
Interfacial Modeling of Fibrinogen Adsorption onto LiNbO3 Single Crystal–Single Domain Surfaces

 ,
, 
Abstract
1. Introduction
2. Results and Discussion
2.1. Experimental Results
2.2. Simulation Results and Discussion
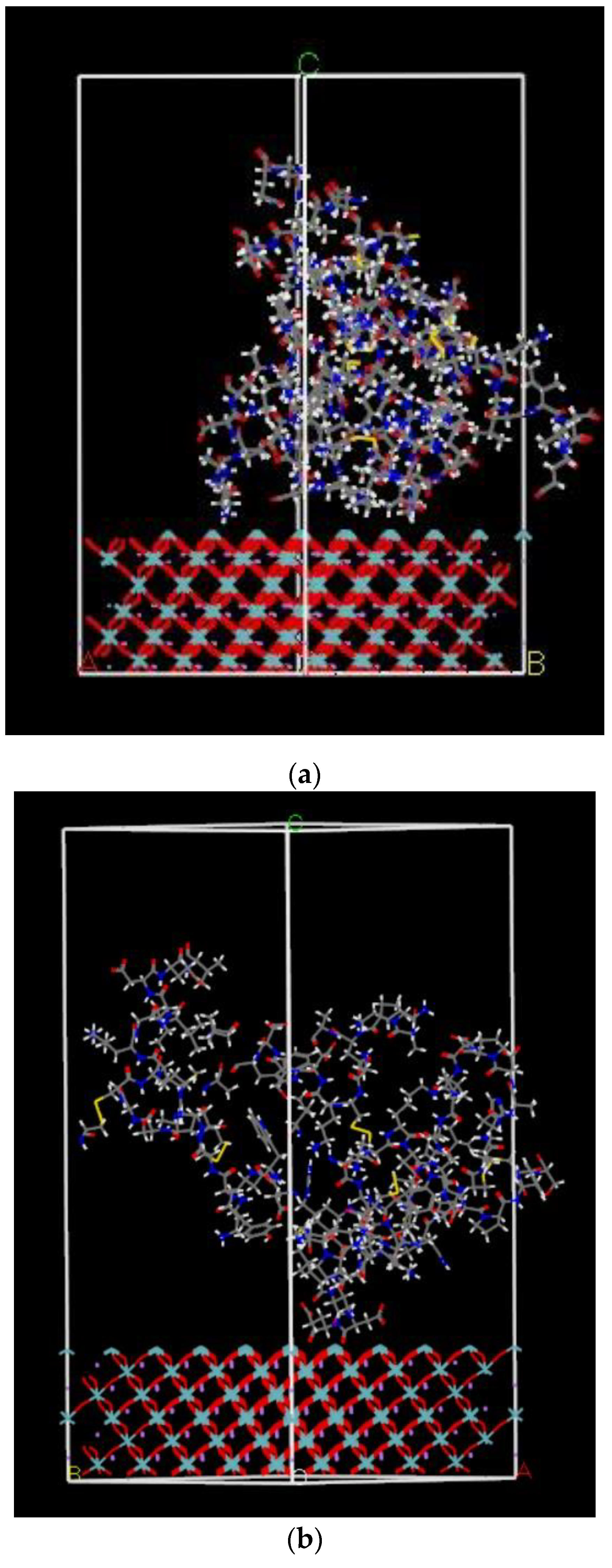
2.2.1. Charge Distribution Calculation for the FBG Protein Structure
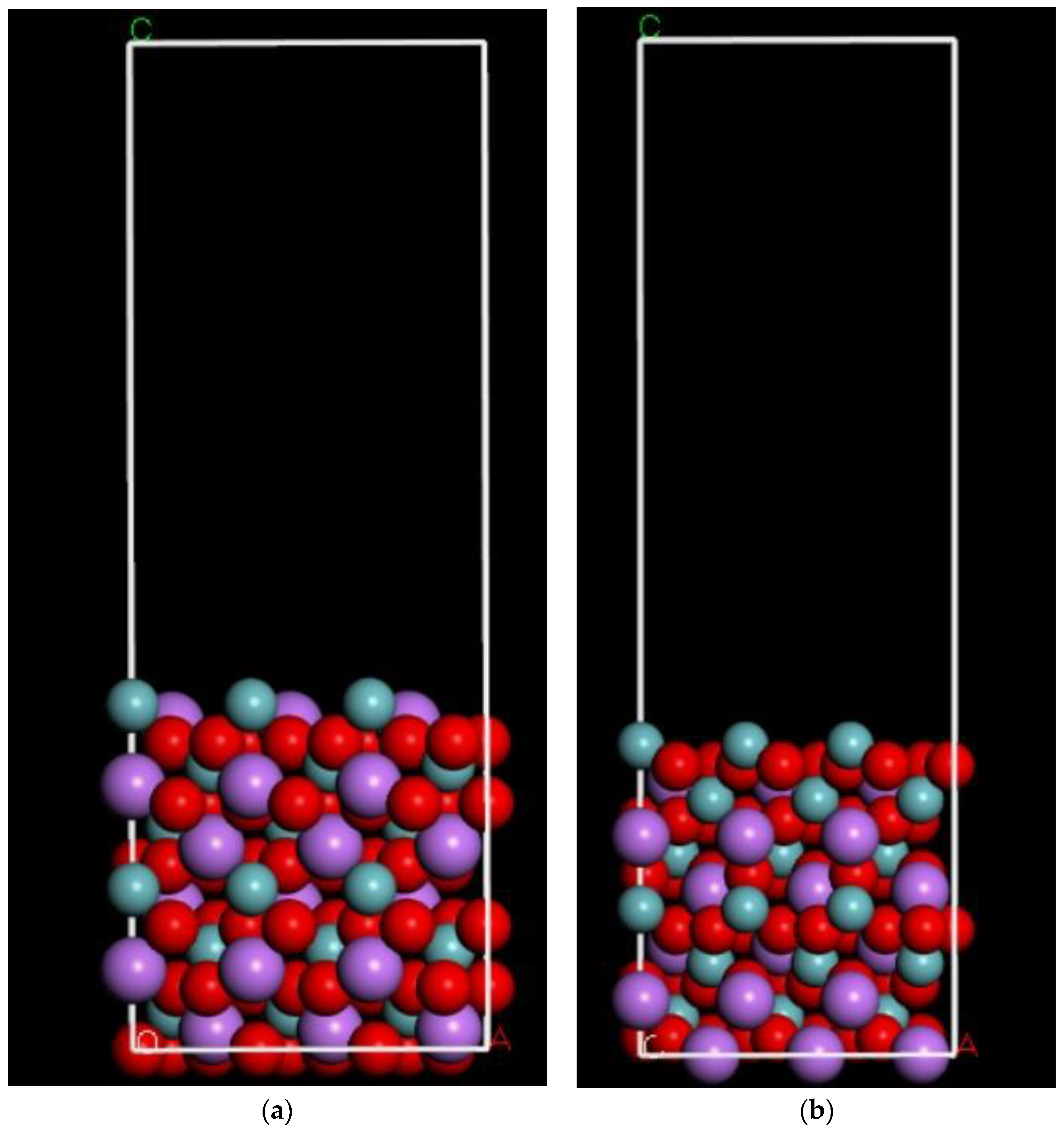
2.2.2. Facet Area Using a Monte Carlo Method to Find the Best Surface Conducive to Crystal Growth
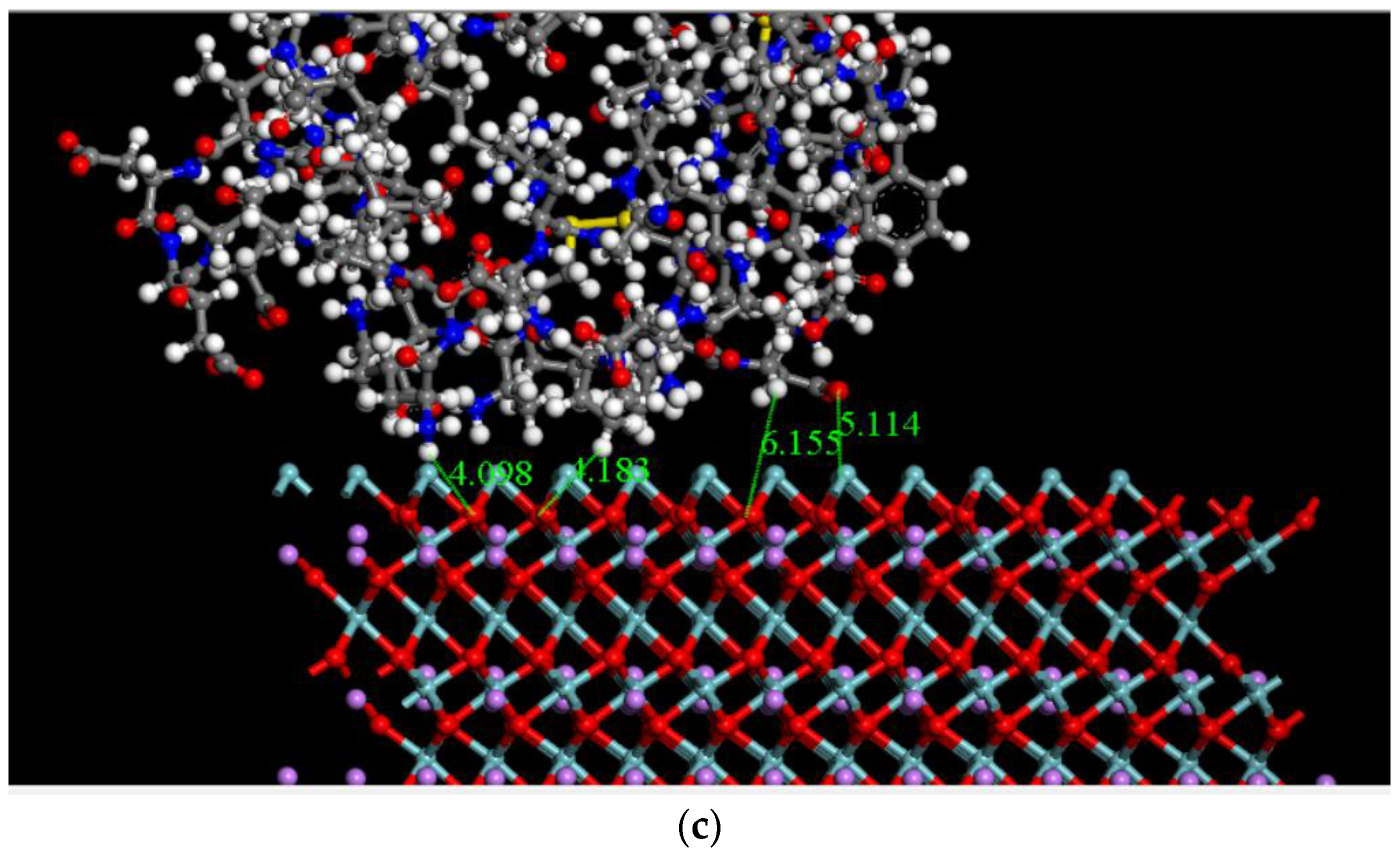
2.2.3. Calculated Adsorption Energy of FBG Fragment on LNO Surface
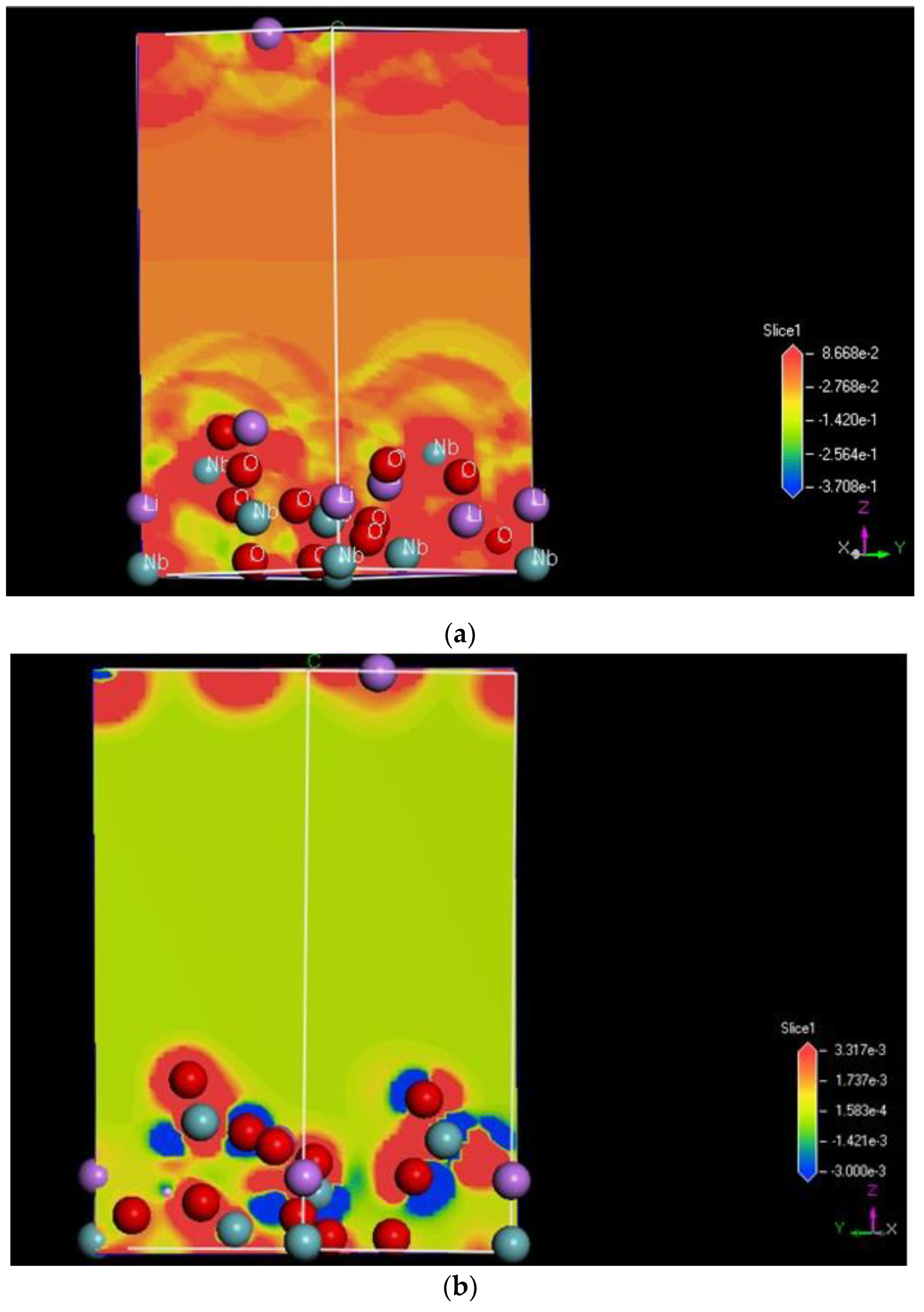
2.2.4. Surface Electrostatic Potential and Charge Calculation Using the DFT Method
2.3. Dynamics Calculation Is Performed Using Molecular Mechanics Method with Water Molecules
3. Experimental Section
3.1. Material Preparation and Interfacial Analysis
3.2. FBG Adsorption
4. Molecular Model and Simulation Methods
4.1. Molecular Model
4.1.1. LNO Surface Simulation
4.1.2. Protein Model
4.2. Simulation Methods
4.2.1. Surface Electrostatic Potential Calculation
4.2.2. Morphology Method
4.2.3. Adsorption Calculation
4.2.4. Amorphous Packing
4.2.5. Molecular Dynamics Method
4.2.6. Density Functional Theory Method
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rocha-Gaso, M.I.; March-Iborra, C.; Montoya-Baides, A.; Arnau-Vives, A. Surface Generated Acoustic Wave Biosensors for the Detection of Pathogens: A Review. Sensors 2009, 9, 5740–5769. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, M.; Zhao, G. Point-of-Care Assessment of Hemostasis with a Love-Mode Surface Acoustic Wave Sensor. ACS Sensors 2020, 5, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Defaÿ, E. Integration of Ferroelectric and Piezoelectric Thin Films: Concepts and Applications for Microsystems, 1st ed.; Wiley-ISTE: London, UK, 2011. [Google Scholar]
