
DNA Homeostasis and Senescence: Lessons from the Naked Mole Rat




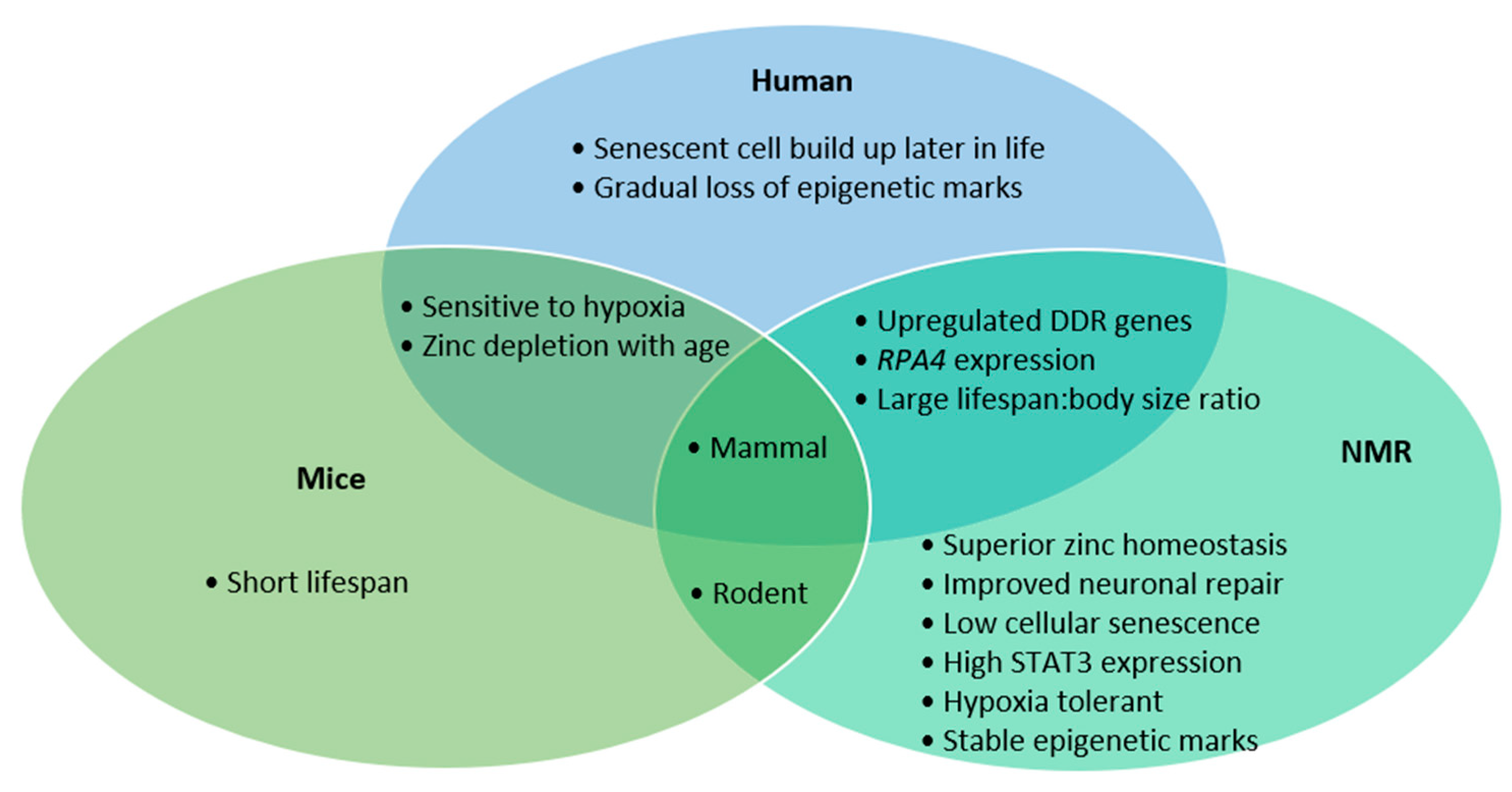{kind=link}
Abstract
1. Introduction
2. DNA Damage Response in NMRs
2.1. DNA Damage Response Upregulation
2.2. Gene and Protein Adaptations
2.3. Antioxidants for DNA Protection
2.4. Epigenetic Stability
3. Preventing and Avoiding Senescence
3.1. Senescence-Associated Secretory Phenotypes
3.2. Avoiding Senescence in Hypoxia
3.3. Avoiding ER Stress-Induced Senescence
3.4. Senescence Cell Death and Unique Senescence
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DDR | DNA Damage Response |
| DSB | Double Strand Break |
| HDR | Homology-Directed Repair |
| MMEJ | Microhomology-Mediated End Joining |
| MMR | Mismatch Repair |
| NHEJ | Non-Homologous End-Joining |
| NMR | Naked Mole Rat |
| PUR | Protein Unfolded Response |
| ROS | Reactive Oxygen Species |
| SASP | Senescence-Associated Secretory Phenotypes |
| SCD | Senescent Cell Death |
References
- Smith, E.S.J.; Schuhmacher, L.-N.; Husson, Z. The naked mole-rat as an animal model in biomedical research: Current perspectives. Open Access Anim. Physiol. 2015, 7, 137. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Zhang, Z.; Gladyshev, V.N.; Vijg, J. Comparative genetics of longevity and cancer: Insights from long-lived rodents. Nat. Rev. Genet. 2014, 15, 531–540. [Google Scholar] [CrossRef]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Buffenstein, R. The Naked Mole-Rat: A New Long-Living Model for Human Aging Research. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 1369–1377. [Google Scholar] [CrossRef]
- Edrey, Y.H.; Medina, D.X.; Gaczynska, M.; Osmulski, P.A.; Oddo, S.; Caccamo, A.; Buffenstein, R. Amyloid beta and the longest-lived rodent: The naked mole-rat as a model for natural protection from Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2352–2360. [Google Scholar] [CrossRef]
- Orr, M.E.; Garbarino, V.R.; Salinas, A.; Buffenstein, R. Sustained high levels of neuroprotective, high molecular weight, phosphorylated tau in the longest-lived rodent. Neurobiol. Aging 2015, 36, 1496–1504. [Google Scholar] [CrossRef]
- Frankel, D.; Davies, M.; Bhushan, B.; Kulaberoglu, Y.; UrriolaMunoz, P.; Bertrand-Michel, J.; Pergande, M.R.; Smith, A.A.; Preet, S.; Park, T.J.; et al. Cholesterol-rich naked mole-rat brain lipid membranes are susceptible to amyloid beta-induced damage in vitro. Aging 2020, 12, 22266–22290. [Google Scholar] [CrossRef]
- Risques, R.A.; Kennedy, S.R. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018, 14, e1007108. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- MacRae, S.L.; Croken, M.M.K.; Calder, R.B.; Aliper, A.; Milholland, B.; White, R.R.; Zhavoronkov, A.; Gladyshev, V.N.; Seluanov, A.; Gorbunova, V.; et al. DNA repair in species with extreme lifespan differences. Aging (Albany NY) 2015, 7, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, A.; Kutuzov, M.; Petruseva, I.; Lukjanchikova, N.; Kashina, E.; Kolova, E.; Zemerova, T.; Romanenko, S.; Perelman, P.; Prokopov, D.; et al. Naked mole rat cells display more efficient excision repair than mouse cells. Aging (Albany NY) 2018, 10, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- MacRae, S.L.; Zhang, Q.; Lemetre, C.; Seim, I.; Calder, R.B.; Hoeijmakers, J.; Suh, Y.; Gladyshev, V.N.; Seluanov, A.; Gorbunova, V.; et al. Comparative analysis of genome maintenance genes in naked mole rat, mouse, and human. Aging Cell 2015, 14, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, A.; Petruseva, I.; Popov, A.; Koval, O.; Lavrik, O. Naked mole rat cells display more efficient DNA excision repair and higher resistance to toxic impacts than mouse cells. BIO Web Conf. 2020, 22, 01017. [Google Scholar] [CrossRef]
