
Cord-Blood-Derived Professional Antigen-Presenting Cells: Functions and Applications in Current and Prospective Cell Therapies


Abstract
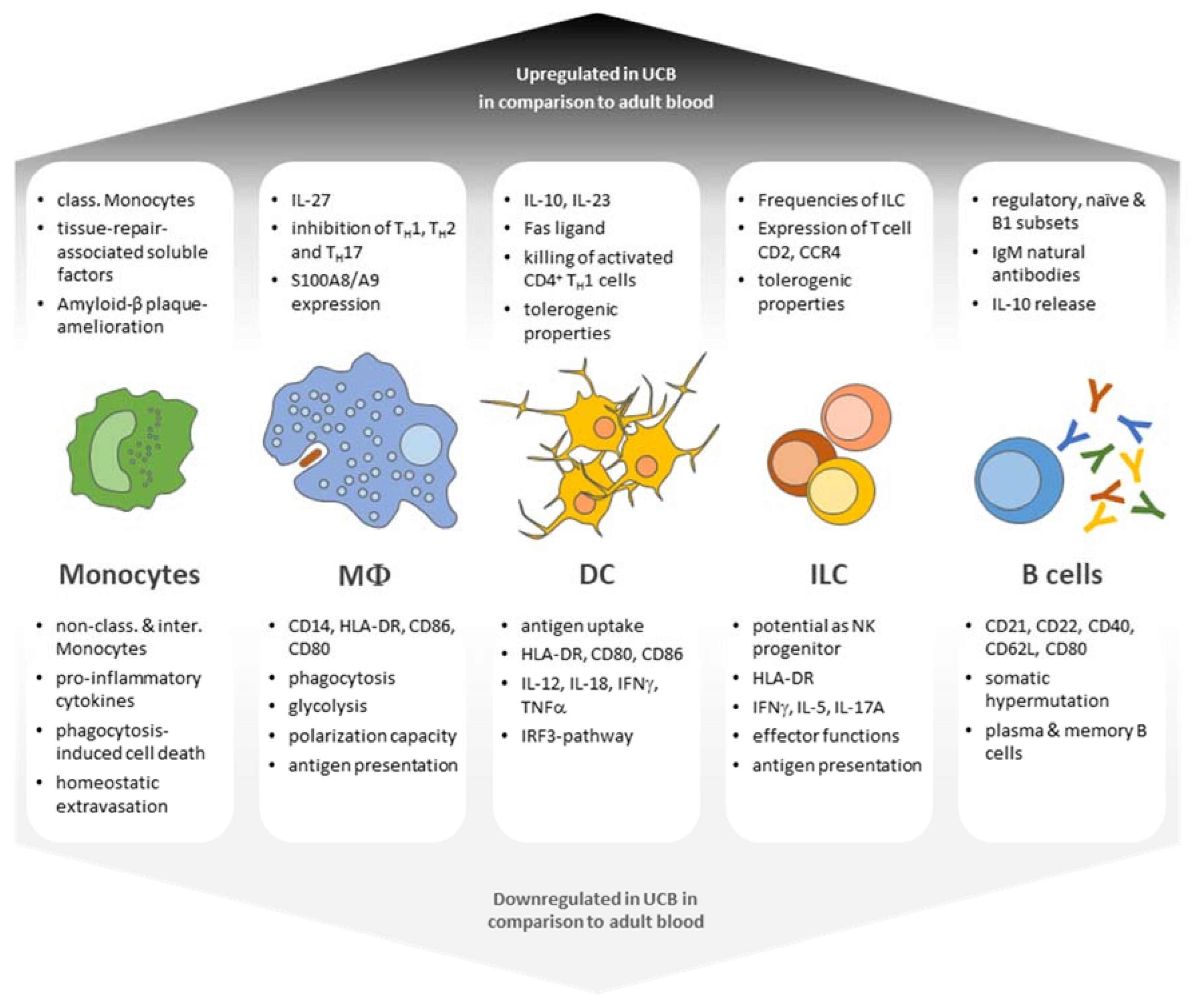
1. Introduction
2. APC Biology in Adult and Umbilical Cord Blood—What Is the Difference?
2.1. Monocytes
2.1.1. Monocytes in Adult Peripheral Blood
2.1.2. Neonatal Monocytes
2.2. DC
2.2.1. Adult DC
2.2.2. Neonatal Dendritic Cells
2.3. Macrophages
2.3.1. Adult MΦ in Peripheral Blood
2.3.2. Neonatal Macrophages
2.4. B Lymphocytes
2.4.1. Adult B Lymphocytes
2.4.2. Neonatal B Cells
2.5. Innate Lymphoid Cells
2.5.1. Adult Innate Lymphoid Cells
2.5.2. Neonatal Innate Lymphoid Cells
3. Current and Prospective UCB-Based APC Cellular Therapies
3.1. UCB-Derived Monocytes in Cell Therapies
3.2. UCB-Derived MΦ in Cell Therapies
3.3. UCB-Derived Dendritic Cells in Cell Therapies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen-presenting cells |
| BCR | B cell receptor |
| BM | Bone marrow |
| CAR | Chimeric antigen receptor |
| CCR2 | CC-chemokine receptor 2 |
| CIK | cytokine-induced killer cell |
| DC | Dendritic cells |
| FasL | Fas ligand |
| GM-CSF | granulocyte colony-stimulating factor |
| GMP | Good manufacturing practice |
| GvHD | Graft-versus-host-disease |
| HLA | human leukocyte antigen |
| HSC | Hematopoietic stem cells |
| IFN | interferon |
| Ig | immunoglobulin |
| IL | interleukine |
| ILC | Innate lymphoid cells |
| IRF | interferon regulatory factor |
| LPS | lipopolysaccharide |
| MoDC | monocyte-derived DC |
| mTOR | mechanistic target of rapamycin |
| MΦ | macrophages |
| NK | Natural killer cells |
| NLRP3 | Nod-Like receptor family pyrin domain containing 3 |
| NOD | nucleotide-binding oligomerization domain-like receptors |
| PBMC | peripheral blood mononuclear cell |
| pDC | Plasmacytoid dendritic cell |
| Pre-B cell | Precursor B cell |
| Pro-B cell | Progenitor B cell |
| sAPPα | amyloid precursor protein metabolite, soluble amyloid precursor protein α |
| SCID | severe combined immunodeficient |
| TGFβ | tumor growth factor β |
| TH | T helper |
| TLR | Toll-like receptor |
| TNFα | tumor necrosis factor α |
| UCB | umbilical cord blood |
| WT1 | Wilms Tumor 1 |
| ZIKV | Zika virus |
References
- Ruggeri, A.; Paviglianiti, A.; Gluckman, E.; Rocha, V. Impact of HLA in cord blood transplantation outcomes. HLA 2016, 87, 413–421. [Google Scholar] [CrossRef]