- Kästner, J.; Thiel, W. Bridging the gap between thermodynamic integration and umbrella sampling provides a novel analysis method: Umbrella integration. J. Chem. Phys. 2005, 123, 144104. [Google Scholar] [CrossRef]
- Geada, I.L.; Ramezani-Dekhel, H.; Jamil, T.; Sulpizi, M.; Heinz, H. Insight into induced charges at metal surfaces and biointerfaces using a polarizable Lennard–Jones potential. Nat. Commun. 2018, 9, 716. [Google Scholar] [CrossRef]
- Yermolenko, I.S.; Lishko, V.K.; Ugarova, T.P.; Magonov, S.N. High-Resolution Visualization of Fibrinogen Molecules and Fibrin Fibers with Atomic Force Microscopy. Biomacromolecules 2011, 12, 370–379. [Google Scholar] [CrossRef]
- Hall, C.E.; Slayter, H.S. The fibrinogen molecule: Its size, shape, and mode of polymerization. J. Cell Biol. 1959, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Friedt, J.M.; Frederix, F.A.; Campitelli, A.; Borghs, G. Simultaneous atomic force microscope and quartz crystal microbalance measurements: Investigation of human plasma fibrinogen adsorption. Appl. Phys. Lett. 2002, 81, 1335. [Google Scholar] [CrossRef]
- Kasemo, B. Biological surface science. Surf. Sci. 2002, 500, 656–677. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Simurda, T.; Brunclikova, M.; Asselta, R.; Caccia, S.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Genetic Variants in the FGB and FGG Genes Map-ping in the Beta and Gamma Nodules of the Fibrinogen Molecule in Congenital Quantitative Fibrinogen Disorders Associated with a Thrombotic Phenotype. Int. J. Mol. Sci. 2020, 21, 4616. [Google Scholar] [CrossRef]
- Höök, F.J.; Voros, J.; Rodahl, M.; Kurrat, R.; Böni, P.J.; Ramsden, J.; Textor, M.; Spencer, N.D.; Tengvall, P.; Gold, J.; et al. A comparative study of protein adsorption on titanium oxide surfaces using in situ elipsometry, optical waveguide light mode spectroscopy, and quartz crystal microbal-ance/dissipation. Colloids Surf. B Biointerfaces 2002, 24, 155. [Google Scholar] [CrossRef]
