
Treatment with a GSK-3β/HDAC Dual Inhibitor Restores Neuronal Survival and Maturation in an In Vitro and In Vivo Model of CDKL5 Deficiency Disorder

 ,
,  ,
,  ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
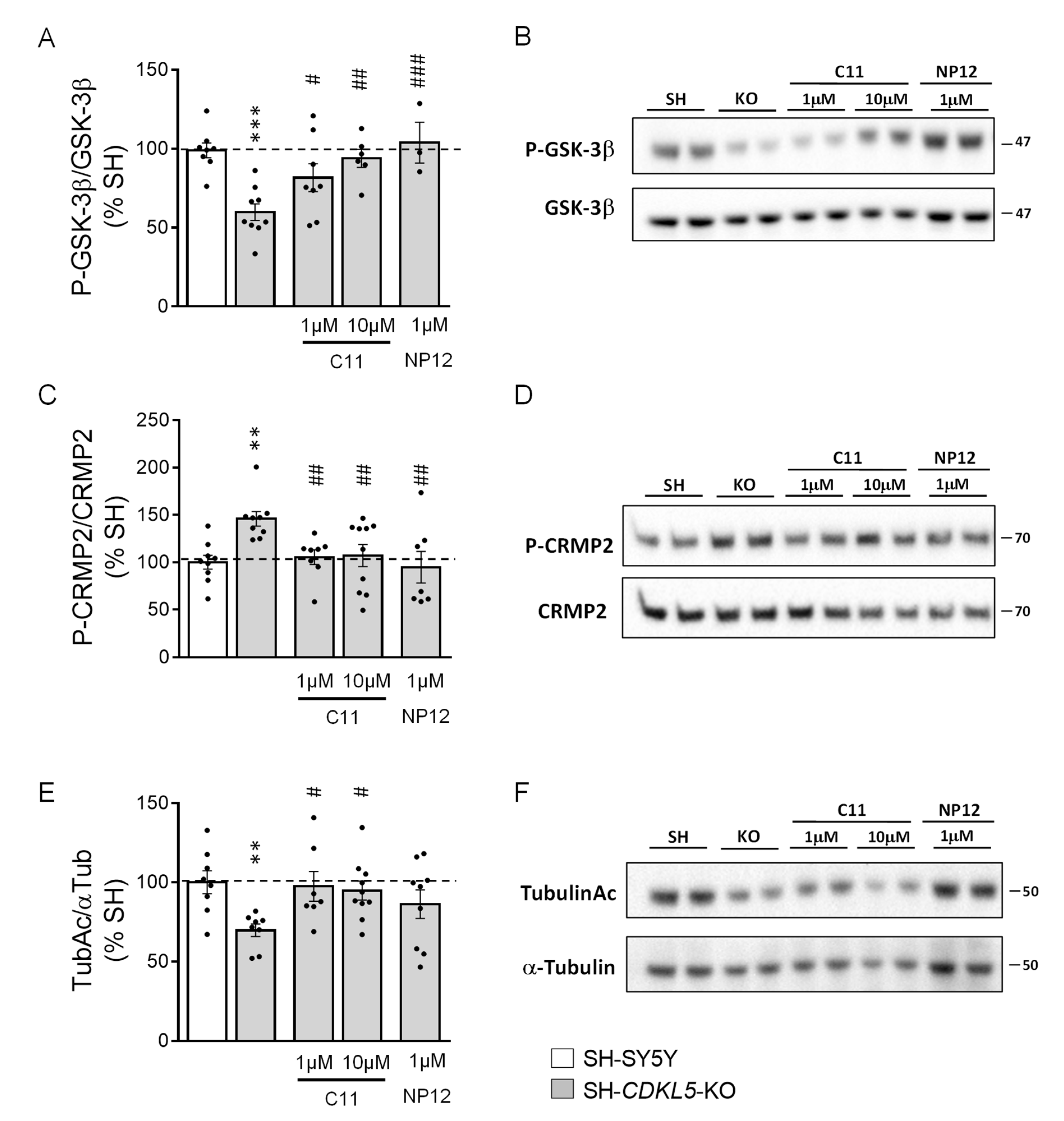
2.1. Treatment with C11 Restores GSK-3β and HDAC6 Activity in a Human Cellular Model of CDKL5 Deficiency
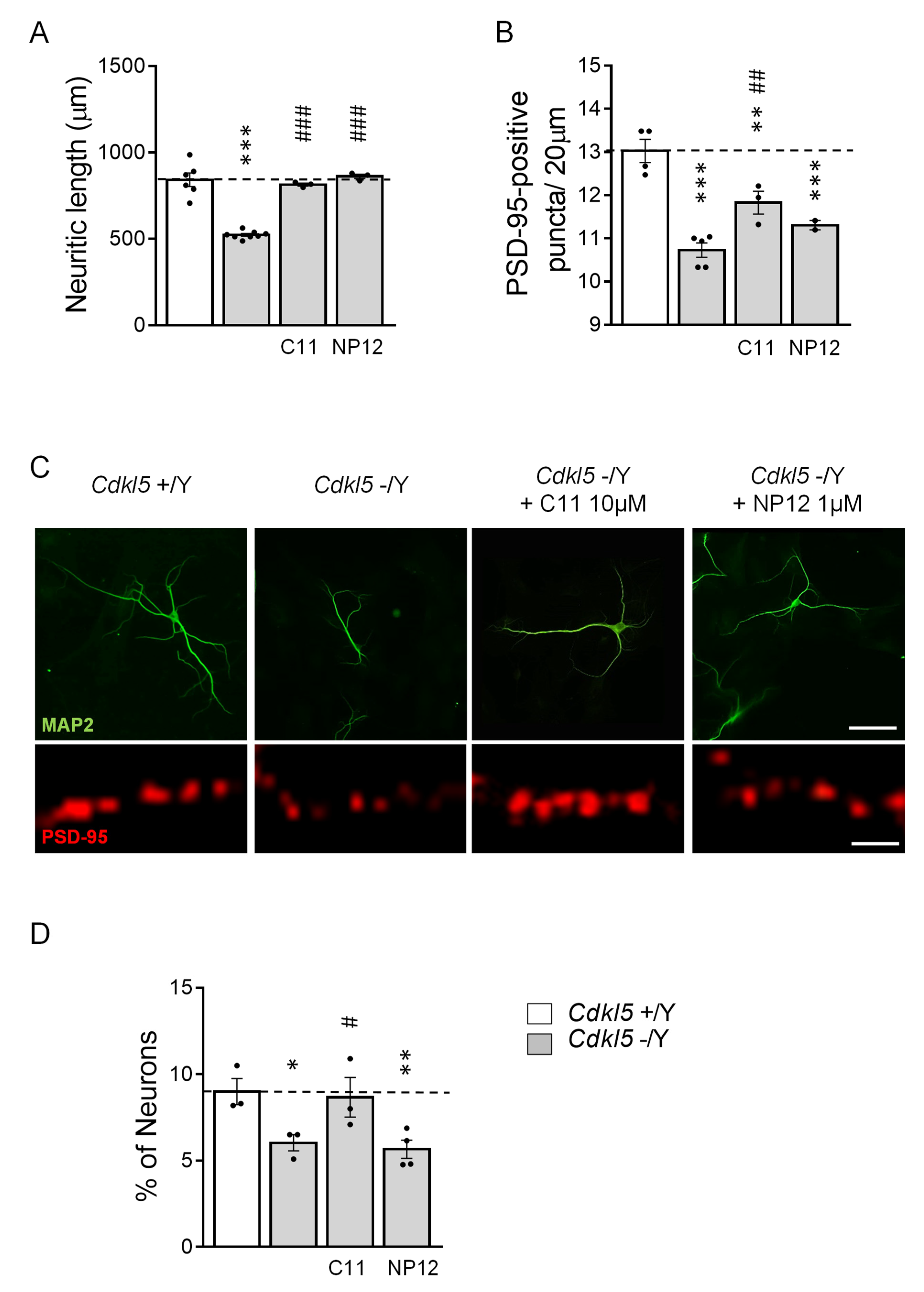
2.2. Treatment with C11 Restores Neuronal Maturation and Survival of a Human Cellular Model of CDKL5 Deficiency
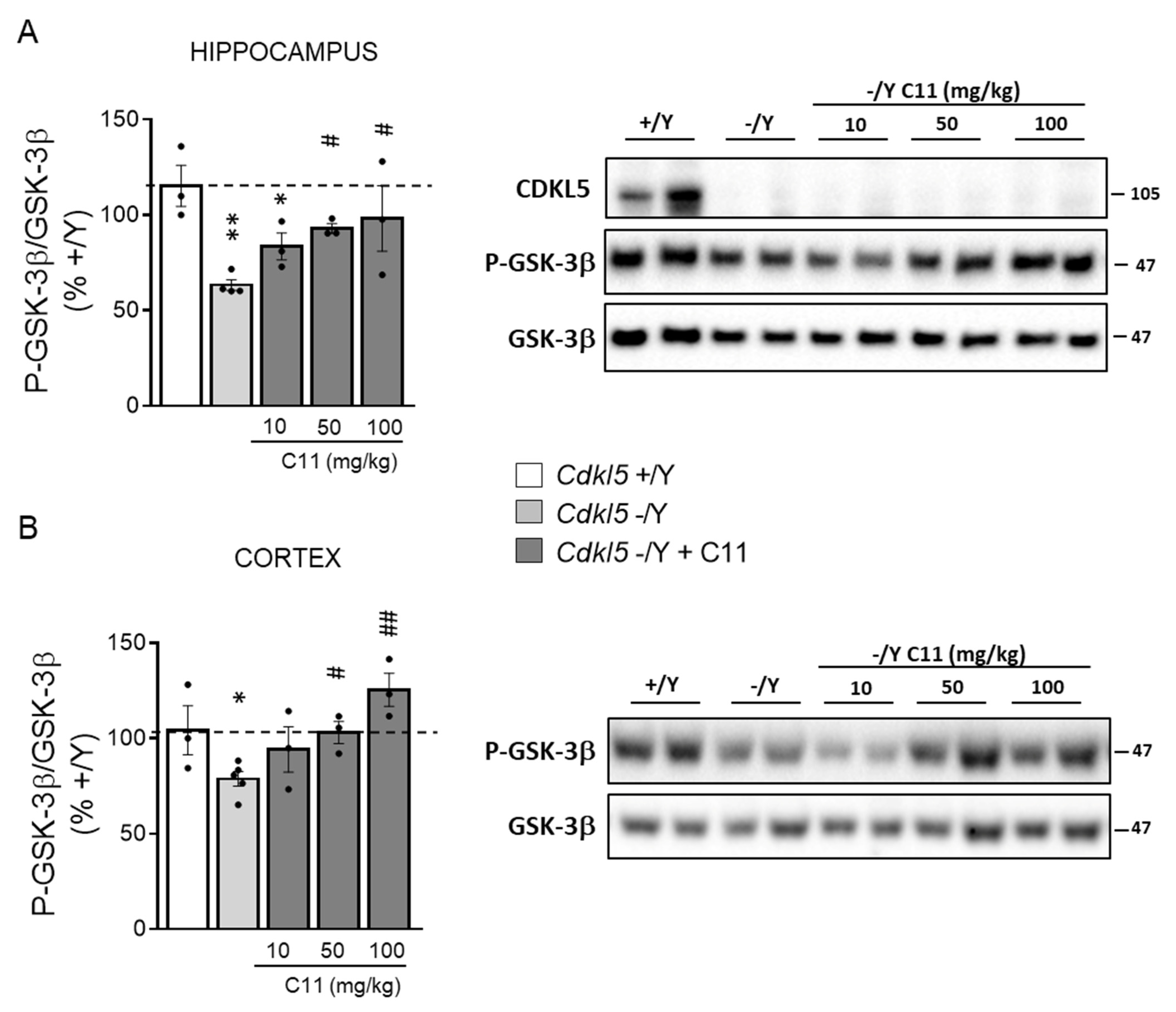
2.3. Treatment with C11 Restores Neuronal Maturation and Survival of Hippocampal Neurons from Cdkl5 KO Mice
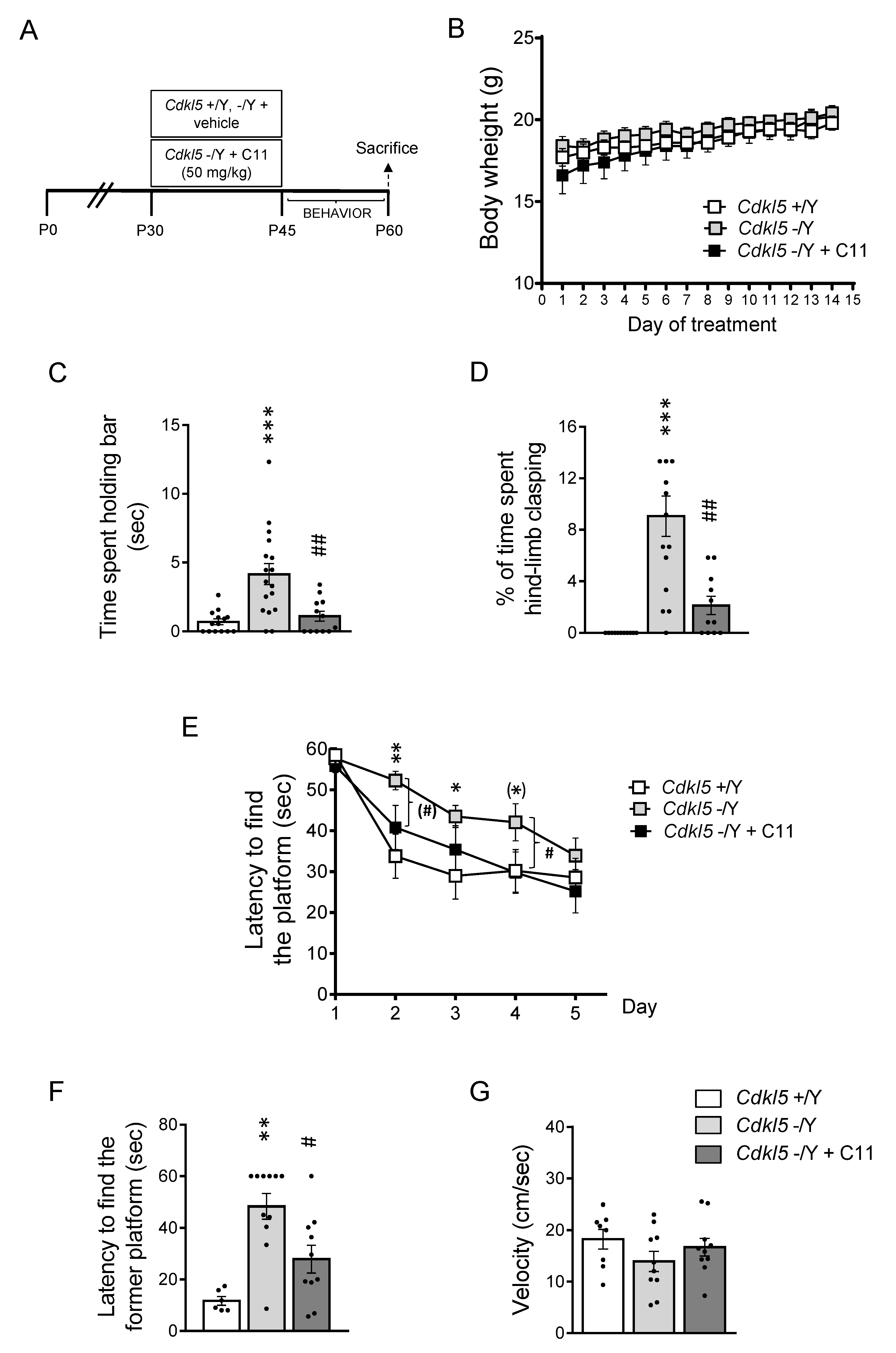
2.4. Effect of Treatment with C11 on Behaviors of Cdkl5 KO Mice
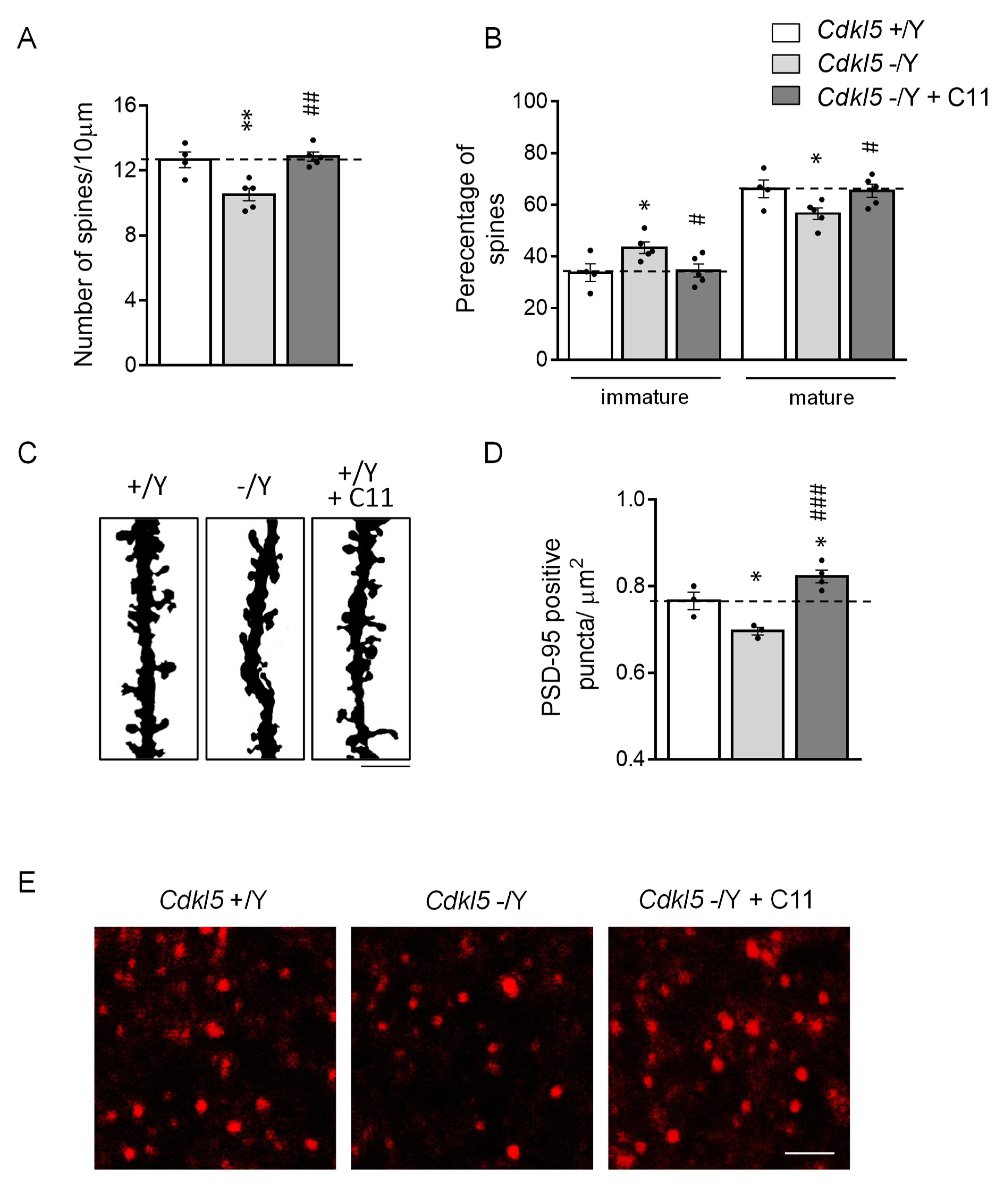
2.5. Effect of Treatment with C11 on Synapse Development in Cdkl5 KO Mice
2.6. Effect of Treatment with C11 on Neuronal Survival in Cdkl5 KO Mice
2.7. Effect of Treatment with C11 on Microglia Over-Activation in Cdkl5 KO Mice
3. Discussion
Conclusions
4. Materials and Methods
4.1. Compound 11 Synthesis
4.2. Cell Lines, Treatments and Measurements
4.2.1. C11 and NP-12 Treatments
4.2.2. Hydrogen Peroxide Treatment
4.2.3. Retinoic Acid Induced Differentiation
4.2.4. Analysis of Neurite Outgrowth
4.2.5. Apoptotic and Mitotic Index
4.3. Colony
4.4. Primary Hippocampal Cultures, Treatments and Measurements
4.4.1. Morphological Analyses
4.4.2. Neuronal Survival
4.5. In Vivo Treatment
4.5.1. Acute C11 Treatment
4.5.2. Chronic C11 Treatment
4.5.3. Chronic NP12 Treatment
4.6. Behavioral Assays
4.6.1. Morris Water Maze
4.6.2. Catalepsy Bar Test
4.6.3. Hind-Limb Clasping
4.7. Western Blot Analysis
4.8. Histological and Immunohistochemistry Procedures
4.8.1. NeuN and AIF-1 Immunohistochemistry and Measurements
4.8.2. PSD-95 Immunohistochemistry and Measurements
4.8.3. Spine Density and Morphology
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.J.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2012, 21, 266–273. [Google Scholar] [CrossRef]