- Deuker, M.M.; Lewis, K.N.; Ingaramo, M.; Kimmel, J.; Buffenstein, R.; Settleman, J. Unprovoked Stabilization and Nuclear Accumulation of the Naked Mole-Rat p53 Protein. Sci. Rep. 2020, 10, 6966. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.-L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of Acute versus Chronic Hypoxia on DNA Damage Responses and Genomic Instability. Cancer Res. 2010, 70, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- León-Ortiz, A.M.; Panier, S.; Sarek, G.; Vannier, J.-B.; Patel, H.; Campbell, P.J.; Boulton, S.J. A Distinct Class of Genome Rearrangements Driven by Heterologous Recombination. Mol. Cell 2018, 69, 292–305.e6. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Eckelmann, B.; Adhikari, S.; Ahmed, K.M.; Sengupta, S.; Pandey, A.; Hegde, P.M.; Tsai, M.-S.; Tainer, J.A.; Weinfeld, M.; et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2016, 45, 2585–2599. [Google Scholar] [CrossRef] [PubMed]
- Rahal, E.A.; Henricksen, L.A.; Li, Y.; Williams, R.S.; Tainer, J.A.; Dixon, K. ATM regulates Mre11-dependent DNA end-degradation and microhomology-mediated end joining. Cell Cycle 2010, 9, 2938–2949. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Caldecott, K.W. XRCC1 protein; Form and function. DNA Repair 2019, 81, 102664. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Berger, S.J.; Berger, N.A. Poly(ADP-ribose) polymerase: A guardian of the genome that facilitates DNA repair by protecting against DNA recombination. In ADP-Ribosylation Reactions: From Bacterial Pathogenesis to Cancer; Springer: Boston, MA, USA, 1999; pp. 23–30. [Google Scholar]
- Simsek, D.; Jasin, M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4–ligase IV during chromosomal translocation formation. Nat. Struct. Mol. Biol. 2010, 17, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.K.; Wywial, E.; Perez, I.V.; Lambert, J.A.; Edrey, H.Y.; Lewis, N.K.; Grimes, K.; Lindsey, L.M.; Brand, D.M.; Buffenstein, R. Walking the Oxidative Stress Tightrope: A Perspective from the Naked Mole-Rat, the Longest-Living Rodent. Curr. Pharm. Des. 2011, 17, 2290–2307. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Andziak, B.; Yang, T.; Buffenstein, R. The Naked Mole-Rat Response to Oxidative Stress: Just Deal with It. Antioxid. Redox Signal. 2013, 19, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of Antioxidants Supplementation on Aging and Longevity. Biomed Res. Int. 2014, 2014, 1–17. [Google Scholar] [CrossRef]
- Huggins, C.J.; Malik, R.; Lee, S.; Salotti, J.; Thomas, S.; Martin, N.; Quiñones, O.A.; Alvord, W.G.; Olanich, M.E.; Keller, J.R.; et al. C/EBPγ Suppresses Senescence and Inflammatory Gene Expression by Heterodimerizing with C/EBPβ. Mol. Cell. Biol. 2013, 33, 3242–3258. [Google Scholar] [CrossRef] [PubMed]
- Mullins, D.N.; Crawford, E.L.; Khuder, S.A.; Hernandez, D.-A.; Yoon, Y.; Willey, J.C. CEBPG transcription factor correlates with antioxidant and DNA repair genes in normal bronchial epithelial cells but not in individuals with bronchogenic carcinoma. BMC Cancer 2005, 5, 141. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of human replication protein A (RPA): From DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef]