- Hemmersam, A.G.; Foss, F.; Chevallier, J.; Besenbacher, F. Adsorption of fibrinogen on tantalum oxide, titanium oxide and gold studied by the QCM-D technique. Colloids Surf. B Biointerfaces 2005, 43, 208. [Google Scholar] [CrossRef] [PubMed]
- Monkawa, A.; Ikoma, T.; Yunoki, S.; Yoshioka, T.; Tanaka, J.; Chakarov, D.; Kasemo, B. Fabrication of hydroxyapatite ultra-thin layer on gold surface and its application for quartz crystal microbalance technique. Biomaterials 2006, 27, 5748. [Google Scholar] [CrossRef]
- Ikoma, T.; Tagaya, M.; Hanagata, N.; Yoshioka, T.; Chakarov, D.; Kasemo, B.; Tanaka, J. Protein adsorption on hydroxyapatite nanosensors with different crystal sizes studied in situ by quartz crystal microbalance with dissipation method. J. Am. Ceram. Soc. 2009, 92, 1125. [Google Scholar] [CrossRef]
- Yoshioka, T.; Ikoma, T.; Monkawa, A.; Tonegawa, T.; Chakarov, D.; Kasemo, B.; Hanagata, N.; Tanaka, M. Proteins adsorption on hydroxyapatite nano-crystals with quartz crystal microbalance technique. Key Eng. Mater. 2008, 361, 1119–1122. [Google Scholar]
- Kharazian, B.; Lohse, S.E.; Ghasemi, F.; Raoufi, M.; Saei, A.A.; Hashemi, F.; Farvadi, F.; Alimohamadi, R.; Jalali, S.A.; Shokrgozar, M.A.; et al. Bare surface of gold nanoparticle induces inflammation through unfolding of plasma fibrinogen. Sci. Rep. 2018, 8, 12557. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.; Grilli, S.; Finizio, A.; Merola, F.; Coppola, S.; Vespini, V. 3D lithography by freezing unstable liquid shapes. SPIE Newsroom 2012. [Google Scholar] [CrossRef]