- Demarest, S.; Pestana-Knight, E.M.; Olson, H.E.; Downs, J.; Marsh, E.D.; Kaufmann, W.E.; Partridge, C.-A.; Leonard, H.; Gwadry-Sridhar, F.; Frame, K.E.; et al. Severity Assessment in CDKL5 Deficiency Disorder. Pediatr. Neurol. 2019, 97, 38–42. [Google Scholar] [CrossRef]
- Demarest, S.T.; Olson, H.E.; Moss, A.; Pestana-Knight, E.; Zhang, X.; Parikh, S.; Swanson, L.C.; Riley, K.D.; Bazin, G.A.; Angione, K.; et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia 2019, 60, 1733–1742. [Google Scholar] [CrossRef]
- Mangatt, M.; Wong, K.; Anderson, B.; Epstein, A.; Hodgetts, S.; Leonard, H.; Downs, J. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J. Rare Dis. 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Kothur, K.; Holman, K.; Farnsworth, E.; Ho, G.; Lorentzos, M.; Troedson, C.; Gupta, S.; Webster, R.; Procopis, P.G.; Menezes, M.P.; et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 2018, 59, 132–140. [Google Scholar] [CrossRef]
- Hector, R.D.; Dando, O.; Landsberger, N.; Kilstrup-Nielsen, C.; Kind, P.C.; Bailey, M.E.S.; Cobb, S.R. Characterisation of CDKL5 Transcript Isoforms in Human and Mouse. PLoS ONE 2016, 11, e0157758. [Google Scholar] [CrossRef]
- Montini, E.; Andolfi, G.; Caruso, A.; Buchner, G.; Walpole, S.M.; Mariani, M.; Consalez, G.G.; Trump, D.; Ballabio, A.; Franco, B. Identification and Characterization of a Novel Serine–Threonine Kinase Gene from the Xp22 Region. Genomics 1998, 51, 427–433. [Google Scholar] [CrossRef]
- Nawaz, M.S.; Giarda, E.; Bedogni, F.; La Montanara, P.; Ricciardi, S.; Ciceri, D.; Alberio, T.; Landsberger, N.; Rusconi, L.; Kilstrup-Nielsen, C. CDKL5 and Shootin1 Interact and Concur in Regulating Neuronal Polarization. PLoS ONE 2016, 11, e0148634. [Google Scholar] [CrossRef]
- Barbiero, I.; Peroni, D.; Tramarin, M.; Chandola, C.; Rusconi, L.; Landsberger, N.; Kilstrup-Nielsen, C. The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5-IQGAP1 complex. Hum. Mol. Genet. 2017, 26, 3520–3530. [Google Scholar] [CrossRef]
- Fuchs, C.; Gennaccaro, L.; Ren, E.; Galvani, G.; Trazzi, S.; Medici, G.; Loi, M.; Conway, E.; Devinsky, O.; Rimondini, R.; et al. Pharmacotherapy with sertraline rescues brain development and behavior in a mouse model of CDKL5 deficiency disorder. Neuropharmacology 2020, 167, 107746. [Google Scholar] [CrossRef]