- Gluckman, E.; Broxmeyer, H.E.; Auerbach, A.D.; Friedman, H.S.; Douglas, G.W.; Devergie, A.; Esperou, H.; Thierry, D.; Socie, G.; Lehn, P.; et al. Hematopoietic Reconstitution in a Patient with Fanconi’s Anemia by Means of Umbilical-Cord Blood from an HLA-Identical Sibling. N. Engl. J. Med. 1989, 321, 1174–1178. [Google Scholar] [CrossRef]
- Gragert, L.; Eapen, M.; Williams, E.; Freeman, J.; Spellman, S.; Baitty, R.; Hartzman, R.; Rizzo, J.D.; Horowitz, M.; Confer, D.; et al. HLA Match Likelihoods for Hematopoietic Stem-Cell Grafts in the U.S. Registry. N. Engl. J. Med. 2014, 371, 339–348. [Google Scholar] [CrossRef]
- Vormoor, J.; Lapidot, T.; Pflumio, F.; Risdon, G.; Patterson, B.; Broxmeyer, H.E.; Dick, J.E. Immature human cord blood progenitors engraft and proliferate to high levels in severe combined immunodeficient mice. Blood 1994, 83, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Bock, T.A.; Orlic, D.; Dunbar, C.E.; Broxmeyer, H.E.; Bodine, D.M. Improved engraftment of human hematopoietic cells in severe combined immunodeficient (SCID) mice carrying human cytokine transgenes. J. Exp. Med. 1995, 182, 2037–2043. [Google Scholar] [CrossRef]
- Erices, A.; Conget, P.; Minguell, J.J. Mesenchymal progenitor cells in human umbilical cord blood. Br. J. Haematol. 2000, 109, 235–242. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef]
- Boyette, L.B.; Macedo, C.; Hadi, K.; Elinoff, B.D.; Walters, J.; Ramaswami, B.; Chalasani, G.; Taboas, J.M.; Lakkis, F.G.; Metes, D.M. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS ONE 2017, 12, e0176460. [Google Scholar] [CrossRef]
- Carlin, L.; Stamatiades, E.G.; Auffray, C.; Hanna, R.N.; Glover, L.; Vizcay-Barrena, G.; Hedrick, C.C.; Cook, H.T.; Diebold, S.; Geissmann, F. Nr4a1-Dependent Ly6Clow Monocytes Monitor Endothelial Cells and Orchestrate Their Disposal. Cell 2013, 153, 362–375. [Google Scholar] [CrossRef]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. New Insights into Tissue Macrophages: From Their Origin to the Development of Memory. Immune Netw. 2015, 15, 167–176. [Google Scholar] [CrossRef]
- Mills, C.D. Macrophage Arginine Metabolism to Ornithine/Urea or Nitric Oxide/Citrulline: A Life or Death Issue. Crit. Rev. Immunol. 2001, 21, 28–425. [Google Scholar] [CrossRef]
- Cros, J.; Cagnard, N.; Woollard, K.; Patey, N.; Zhang, S.-Y.; Senechal, B.; Puel, A.; Biswas, S.K.; Moshous, D.; Picard, C.; et al. Human CD14dim Monocytes Patrol and Sense Nucleic Acids and Viruses via TLR7 and TLR8 Receptors. Immunity 2010, 33, 375–386. [Google Scholar] [CrossRef]