- Falk, G.; Borlaf, M.; Bendo, T.; de Oliveira, A.P.N.; Neto, J.B.R.; Moreno, R. Colloidal Sol–Gel Synthesis and Photocatalytic Activity of Nanoparticulate Nb2O5 Sols. J. Am. Ceram. Soc. 2016, 99, 1968–1973. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta B 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Marucco, A.; Fenoglio, I.; Turci, F.; Fubini, B. Interaction of fibrinogen and albumin with titanium dioxide nanoparticles of different crystalline phases. J. Phys. Conf. Ser. 2013, 429, 012014. [Google Scholar] [CrossRef]
- Roach, P.; Farrar, D.; Perry, C. Interpretation of protein adsorption: Surface-induced conformational changes. J. Am. Chem. Soc. 2005, 127, 8168. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Dobortova, M.; Skornova, I.; Brunclíkova, M.; Grendar, M.; Lasabova, Z.; Stasko, J.; et al. Comparison of clinical phenotype with genetic and laboratory results in 31 patients with congenital dysfibrinogenemia in northern Slovakia. Int. J. Hematol. 2020, 111, 795. [Google Scholar] [CrossRef]
- Ke, C.; Wang, X.; Hu, X.P.; Zhu, S.N.; Qi, M. Nanoparticle decoration of ferroelectric domain patterns in LiNbO3 crystal. Appl. Phys. 2007, 101, 064107. [Google Scholar] [CrossRef]
- Li, Y.; Golding, G.B.; Ilie, L. DELPHI: Accurate deep ensemble model for protein interaction sites prediction. Bioiformatics 2021, 37, 896. [Google Scholar] [CrossRef] [PubMed]
- Ferjani, H. Structural, Hirshfeld Surface Analysis, Morphological Approach, and Spectroscopic Study of New Hybrid Iodobismuthate Containing Tetranuclear 0D Cluster Bi4I16·4(C6H9N2) 2(H2O). Crystals 2020, 10, 397. [Google Scholar] [CrossRef]
- Cordero-Edwards, K. Water Affinity and Surface Charging at the z-Cut and y-Cut LNO Surfaces: An Ambient Pressure X-ray Photoelectron Spectroscopy Study. J. Phys. Chem. C 2016, 120, 24048–24055. [Google Scholar] [CrossRef]