- Trazzi, S.; Fuchs, C.; Viggiano, R.; De Franceschi, M.; Valli, E.; Jedynak, P.; Hansen, F.K.; Perini, G.; Rimondini, R.; Kurz, T.; et al. HDAC4: A key factor underlying brain developmental alterations in CDKL5 disorder. Hum. Mol. Genet. 2016, 25, 3887–3907. [Google Scholar] [CrossRef]
- Trovò, L.; Fuchs, C.; De Rosa, R.; Barbiero, I.; Tramarin, M.; Ciani, E.; Rusconi, L.; Kilstrup-Nielsen, C. The green tea polyphenol epigallocatechin-3-gallate (EGCG) restores CDKL5-dependent synaptic defects in vitro and in vivo. Neurobiol. Dis. 2020, 138, 104791. [Google Scholar] [CrossRef]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D.; et al. Mapping Pathological Phenotypes in a Mouse Model of CDKL5 Disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef]
- Fuchs, C.; Gennaccaro, L.; Trazzi, S.; Bastianini, S.; Bettini, S.; Martire, V.L.; Ren, E.; Medici, G.; Zoccoli, G.; Rimondini, R.; et al. Heterozygous CDKL5 Knockout Female Mice Are a Valuable Animal Model for CDKL5 Disorder. Neural Plast. 2018, 2018, 1–18. [Google Scholar] [CrossRef]
- Lupori, L.; Sagona, G.; Fuchs, C.; Mazziotti, R.; Stefanov, A.; Putignano, E.; Napoli, D.; Strettoi, E.; Ciani, E.; Pizzorusso, T. Site-specific abnormalities in the visual system of a mouse model of CDKL5 deficiency disorder. Hum. Mol. Genet. 2019, 28, 2851–2861. [Google Scholar] [CrossRef]
- Ren, E.; Roncacé, V.; Trazzi, S.; Fuchs, C.; Medici, G.; Gennaccaro, L.; Loi, M.; Galvani, G.; Ye, K.; Rimondini, R.; et al. Functional and Structural Impairments in the Perirhinal Cortex of a Mouse Model of CDKL5 Deficiency Disorder Are Rescued by a TrkB Agonist. Front. Cell. Neurosci. 2019, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, G.; Putignano, E.; Chelini, G.; Melani, R.; Calcagno, E.; Ratto, G.M.; Amendola, E.; Gross, C.T.; Giustetto, M.; Pizzorusso, T. Dendritic Spine Instability in a Mouse Model of CDKL5 Disorder Is Rescued by Insulin-like Growth Factor 1. Biol. Psychiatry 2016, 80, 302–311. [Google Scholar] [CrossRef]
