I.; Andrés-Benito, P.; Zelaya, M.V.; Aguirre, M.E.E.; Carmona, M.; Ausín, K.; Lachén-Montes, M.; Fernández-Irigoyen, J.; Santamaría, E.; Del Rio, J.A. Familial globular glial tauopathy linked to MAPT mutations: Molecular neuropathology and seeding capacity of a prototypical mixed neuronal and glial tauopathy. Acta Neuropathol. 2020, 139, 735–771. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; García, M.A.; González, I.L.; Lucena, D.D.; Villalonga, A.R.; Tech, M.C.; Llorens, F.; Garcia-Esparcia, P.; Martinez-Maldonado, A.; Mendez, M.F.; et al. Aging-related tau astrogliopathy (ARTAG): Not only tau phosphorylation in astrocytes. Brain Pathol. 2018, 28, 965–985. [Google Scholar] [CrossRef]
- Ferrer, I.; García, M.A.; Carmona, M.; Andrés-Benito, P.; Torrejón-Escribano, B.; Garcia-Esparcia, P.; Del Rio, J. Involvement of oligodendrocytes in tau seeding and spreading in tauopathies. Front. Aging Neurosci. 2019, 11, 112. [Google Scholar] [CrossRef]
- Ferrer, I.; Zelaya, M.V.; Aguiló García, M.; Carmona, M.; López-González, I.; Andrés-Benito, P.; Lidón, L.; Gavín, R.; Garcia-Esparcia, P.; Del Rio, J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol. 2020, 30, 298–318. [Google Scholar] [CrossRef]
- Ferrer, I.; Andrés-Benito, P.; Sala-Jarque, J.; Gil, V.; del Rio, J.A. Capacity for Seeding and Spreading of Argyrophilic Grain Disease in a Wild-Type Murine Model; Comparisons with Primary Age-Related Tauopathy. Front. Mol. Neurosci. 2020, 13, 101. [Google Scholar] [CrossRef]
- Hernández, F.; Merchán-Rubira, J.; Vallés-Saiz, L.; Rodríguez-Matellán, A.; Avila, J. Differences between Human and Murine Tau at the N-terminal End. Front. Aging Neurosci. 2020, 12, 11. [Google Scholar] [CrossRef]
- Uchihara, T. Pretangles and neurofibrillary changes: Similarities and differences between AD and CBD based on molecular and morphological evolution. Neuropathology 2014, 34, 571–577. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, X.; Tung, Y.C.; Xie, S.; Liu, F.; Iqbal, K. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimer Dement. 2016, 12, 1066–1077. [Google Scholar] [CrossRef]
- Miao, J.; Shi, R.; Li, L.; Chen, F.; Zhou, Y.; Tung, Y.C.; Hu, W.; Gong, C.-X.; Iqbal, K.; Liu, F. Pathological Tau From Alzheimer’s Brain Induces Site-Specific Hyperphosphorylation and SDS- and Reducing Agent-Resistant Aggregation of Tau in vivo. Front. Aging Neurosci. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Zareba-Paslawska, J.; Patra, K.; Kluzer, L.; Revesz, T.; Svenningsson, P. Tau Isoform-Driven CBD Pathology Transmission in Oligodendrocytes in Humanized Tau Mice. Front. Neurol. 2021, 11, 589471. [Google Scholar] [CrossRef]
- Xu, H.; O’Reilly, M.; Gibbons, G.S.; Changolkar, L.; McBride, J.D.; Riddle, D.M.; Zhang, B.; Stieber, A.; Nirschl, J.; Kim, S.-J.; et al. In vitro amplification of pathogenic tau conserves disease-specific bioactive characteristics. Acta Neuropathol. 2021, 141, 193–215. [Google Scholar] [CrossRef]
- Weitzman, S.A.; Narasimhan, S.; He, Z.; Changolkar, L.; McBride, J.D.; Zhang, B.; Schellenberg, G.D.; Trojanowski, J.W.; Lee, V.M.Y. Insoluble Tau From Human FTDP-17 Cases Exhibit Unique Transmission Properties In Vivo. J. Neuropathol. Exp. Neurol. 2020, 79, 941–949. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Gerson, J.E.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Kayed, R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J. Alzheimer Dis. 2014, 40, S97–S111. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular A and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef]