- Dubrovin, E.V.; Barinov, N.A.; Schäffer, T.E. In Situ Single-Molecule AFM Investigation of Surface-Induced Fibrinogen Unfolding on Graphite. Langmuir 2019, 35, 9732–9739. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Vilar, R.; Zolkova, J.; Ceznerova, E.; Kolkova, Z. A Novel Nonsense Mutation in FGB (c. 1421G> A; p. Trp474Ter) in the Beta Chain of Fibrinogen Causing Hypofibrinogenemia with Bleeding Phenotype. Biomedicines 2020, 8, 605. [Google Scholar] [CrossRef]
- Nicholls, A.; Honig, B. A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson–Boltzmann equation. J. Comput. Chem. 1991, 12, 435–445. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Bravais, A. Etudes Crystallographiques; Academie Des Sciences: Paris, France, 1913. [Google Scholar]
- Friedel, G. Études sur la loi de Bravais. Bull. Soc. Minéralogie 1907, 30, 326–332. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1111. [Google Scholar] [CrossRef]
- Cerný, V. A thermodynamical approach to the travelling salesman problem: An efficient simulation algorithm. J. Optim. Theor. Appl. 1985, 45, 41–51. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10032. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 1987. [Google Scholar]
- Donnay, J.D.H.; Harker, D. A new law of crystal morphology extending the Law of Bravais. Am. Miner. 1937, 22, 446–467. [Google Scholar]
- Abe, A.; Jernigan, R.L.; Flory, P.J. Conformational Energies of n-Alkanes and the Random Configuration of Higher Homologs Including Polymethylene. J. Am. Chem. Soc. 1966, 88, 631–639. [Google Scholar] [CrossRef]
- Verlet, L. Computer experiments on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Samoletov, A.A.; Dettmann, C.P.; Chaplain, M.A.J. Thermostats for “slow” configurational modes. J. Stat. Phys. 2007, 128, 1321–1336. [Google Scholar] [CrossRef]
- Leimkuhler, B.; Noorizadeh, E.; Penrose, O. Comparing the efficiencies of stochastic isothermal molecular dynamics methods. J. Stat. Phys. 2011, 143, 921–942. [Google Scholar] [CrossRef]