- Pizzo, R.; Gurgone, A.; Castroflorio, E.; Amendola, E.; Gross, C.; Sassoè-Pognetto, M.; Giustetto, M. Lack of Cdkl5 Disrupts the Organization of Excitatory and Inhibitory Synapses and Parvalbumin Interneurons in the Primary Visual Cortex. Front. Cell. Neurosci. 2016, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Trazzi, S.; Fuchs, C.; Galvani, G.; Medici, G.; Gennaccaro, L.; Tassinari, M.; Ciani, E. Increased DNA Damage and Apoptosis in CDKL5-Deficient Neurons. Mol. Neurobiol 2020, 57, 2244–2262. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Medici, G.; Trazzi, S.; Gennaccaro, L.; Galvani, G.; Berteotti, C.; Ren, E.; Loi, M.; Ciani, E. CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling. Brain Pathol. 2019, 29, 658–674. [Google Scholar] [CrossRef]
- Fuchs, C.; Trazzi, S.; Torricella, R.; Viggiano, R.; De Franceschi, M.; Amendola, E.; Gross, C.; Calza, L.; Bartesaghi, R.; Ciani, E. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef]
- Gennaccaro, L.; Fuchs, C.; Loi, M.; Roncacè, V.; Trazzi, S.; Ait-Bali, Y.; Galvani, G.; Berardi, A.C.; Medici, G.; Tassinari, M.; et al. A GABAB receptor antagonist rescues functional and structural impairments in the perirhinal cortex of a mouse model of CDKL5 deficiency disorder. Neurobiol. Dis. 2021, 153, 105304. [Google Scholar] [CrossRef]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef]
- Salcedo-Tello, P.; Ortiz-Matamoros, A.; Arias, C. GSK3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. Int. J. Alzheimer’s Dis. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Dorszewska, J.; Kozubski, W.; Waleszczyk, W.; Zabel, M.; Ong, K. Neuroplasticity in the Pathology of Neurodegenerative Diseases. Neural Plast. 2020, 2020, 1–2. [Google Scholar] [CrossRef]
- Fuchs, C.; Fustini, N.; Trazzi, S.; Gennaccaro, L.; Rimondini, R.; Ciani, E. Treatment with the GSK3-beta inhibitor Tideglusib improves hippocampal development and memory performance in juvenile, but not adult, Cdkl5 knockout mice. Eur. J. Neurosci. 2018, 47, 1054–1066. [Google Scholar] [CrossRef]
- Fuchs, C.; Rimondini, R.; Viggiano, R.; Trazzi, S.; De Franceschi, M.; Bartesaghi, R.; Ciani, E. Inhibition of GSK3beta rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol. Dis. 2015, 82, 298–310. [Google Scholar] [CrossRef]