- Lam, S.; Boluda, S.; Hérard, A.-S.; Petit, F.; Eddarkaoui, S.; Cambon, K.; Picq, J.L.; Buee, L.; Duyckaerts, C.; Haik, S.; et al. Alzheimer’s brain inoculation in Aß-plaque bearing mice: Synaptic loss is linked to tau seeding and low microglial activity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Houben, S.; de Fisenne, M.A.; Ando, K.; vanden Dries, V.; Poncelet, L.; Yilmaz, Z.; Mansour, S.; De Decker, R.; Brion, J.-P.; Leroy, K. Intravenous Injection of PHF-Tau Proteins From Alzheimer Brain Exacerbates Neuroinflammation, Amyloid Beta, and Tau Pathologies in 5XFAD Transgenic Mice. Front. Mol. Neurosci. 2020, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, B.T.; Dooley, P.M.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, V.I.; van Ziel, A.M.; Vazquez-Sanchez, S.; Nölle, A.; Berenjeno-Correa, E.; Bonaterra-Pastra, A.; Clavaguera, F.; Tolnay, M.; Musters, R.J.P.; van Weering, J.R.t.; et al. Granulovacuolar degeneration bodies are neuron-selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathol. 2019, 138, 943–970. [Google Scholar] [CrossRef] [PubMed]
- Darwich, N.F.; Phan, J.M.; Kim, B.; Suh, E.; Papatriantafyllou, J.D.; Changolkar, L.; Nguyen, A.T.; O’Rourke, C.M.; He, Z.; Porta, S.; et al. Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science 2020, 370, eaay8826. [Google Scholar] [CrossRef]
- Narasimhan, S.; Changolkar, L.; Riddle, D.M.; Kats, A.; Stieber, A.; Weitzman, S.A.; Zhang, B.; Li, Z.; Roberson, E.D.; Trojanowski, J.Q.; et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med. 2020, 217, e20190783. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.W.; Bajaj, L.; Bader, D.A.; Lee, V.M.Y.; Trojanowski, J.Q.; Liu, Z.; et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 645233. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Felsky, D.; Roostaei, T.; Nho, K.; Risacher, S.L.; Bradshaw, E.M.; Petyuk, V.; Schneider, J.A.; Saykin, A.; Bennett, D.A.; De Jager, P.L. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat. Commun. 2019, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Dejanovic, B.; Huntley, M.A.; de Mazière, A.; Meilandt, W.J.; Wu, T.; Srinivasan, K.; Jiang, J.; Gandham, V.; Friedman, B.A.; Ngu, H.; et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 2018, 100, 1322–1336. [Google Scholar] [CrossRef]
- Brelstaff, J.; Tolkovsky, A.M.; Ghetti, B.; Goedert, M.; Spillantini, M.G. Living Neurons with Tau Filaments Aberrantly Expose Phosphatidylserine and Are Phagocytosed by Microglia. Cell Rep. 2018, 24, 1939–1948. [Google Scholar] [CrossRef]
- Vogels, T.; Murgoci, A.N.; Hromádka, T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol. Commun. 2019, 7, 109. [Google Scholar] [CrossRef]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; Devos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezy, T. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, S.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Wang, C.; Fan, L.; Zhan, L.; Kodama, L.; Liu, B.; Chin, M.Y.; Li, Y.; Le, D.; Zhou, Y.; Condello, C.; et al. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Henderson, M.X.; Changolkar, L.; Trojanowski, J.Q.; Lee, V.M.Y. LRRK2 Kinase Activity Does Not Alter Cell-Autonomous Tau Pathology Development in Primary Neurons. J. Parkinson Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Cornblath, E.J.; Li, H.L.; Changolkar, L.; Zhang, B.; Brown, H.J.; Gathagan, R.J.; Olufemi, M.F.; Bassett, D.S.; Trojanowski, J.Q.; et al. Tau pathology spreads between anatomically-connected regions of the brain and is modulated by a LRRK2 mutation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Healy, D.G.; Falchi, M.; O’sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, Genotype, and Worldwide Genetic Penetrance of LRRK2-Associated Parkinson’s Disease: A Case-Control Study. 2008. Available online: http://neurology.thelancet.com (accessed on 12 April 2021).