- Haring, S.J.; Humphreys, T.D.; Wold, M.S. A naturally occurring human RPA subunit homolog does not support DNA replication or cell-cycle progression. Nucleic Acids Res. 2010, 38, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Hypoxia, anoxia, and O2 sensing: The search continues. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L918–L921. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Waypa, G.B.; Schumacker, P.T. O(2) sensing in hypoxic pulmonary vasoconstriction: The mitochondrial door re-opens. Respir. Physiol. Neurobiol. 2002, 132, 81–91. [Google Scholar] [CrossRef]
- Tew, K.D.; Ronai, Z. GST function in drug and stress response. Drug Resist. Updat. 1999, 2, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.D.; Ullrich, M.S.; Calvo, J.S.; Behnia, F.; Meloni, G.; Winkler, D.D. Mutations in superoxide dismutase 1 (SOD1) linked to familial amyotrophic lateral sclerosis can disrupt high-affinity zinc-binding promoted by the copper chaperone for SOD1 (CCS). Molecules 2020, 25, 1086. [Google Scholar] [CrossRef]
- Seetharaman, S.V.; Winkler, D.D.; Taylor, A.B.; Cao, X.; Whitson, L.J.; Doucette, P.A.; Valentine, J.S.; Schirf, V.; Demeler, B.; Carroll, M.C.; et al. Disrupted Zinc-Binding Sites in Structures of Pathogenic SOD1 Variants D124V and H80R. Biochemistry 2010, 49, 5714–5725. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Costarelli, L.; Giacconi, R.; Cipriano, C.; Muti, E.; Rink, L.; Malavolta, M. Zinc Homeostasis in Aging: Two Elusive Faces of the Same “Metal.”. Rejuvenation Res. 2006, 9, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Borth, W. α 2 “Macroglobulin, a multifunctional binding protein with targeting characteristics. FASEB J. 1992, 6, 3345–3353. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Li, Y.; Holmes, A.; Szafranski, K.; Faulkes, C.G.; Coen, C.W.; Buffenstein, R.; Platzer, M.; de Magalhães, J.P.; Church, G.M. RNA Sequencing Reveals Differential Expression of Mitochondrial and Oxidation Reduction Genes in the Long-Lived Naked Mole-Rat When Compared to Mice. PLoS ONE 2011, 6, e26729. [Google Scholar] [CrossRef]
- Lewis, K.N.; Mele, J.; Hornsby, P.J.; Buffenstein, R. Stress Resistance in the Naked Mole-Rat: The Bare Essentials—A Mini Review. Gerontology 2012, 58, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Walsh, M.; Bokov, A.; Ikeno, Y.; Jang, Y.C.; Perez, V.I.; Van Remmen, H.; Richardson, A. Liver specific expression of Cu/ZnSOD extends the lifespan of Sod1 null mice. Mech. Ageing Dev. 2016, 154, 1–8. [Google Scholar] [CrossRef]
- Kaeberlein, M. Longevity and aging. F1000Prime Rep. 2013, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.; Craig, T.; Alföldi, J.; Berlin, A.M.; Johnson, J.; Seluanov, A.; Gorbunova, V.; Di Palma, F.; Lindblad-Toh, K.; Church, G.M.; et al. The Naked Mole Rat Genome Resource: Facilitating analyses of cancer and longevity-related adaptations. Bioinformatics 2014, 30, 3558–3560. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Sierra, M.; Fernández, A.; Fraga, M. Epigenetics of Aging. Curr. Genom. 2015, 16, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.M.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.A.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Ke, Z.; Tombline, G.; Macoretta, N.; Hayes, K.; Tian, X.; Lv, R.; Ablaeva, J.; Gilbert, M.; Bhanu, N.V.; et al. Naked Mole Rat Cells Have a Stable Epigenome that Resists iPSC Reprogramming. Stem Cell Rep. 2017, 9, 1721–1734. [Google Scholar] [CrossRef]