- Sharma, S.; Taliyan, R. Synergistic effects of GSK-3beta and HDAC inhibitors in intracerebroventricular streptozotocin-induced cognitive deficits in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 337–349. [Google Scholar] [CrossRef]
- De Simone, A.; La Pietra, V.; Betari, N.; Petragnani, N.; Conte, M.; Daniele, S.; Pietrobono, D.; Martini, C.; Petralla, S.; Casadei, R.; et al. Discovery of the First-in-Class GSK-3beta/HDAC Dual Inhibitor as Disease-Modifying Agent To Combat Alzheimer’s Disease. ACS Med. Chem. Lett. 2019, 10, 469–474. [Google Scholar] [CrossRef]
- Rivieccio, M.A.; Brochier, C.; Willis, D.E.; Walker, B.A.; D’Annibale, M.A.; McLaughlin, K.; Siddiq, A.; Kozikowski, A.P.; Jaffrey, S.R.; Twiss, J.L.; et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 19599–19604. [Google Scholar] [CrossRef]
- Luo, J. The role of GSK3beta in the development of the central nervous system. Front. Biol. 2012, 7, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Luna-Medina, R.; Alonso-Gil, S.; Sanz-SanCristobal, M.; Palomo, V.; Gil, C.; Santos, A.; Martinez, A.; Perez-Castillo, A. Glycogen Synthase Kinase 3 Inhibition Promotes Adult Hippocampal Neurogenesis in Vitro and in Vivo. ACS Chem. Neurosci. 2012, 3, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131 Pt 10, 2647–2661. [Google Scholar] [CrossRef]
- Banasikowski, T.J.; Beninger, R.J. Haloperidol conditioned catalepsy in rats: A possible role for D1-like receptors. Int. J. Neuropsychopharmacol. 2011, 15, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, S.; Ungaro, F.; Hambrock, M.; Rademacher, N.; Stefanelli, G.; Brambilla, D.; Sessa, A.; Magagnotti, C.; Bachi, A.; Giarda, E.; et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 2012, 14, 911–923. [Google Scholar] [CrossRef]
- Taft, C.E.; Turrigiano, G.G. PSD-95 promotes the stabilization of young synaptic contacts. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130134. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; De Franceschi, M.; Fuchs, C.; Bastianini, S.; Viggiano, R.; Lupori, L.; Mazziotti, R.; Medici, G.; Martire, V.L.; Ren, E.; et al. CDKL5 protein substitution therapy rescues neurological phenotypes of a mouse model of CDKL5 disorder. Hum. Mol. Genet. 2018, 27, 1572–1592. [Google Scholar] [CrossRef]