- Blauwendraat, C.; Pletnikova, O.; Geiger, J.T.; Murphy, N.A.; Abramzon, Y.; Rudow, G.; Mamais, A.; Sabir, M.S.; Crain, C.; Ahmed, S.; et al. Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol. Aging 2019, 76, 214.e1–214.e9. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Sengupta, M.; Trojanowski, J.Q.; Lee, V.M.Y. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 2019, 7, 183. [Google Scholar] [CrossRef]
- Coughlin, D.; Xie, S.X.; Liang, M.; Williams, A.; Peterson, C.; Weintraub, D.; McMillan, C.T.; Wolk, D.A.; Akhtar, R.S.; Hurtig, H.I.; et al. Cognitive and Pathological Influences of Tau Pathology in Lewy Body Disorders. Ann. Neurol. 2019, 85, 259–271. [Google Scholar] [CrossRef]
- Marui, W.; Iseki, E.; Ueda, K.; Kosaka, K. Occurrence of human a-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer’s disease. J. Neurol. Sci. 2000, 174, 81–84. [Google Scholar] [CrossRef]
- Arai, Y.; Yamazaki, M.; Mori, O.; Muramatsu, H.; Asano, G.; Katayama, Y. α-Synuclein-positive structures in cases with sporadic Alzheimer’s disease: Morphology and its relationship to tau aggregation. Brain Res. 2001, 888, 287–296. [Google Scholar] [CrossRef]
- Bassil, F.; Meymand, E.S.; Brown, H.J.; Xu, H.; Cox, T.O.; Pattabhiraman, S.; Maghames, C.M.; Wu, Q.; Zhang, B.; Trojanowski, J.Q.; et al. α-Synuclein modulates tau spreading in mouse brains. J. Exp. Med. 2021, 218, e20192193. [Google Scholar] [CrossRef]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell 2013, 154, 103. [Google Scholar] [CrossRef]
- Vandermeeren, M.; Borgers, M.; van Kolen, K.; Theunis, C.; Vasconcelos, B.; Bottelbergs, A.; Wintmolders, C.; Daneels, G.; Willems, R.; Dockx, K.; et al. Anti-tau monoclonal antibodies derived from soluble and filamentous tau show diverse functional properties in vitro and in vivo. J. Alzheimer Dis. 2018, 65, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Courade, J.P.; Angers, R.; Mairet-Coello, G.; Pacico, N.; Tyson, K.; Lightwood, D.; Munro, R.; McMillan, D.; Griffin, R.; Baker, T.; et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018, 136, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Nobuhara, C.K.; DeVos, S.L.; Commins, C.; Wegmann, S.; Moore, B.D.; Roe, A.D.; Constantino, I.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Tau Antibody Targeting Pathological Species Blocks Neuronal Uptake and Interneuron Propagation of Tau in Vitro. Am. J. Pathol. 2017, 187, 1399–1412. [Google Scholar] [CrossRef]
- Rosenqvist, N.; Asuni, A.A.; Andersson, C.R.; Christensen, S.; Daechsel, J.A.; Egebjerg, J.; Falsig, J.; Helboe, L.; Jul, P.; Kartberg, F.; et al. Highly specific and selective anti-pS396-tau antibody C10.2 targets seeding-competent tau. Alzheimer Dement. Transl. Res. Clin. Interv. 2018, 4, 521–534. [Google Scholar] [CrossRef]
- Dai, C.L.; Chen, X.; Kazim, S.F.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J. Neural Transm. 2015, 122, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.L.; Hu, W.; Tung, Y.C.; Liu, F.; Gong, C.X.; Iqbal, K. Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 × Tg-AD mice. Alzheimer Res. Ther. 2018, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.; Mairet-Coello, G.; Danis, C.; Lieger, S.; Caillierez, R.; Carrier, S.; Skrobala, E.; Landrieu, I.; Michel, A.; Schmitt, M.; et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain 2019, 142, 1736–1750. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Kim, S.J.; Wu, Q.; Riddle, D.M.; Leight, S.N.; Changolkar, L.; Xu, H.; Meymand, E.S.; O’Reilly, M.; Zhang, B.; et al. Conformation-selective tau monoclonal antibodies inhibit tau pathology in primary neurons and a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 64. [Google Scholar] [CrossRef]