- Seluanov, A.; Hine, C.; Azpurua, J.; Feigenson, M.; Bozzella, M.; Mao, Z.; Catania, K.C.; Gorbunova, V. Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc. Natl. Acad. Sci. USA 2009, 106, 19352–19357. [Google Scholar] [CrossRef]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.-F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.Z.; O’Brien, M.; et al. Inhibition of Pluripotency Networks by the Rb Tumor Suppressor Restricts Reprogramming and Tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; González-Ramírez, M.; Alcaine-Colet, A.; Aranda, S.; Di Croce, L. The Bivalent Genome: Characterization, Structure, and Regulation. Trends Genet. 2020, 36, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W.-W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukášová, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to γ-radiation. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tollefsbol, T.O. Age-related epigenetic drift and phenotypic plasticity loss: Implications in prevention of age-related human diseases. Epigenomics 2016, 8, 1637–1651. [Google Scholar] [CrossRef]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.-Y.; Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, A.R.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef]
- Buffenstein, R. Negligible senescence in the longest living rodent, the naked mole-rat: Insights from a successfully aging species. J. Comp. Physiol. B 2008, 178, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Jill Saffrey, M.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef]
- Banasiak, K.J.; Xia, Y.; Haddad, G.G. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol. 2000, 62, 215–249. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Park, K.K.; Luo, X.; Mooney, S.J.; Yungher, B.J.; Belin, S.; Wang, C.; Holmes, M.M.; He, Z. Retinal ganglion cell survival and axon regeneration after optic nerve injury in naked mole-rats. J. Comp. Neurol. 2017, 525, 380–388. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Salzer, J.L. Schwann Cell Myelination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020529. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R.; Lloyd, A.C. Schwann Cells: Development and Role in Nerve Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a020487. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Front. Cell. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Davis, C.M.; Gomez-Sanchez, J.A.; Turmaine, M.; Meijer, D.; Poli, V.; Mirsky, R.; Jessen, K.R. STAT3 Controls the Long-Term Survival and Phenotype of Repair Schwann Cells during Nerve Regeneration. J. Neurosci. 2017, 37, 4255–4269. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Andereggen, L.; Yuki, K.; Omura, K.; Yin, Y.; Gilbert, H.-Y.; Erdogan, B.; Asdourian, M.S.; Shrock, C.; de Lima, S.; et al. Mobile zinc increases rapidly in the retina after optic nerve injury and regulates ganglion cell survival and optic nerve regeneration. Proc. Natl. Acad. Sci. USA 2017, 114, E209–E218. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.A.; Adamson, D.C. Neuronal-Astrocyte Metabolic Interactions: Understanding the Transition into Abnormal Astrocytoma Metabolism. J. Neuropathol. Exp. Neurol. 2011, 70, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, Y.; Inagaki, Y.; Logan, S.; Bosnjak, Z.J.; Bai, X. Insufficient Astrocyte-Derived Brain-Derived Neurotrophic Factor Contributes to Propofol-Induced Neuron Death Through Akt/Glycogen Synthase Kinase 3β/Mitochondrial Fission Pathway. Anesth. Analg. 2017, 125, 241–254. [Google Scholar] [CrossRef]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front. Mol. Neurosci. 2017, 10, 88. [Google Scholar] [CrossRef]