- Gennaccaro, L.; Fuchs, C.; Loi, M.; Pizzo, R.; Alvente, S.; Berteotti, C.; Lupori, L.; Sagona, G.; Galvani, G.; Gurgone, A.; et al. Age-Related Cognitive and Motor Decline in a Mouse Model of CDKL5 Deficiency Disorder is Associated with Increased Neuronal Senescence and Death. Aging Dis. 2021, 12, 764–785. [Google Scholar] [CrossRef]
- Galvani, G.; Mottolese, N.; Gennaccaro, L.; Loi, M.; Medici, G.; Tassinari, M.; Fuchs, C.; Ciani, E.; Trazzi, S. Inhibition of Microglia Over-activation Restores Neuronal Survival in a Mouse Model of CDKL5 Deficent Disorder. J. Neuroinflamm. 2021, preprints. [Google Scholar]
- Torres-Platas, S.G.; Comeau, S.; Rachalski, A.; Bo, G.D.; Cruceanu, C.; Turecki, G.; Giros, B.; Mechawar, N. Morphometric characterization of microglial phenotypes in human cerebral cortex. J. Neuroinflamm. 2014, 11, 12. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.-L.; Chua, K.; et al. Mice Lacking Histone Deacetylase 6 Have Hyperacetylated Tubulin but Are Viable and Develop Normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2012, 280, 775–793. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Makarenkova, H.; Edelman, D.B. HDAC6 Regulates Mitochondrial Transport in Hippocampal Neurons. PLoS ONE 2010, 5, e10848. [Google Scholar] [CrossRef] [PubMed]
- Mazanetz, M.P.; Fischer, P. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007, 6, 464–479. [Google Scholar] [CrossRef]
- Lovestone, S.; Killick, R.; Di Forti, M.; Murray, R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007, 30, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Roh, M.S. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr. Drug Targets 2006, 7, 1421–1434. [Google Scholar] [CrossRef]
- D’Mello, S.R. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009, 22, 513–524. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef]
- Sleiman, S.F.; Basso, M.; Mahishi, L.; Kozikowski, A.P.; Donohoe, M.E.; Langley, B.; Ratan, R.R. Putting the ‘HAT’ back on survival signalling: The promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin. Investig. Drugs 2009, 18, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A. Small-molecule Modulation of HDAC6 Activity: The Propitious