- Baerends, E.J.; Delley, B.; Ellis, D.E.; Freeman, A.J.; Post, D. Binding energy and electronic structure of small copper particles. Phys. Rev. B Condens. Matter. 1983, 27, 2132–2144. [Google Scholar]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Delley, B. Analytical energy derivatives in the numerical local density functional approach. J. Chem. Phys. 1991, 94, 7245–7250. [Google Scholar] [CrossRef]
- Pulay, P. Improved SCF convergence acceleration. J. Comput. Chem. 1982, 3, 556–560. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| h k l | Multiplicity | D hkl (Å) | % Total Facet Area |
|---|---|---|---|
| 0 0 1 | 1 | 2.3105 | 49.5433 |
| 0 0 −1 | 1 | 2.3105 | 49.5433 |
| −1 1 −2 | 3 | 3.7498 | 0 |
| 1 −1 2 | 3 | 3.7498 | 0 |
| −3 2 1 | 6 | 1.6728 | 0.456671 |
| −3 2 2 | 6 | 1.6374 | 0.456671 |
| Fragment Nature | Adsorption Energies (kcal/mol) | Average Adsorption Energy/Per Atom for Protein Fragment (kcal/mol) |
|---|---|---|
| Positive | −906.3 | −4.89 (185) |
| Negative | −718.1 | −4.10 (175) |
| Fragment Nature | Adsorption Energies (kcal/mol) | Average Adsorption Energy/Per Atom of Protein Fragment (kcal/mol) |
|---|---|---|
| Positive | −713.2 | −3.85 (185) |
| Negative | −856.1 | −4.89 (175) |
| System | Total Energy (kcal/mol) | Average Binding Energy of Protein Fragment ΔE (kcal/mol) |
|---|---|---|
| Blank (001) surface | −13,428.29 | |
| Blank (00−1) surface | −13,425.67 | |
| Positive Protein Fragment | −10,651.34 | |
| Negative Protein fragment | −10,548.28 | |
| Positive fragment + (001) | −24,085.78 | −6.15 |
| Negative fragment + (001) | −23,980.18 | −3.61 |
| Positive fragment + (00−1) | −24,080.34 | −3.33 |
| Negative fragment + (00−1) | −23,978.23 | −4.28 |
| System | Total Energy (kcal/mol) | Average Binding Energy of Protein Fragment ΔE (kcal/mol) |