- Park, T.J.; Reznick, J.; Peterson, B.L.; Blass, G.; Omerbašić, D.; Bennett, N.C.; Kuich, P.H.J.L.; Zasada, C.; Browe, B.M.; Hamann, W.; et al. Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science 2017, 356, 307–311. [Google Scholar] [CrossRef]
- Mason, S. Lactate Shuttles in Neuroenergetics—Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.-C.; Schuman, E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: Structure and function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Du, Z.; Chakrabarti, S.; Kulaberoglu, Y.; Smith, E.S.J.; Dobson, C.M.; Itzhaki, L.S.; Kumita, J.R. Probing the unfolded protein response in long-lived naked mole-rats. Biochem. Biophys. Res. Commun. 2020, 529, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.A.; Osmulski, P.A.; Pierce, A.; Weintraub, S.T.; Gaczynska, M.; Buffenstein, R. A cytosolic protein factor from the naked mole-rat activates proteasomes of other species and protects these from inhibition. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Srivastava, P. Heat-Shock Proteins. Curr. Protoc. Immunol. 2003, 58, A.1T.1–A.1T.6. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef]
- Azpurua, J.; Ke, Z.; Chen, I.X.; Zhang, Q.; Ermolenko, D.N.; Zhang, Z.D.; Gorbunova, V.; Seluanov, A. Naked mole-rat has increased translational fidelity compared with the mouse, as well as a unique 28S ribosomal RNA cleavage. Proc. Natl. Acad. Sci. USA 2013, 110, 17350–17355. [Google Scholar] [CrossRef]
- Von Der Haar, T.; Leadsham, J.E.; Sauvadet, A.; Tarrant, D.; Adam, I.S.; Saromi, K.; Laun, P.; Rinnerthaler, M.; Breitenbach-Koller, H.; Breitenbach, M.; et al. The control of translational accuracy is a determinant of healthy ageing in yeast. Open Biol. 2017, 7, 160291. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, L.M.; Tandoc, K.; Topisirovic, I.; Furic, L. Cross-talk between protein synthesis, energy metabolism and autophagy in cancer. Curr. Opin. Genet. Dev. 2018, 48, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Wang, S.; Yang, G.; Sun, X.; Zhao, S.; Lin, L.; Cheng, J.; Yang, W.; Cong, W.; Sun, W.; et al. HIF-1α contributes to hypoxia adaptation of the naked mole rat. Oncotarget 2017, 8, 109941–109951. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of Mammalian O 2 Homeostasis by Hypoxia-Inducible Factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.B.; Fang, X.; Fushan, A.A.; Huang, Z.; Lobanov, A.V.; Han, L.; Marino, S.M.; Sun, X.; Turanov, A.A.; Yang, P.; et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 2011, 479, 223–227. [Google Scholar] [CrossRef]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1 is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.A.; Edrey, Y.H.; Osmulski, P.; Gaczynska, M.; Buffenstein, R. Altered Composition of Liver Proteasome Assemblies Contributes to Enhanced Proteasome Activity in the Exceptionally Long-Lived Naked Mole-Rat. PLoS ONE 2012, 7, e35890. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.V.; Kim, E.R.; Ovchinnikov, L.P. Proteasome system of protein degradation and processing. Biochemistry 2009, 74, 1411–1442. [Google Scholar] [CrossRef]
- Petruseva, I.O.; Evdokimov, A.N.; Lavrik, O.I. Genome Stability Maintenance in Naked Mole-Rat. Acta Nat. 2017, 9, 31–41. [Google Scholar] [CrossRef]