Therapeutic Strategy to Vanquish Neurodegenerative Disorders. Curr. Med. Chem. 2017, 24, 4104–4120. [Google Scholar] [CrossRef]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An Acetylation Site in the Middle Domain of Hsp90 Regulates Chaperone Function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Takaki, E.; Hayashi, T.; Kitaura, Y.; Tanaka, Y.; Inouye, S.; Nakai, A. Active HSF1 Significantly Suppresses Polyglutamine Aggregate Formation in Cellular and Mouse Models. J. Biol. Chem. 2005, 280, 34908–34916. [Google Scholar] [CrossRef]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Green, H.F.; Nolan, Y.M. GSK-3 mediates the release of IL-1beta, TNF-alpha and IL-10 from cortical glia. Neurochem. Int. 2012, 61, 666–671. [Google Scholar] [CrossRef]
- Zain, Z.M.; Vidyadaran, S.; Hassan, M. GSK3 Inhibition Reduces Inflammatory Responses of Microglia and Upregulates Il-10 Production. Mal. J. Med. Health Sci. 2017, 13, 8. [Google Scholar]
- Suuronen, T.; Huuskonen, J.; Pihlaja, R.; Kyrylenko, S.; Salminen, A. Regulation of microglial inflammatory response by histone deacetylase inhibitors. J. Neurochem. 2003, 87, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Kannan, V.; Brouwer, N.; Hanisch, U.-K.; Regen, T.; Eggen, B.J.; Boddeke, H.W. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J. Neurosci. Res. 2013, 91, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Schluesener, H.J. Oral Administration of Histone Deacetylase Inhibitor MS-275 Ameliorates Neuroinflammation and Cerebral Amyloidosis and Improves Behavior in a Mouse Model. J. Neuropathol. Exp. Neurol. 2013, 72, 178–185. [Google Scholar] [CrossRef]
- Shein, N.A.; Shohami, E. Histone Deacetylase Inhibitors as Therapeutic Agents for Acute Central Nervous System Injuries. Mol. Med. 2011, 17, 448–456. [Google Scholar] [CrossRef]
- Bruserud, O.; Stapnes, C.; Ersvaer, E.; Gjertsen, B.T.; Ryningen, A. Histone deacetylase inhibitors in cancer treatment: A review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr. Pharm. Biotechnol. 2007, 8, 388–400. [Google Scholar] [CrossRef]
- Beaudoin, G.M., 3rd; Lee, S.H.; Singh, D.; Yuan, Y.; Ng, Y.G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef]