|---|---|---|
| Blank (001) surface | −13,428.29 | |
| Blank (00−1) surface | −13,425.67 | |
| Positive Protein Fragment | −10,651.34 | |
| Negative Protein fragment | −10,548.28 | |
| 1701 Water Molecules | −85,896.09 | |
| Positive fragment + (001) | −109,993.21 | −17.49 |
| Negative fragment + (001) | −109,879.38 | −6.72 |
| Positive fragment + (00−1) | −109,980.21 | −7.11 |
| Negative fragment + (00−1) | −109,878.12 | −8.08 |
| System | Diffusion Coefficient A^2/ps | R^2 |
|---|---|---|
| Protein Fragment with Nb rich surface (00−1) | 3.634 × 10−6 | 0.9972 |
| Protein Fragment with Nb poor surface (001) | 2.284 × 10−6 | 0.9965 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cross, J.S.; Kubota, Y.; Chatterjee, A.; Unni, S.; Ikoma, T.; Tagaya, M. Interfacial Modeling of Fibrinogen Adsorption onto LiNbO3 Single Crystal–Single Domain Surfaces. Int. J. Mol. Sci. 2021, 22, 5946. https://doi.org/10.3390/ijms22115946
Cross JS, Kubota Y, Chatterjee A, Unni S, Ikoma T, Tagaya M. Interfacial Modeling of Fibrinogen Adsorption onto LiNbO3 Single Crystal–Single Domain Surfaces. International Journal of Molecular Sciences. 2021; 22(11):5946. https://doi.org/10.3390/ijms22115946
Chicago/Turabian StyleCross, Jeffrey S., Yasuhiro Kubota, Abhijit Chatterjee, Samir Unni, Toshiyuki Ikoma, and Motohiro Tagaya. 2021. "Interfacial Modeling of Fibrinogen Adsorption onto LiNbO3 Single Crystal–Single Domain Surfaces" International Journal of Molecular Sciences 22, no. 11: 5946. https://doi.org/10.3390/ijms22115946
APA StyleCross, J. S., Kubota, Y., Chatterjee, A., Unni, S., Ikoma, T., & Tagaya, M. (2021). Interfacial Modeling of Fibrinogen Adsorption onto LiNbO3 Single Crystal–Single Domain Surfaces. International Journal of Molecular Sciences, 22(11), 5946. https://doi.org/10.3390/ijms22115946