- DeVos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Yamashita, S.; Fukuda, T.; Park, J.M.; Murayama, M.; Mizoroki, T.; Yoshiike, Y.; Sahara, N.; Takashima, A. Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J. 2007, 26, 5143–5152. [Google Scholar] [CrossRef]
- Miller, L.V.C.; Mukadam, A.S.; Durrant, C.S.; Vaysburd, M.J.; Katsinelos, T.; Tuck, B.J.; Sanford, S.; Sheppard, O.; Knox, C.; Cheng, S.; et al. Tau assemblies do not behave like independently acting prion-like particles in mouse neural tissue. Acta Neuropathol. Commun. 2021, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Jebarupa, B.; Muralidharan, M.; Arun, A.; Mandal, A.K.; Mitra, G. Conformational heterogeneity of tau: Implication on intrinsic disorder, acid stability and fibrillation in Alzheimer’s disease. Biophys. Chem. 2018, 241, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Smolek, T.; Cubinkova, V.; Brezovakova, V.; Valachova, B.; Szalay, P.; Zilka, N.; Jadhav, S. Genetic Background Influences the Propagation of Tau Pathology in Transgenic Rodent Models of Tauopathy. Front. Aging Neurosci. 2019, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- Smolek, T.; Jadhav, S.; Brezovakova, V.; Cubinkova, V.; Valachova, B.; Novak, P.; Zilka, N. First-in-Rat Study of Human Alzheimer’s Disease Tau Propagation. Mol. Neurobiol. 2019, 56, 621–631. [Google Scholar] [CrossRef]


{kind=link}
| Tg Line | Tau Isoform | Tau Overexpression | Key Study | ||
|---|---|---|---|---|---|
| Wild-type mice | Mice expressing wild-type mouse tau | 4R | No | Lasagna-Reeves et al., 2012 Guo et al., 2016 Ferrer et al., 2018, 2019, 2020, 2020, 2020 Narasimhan et al., 2017 Henderson et al., 2020 | |
| Tauopathy mouse models | Tg mice overexpressing human tau with FTD-linked mutation | PS19 (P301S MAPT mutation) | 4R | Yes | Boluda et al., 2015 |
| T40PL-GFP (P301L MAPT mutation + GFP) | 4R | Yes | Gibbons et al., 2017 | ||
| Tg mice overexpressing WT human tau | ALZ17 | 4R | Yes | Clavaguera et al., 2013 | |
| Mice overexpressing humanized MAPT in the absence of mouse tau | hTau | 3R > 4R | Yes | Hu et al., 2016 Miao et al., 2019 | |
| Mice overexpressing all 6 human tau isoforms in the absence of mouse tau | 6hTau | 3R = 4R | Yes | He et al., 2020 Xu et al., 2021 | |
| Amyloid mouse models | Mice expressing wild-type mouse tau | 5xFAD | No | He et al., 2018 Vergara et al., 2019 Xu et al., 2021 | |
| APP KI | No | He et al., 2018 | |||
| Mice with targeted replacement of the mouse MAPT sequence | MAPT/APP double KI | 3R > 4R | No | Saito et al., 2019 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robert, A.; Schöll, M.; Vogels, T. Tau Seeding Mouse Models with Patient Brain-Derived Aggregates. Int. J. Mol. Sci. 2021, 22, 6132. https://doi.org/10.3390/ijms22116132
Robert A, Schöll M, Vogels T. Tau Seeding Mouse Models with Patient Brain-Derived Aggregates. International Journal of Molecular Sciences. 2021; 22(11):6132. https://doi.org/10.3390/ijms22116132
Chicago/Turabian StyleRobert, Aiko, Michael Schöll, and Thomas Vogels. 2021. "Tau Seeding Mouse Models with Patient Brain-Derived Aggregates" International Journal of Molecular Sciences 22, no. 11: 6132. https://doi.org/10.3390/ijms22116132
APA StyleRobert, A., Schöll, M., & Vogels, T. (2021). Tau Seeding Mouse Models with Patient Brain-Derived Aggregates. International Journal of Molecular Sciences, 22(11), 6132. https://doi.org/10.3390/ijms22116132