- Finlay, B.L.; Darlington, R.B. Linked regularities in the development and evolution of mammalian brains. Science 1995, 268, 1578–1584. [Google Scholar] [CrossRef]
- Zhou, A.; Lin, K.; Zhang, S.; Chen, Y.; Zhang, N.; Xue, J.; Wang, Z.; Aldape, K.D.; Xie, K.; Woodgett, J.R.; et al. Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 2016, 18, 954–966. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Grubbs, F.E. Procedures for Detecting Outlying Observations in Samples. Technometrics 1969, 11, 21. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loi, M.; Gennaccaro, L.; Fuchs, C.; Trazzi, S.; Medici, G.; Galvani, G.; Mottolese, N.; Tassinari, M.; Rimondini Giorgini, R.; Milelli, A.; et al. Treatment with a GSK-3β/HDAC Dual Inhibitor Restores Neuronal Survival and Maturation in an In Vitro and In Vivo Model of CDKL5 Deficiency Disorder. Int. J. Mol. Sci. 2021, 22, 5950. https://doi.org/10.3390/ijms22115950
Loi M, Gennaccaro L, Fuchs C, Trazzi S, Medici G, Galvani G, Mottolese N, Tassinari M, Rimondini Giorgini R, Milelli A, et al. Treatment with a GSK-3β/HDAC Dual Inhibitor Restores Neuronal Survival and Maturation in an In Vitro and In Vivo Model of CDKL5 Deficiency Disorder. International Journal of Molecular Sciences. 2021; 22(11):5950. https://doi.org/10.3390/ijms22115950
Chicago/Turabian StyleLoi, Manuela, Laura Gennaccaro, Claudia Fuchs, Stefania Trazzi, Giorgio Medici, Giuseppe Galvani, Nicola Mottolese, Marianna Tassinari, Roberto Rimondini Giorgini, Andrea Milelli, and et al. 2021. "Treatment with a GSK-3β/HDAC Dual Inhibitor Restores Neuronal Survival and Maturation in an In Vitro and In Vivo Model of CDKL5 Deficiency Disorder" International Journal of Molecular Sciences 22, no. 11: 5950. https://doi.org/10.3390/ijms22115950
APA StyleLoi, M., Gennaccaro, L., Fuchs, C., Trazzi, S., Medici, G., Galvani, G., Mottolese, N., Tassinari, M., Rimondini Giorgini, R., Milelli, A., & Ciani, E. (2021). Treatment with a GSK-3β/HDAC Dual Inhibitor Restores Neuronal Survival and Maturation in an In Vitro and In Vivo Model of CDKL5 Deficiency Disorder. International Journal of Molecular Sciences, 22(11), 5950. https://doi.org/10.3390/ijms22115950