- Prabhu, S.B.; Rathore, D.K.; Nair, D.; Chaudhary, A.; Raza, S.; Kanodia, P.; Sopory, S.; George, A.; Rath, S.; Bal, V.; et al. Comparison of Human Neonatal and Adult Blood Leukocyte Subset Composition Phenotypes. PLoS ONE 2016, 11, e0162242. [Google Scholar] [CrossRef] [PubMed]
- Sohlberg, E.; Saghafian-Hedengren, S.; Bremme, K.; Sverremark-Ekström, E. Cord blood monocyte subsets are similar to adult and show potent peptidoglycan-stimulated cytokine responses. Immunology 2011, 133, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Flower, R.; Han, P. Cord Blood Leucocyte Expression of Functionally Significant Molecules Involved in the Regulation of Cellular Immunity. Scand. J. Immunol. 2001, 53, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Pettengill, M.A.; van Haren, S.; Levy, O. Soluble Mediators Regulating Immunity in Early Life. Front. J. Immunol. 2014, 5, 457. [Google Scholar] [CrossRef]
- Levy, O.; Suter, E.E.; Miller, R.L.; Wessels, M.R. Unique efficacy of Toll-like receptor 8 agonists in activating human neonatal antigen-presenting cells. Blood 2006, 108, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Philbin, V.J.; Dowling, D.J.; Gallington, L.C.; Cortés, G.; Tan, Z.; Suter, E.E.; Chi, K.W.; Shuckett, A.; Stoler-Barak, L.; Tomai, M.; et al. Imidazoquinoline Toll-like receptor 8 agonists activate human newborn monocytes and dendritic cells through adenosine-refractory and caspase-1–dependent pathways. J. Allergy Clin. Immunol. 2012, 130, 195–204.e9. [Google Scholar] [CrossRef]
- Kollmann, T.R.; Crabtree, J.; Rein-Weston, A.; Blimkie, D.; Thommai, F.; Wang, X.Y.; Lavoie, P.M.; Furlong, J.; Fortuno, E.S.; Hajjar, A.; et al. Neonatal Innate TLR-Mediated Responses Are Distinct from Those of Adults. J. Immunol. 2009, 183, 7150–7160. [Google Scholar] [CrossRef] [PubMed]
- De Wit, D.; Tonon, S.; Olislagers, V.; Goriely, S.; Boutriaux, M.; Goldman, M.; Willems, F. Impaired responses to toll-like receptor 4 and toll-like receptor 3 ligands in human cord blood. J. Autoimmun. 2003, 21, 277–281. [Google Scholar] [CrossRef]
- Li, Y.P.; Yu, S.L.; Huang, Z.J.; Huang, J.; Pan, J.; Feng, X.; Zhang, X.G.; Wang, J.H.; Wang, J. An Impaired Inflammatory Cytokine Response to Gram-Negative LPS in Human Neonates is Associated with the Defective TLR-Mediated Signaling Pathway. J. Clin. Immunol. 2015, 35, 218–226. [Google Scholar] [CrossRef]