- Triplett, J.C.; Tramutola, A.; Swomley, A.; Kirk, J.; Grimes, K.; Lewis, K.; Orr, M.; Rodriguez, K.; Cai, J.; Klein, J.B.; et al. Age-related changes in the proteostasis network in the brain of the naked mole-rat: Implications promoting healthy longevity. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2213–2224. [Google Scholar] [CrossRef]
- Correale, S.; de Paola, I.; Morgillo, C.M.; Federico, A.; Zaccaro, L.; Pallante, P.; Galeone, A.; Fusco, A.; Pedone, E.; Luque, F.J.; et al. Structural Model of the hUbA1-UbcH10 Quaternary Complex: In Silico and Experimental Analysis of the Protein-Protein Interactions between E1, E2 and Ubiquitin. PLoS ONE 2014, 9, e112082. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Long, M.J.C.; Wang, Y.; Zhang, S.; Aye, Y. Ube2V2 Is a Rosetta Stone Bridging Redox and Ubiquitin Codes, Coordinating DNA Damage Responses. ACS Cent. Sci. 2018, 4, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.K.; Brun, J.; Ramaekers, C.; Theys, J.; Weng, L.; Lambin, P.; Gray, D.A.; Wouters, B.G. Lysine 63-Polyubiquitination Guards against Translesion Synthesis–Induced Mutations. PLoS Genet. 2006, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Jariel-Encontre, I.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta Rev. Cancer 2008, 1786, 153–177. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.I.; Buffenstein, R.; Masamsetti, V.; Leonard, S.; Salmon, A.B.; Mele, J.; Andziak, B.; Yang, T.; Edrey, Y.; Friguet, B.; et al. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc. Natl. Acad. Sci. USA 2009, 106, 3059–3064. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Mallis, R.J. Aging and oxidation of reactive protein sulfhydryls. Exp. Gerontol. 2001, 36, 1519–1526. [Google Scholar] [CrossRef]
- Grune, T.; Shringarpure, R.; Sitte, N.; Davies, K. Age-Related Changes in Protein Oxidation and Proteolysis in Mammalian Cells. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, B459–B467. [Google Scholar] [CrossRef]
- Kawamura, Y.; Oka, K.; Takamori, M.; Sugiura, Y.; Oiwa, Y.; Fujioka, S.; Homma, S.; Miyawaki, S.; Narita, M.; Fukuda, T.; et al. Senescent cell death as an aging resistance mechanism in naked mole-rat. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kirby, A.M.; Fairman, G.D.; Pamenter, M.E. Atypical behavioural, metabolic and thermoregulatory responses to hypoxia in the naked mole rat (Heterocephalus glaber). J. Zool. 2018, 305, 106–115. [Google Scholar] [CrossRef]
- Zhao, Y.; Tyshkovskiy, A.; Muñoz-Espín, D.; Tian, X.; Serrano, M.; de Magalhaes, J.P.; Nevo, E.; Gladyshev, V.N.; Seluanov, A.; Gorbunova, V. Naked mole rats can undergo developmental, oncogene-induced and DNA damage-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2018, 115, 1801–1806. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boughey, H.; Jurga, M.; El-Khamisy, S.F. DNA Homeostasis and Senescence: Lessons from the Naked Mole Rat. Int. J. Mol. Sci. 2021, 22, 6011. https://doi.org/10.3390/ijms22116011
Boughey H, Jurga M, El-Khamisy SF. DNA Homeostasis and Senescence: Lessons from the Naked Mole Rat. International Journal of Molecular Sciences. 2021; 22(11):6011. https://doi.org/10.3390/ijms22116011
Chicago/Turabian StyleBoughey, Harvey, Mateusz Jurga, and Sherif F. El-Khamisy. 2021. "DNA Homeostasis and Senescence: Lessons from the Naked Mole Rat" International Journal of Molecular Sciences 22, no. 11: 6011. https://doi.org/10.3390/ijms22116011
APA StyleBoughey, H., Jurga, M., & El-Khamisy, S. F. (2021). DNA Homeostasis and Senescence: Lessons from the Naked Mole Rat. International Journal of Molecular Sciences, 22(11), 6011. https://doi.org/10.3390/ijms22116011