- Namakula, R.; De Bree, L.C.J.; Tvedt, T.H.A.; Netea, M.G.; Cose, S.; Hanevik, K. Monocytes from neonates and adults have a similar capacity to adapt their cytokine production after previous exposure to BCG and β-glucan. PLoS ONE 2020, 15, e0229287. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.-C.; Ratter, J.; Berentsen, K.; Van Der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Smith, S.G.; Kleinnijenhuis, J.; Netea, M.G.; Dockrell, H.M. Whole Blood Profiling of Bacillus Calmette–Guérin-Induced Trained Innate Immunity in Infants Identifies Epidermal Growth Factor, IL-6, Platelet-Derived Growth Factor-AB/BB, and Natural Killer Cell Activation. Front. Immunol. 2017, 8, 644. [Google Scholar] [CrossRef]
- Bermick, J.R.; Lambrecht, N.; Dendekker, A.D.; Kunkel, S.L.; Lukacs, N.W.; Hogaboam, C.M.; Schaller, M.A. Neonatal monocytes exhibit a unique histone modification landscape. Clin. Epigenetics 2016, 8, 1–15. [Google Scholar] [CrossRef]
- Yoshikawa, F.S.Y.; Pietrobon, A.J.; Branco, A.C.C.C.; Pereira, N.Z.; Oliveira, L.; Machado, C.M.; Duarte, A.J.D.S.; Sato, M.N. Zika Virus Infects Newborn Monocytes without Triggering a Substantial Cytokine Response. J. Infect. Dis. 2019, 220, 32–40. [Google Scholar] [CrossRef]
- Slavica, L.; Nordström, I.; Karlsson, M.N.; Valadi, H.; Kacerovsky, M.; Jacobsson, B.; Eriksson, K. TLR3 impairment in human newborns. J. Leukoc. Biol. 2013, 94, 1003–1011. [Google Scholar] [CrossRef]
- Dreschers, S.; Gille, C.; Haas, M.; Seubert, F.; Platen, C.; Orlikowsky, T.W. Reduced internalization of TNF-ɑ/TNFR1 down-regulates caspase dependent phagocytosis induced cell death (PICD) in neonatal monocytes. PLoS ONE 2017, 12, e0182415. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Coopersmith, C.M.; McDunn, J.; Ferguson, T.A. The sepsis seesaw: Tilting toward immunosuppression. Nat. Med. 2009, 15, 496–497. [Google Scholar] [CrossRef]
- Sanchez-Schmitz, G.; Morrocchi, E.; Cooney, M.; Soni, D.; Khatun, R.; Palma, P.; Dowling, D.J.; Levy, O. Neonatal monocytes demonstrate impaired homeostatic extravasation into a microphysiological human vascular model. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Shang, X.-Z.; Issekutz, A.C. Contribution of CD11a/CD18, CD11b/CD18, ICAM-1 (CD54) and −2 (CD102) to human monocyte migration through endothelium and connective tissue fibroblast barriers. Eur. J. Immunol. 1998, 28, 1970–1979. [Google Scholar] [CrossRef]
- Willems, F.; Vollstedt, S.; Suter, M. Phenotype and function of neonatal DC. Eur. J. Immunol. 2009, 39, 26–35. [Google Scholar] [CrossRef]
- Schüller, S.S.; Sadeghi, K.; Wisgrill, L.; Dangl, A.; Diesner, S.C.; Prusa, A.R.; Klebermasz-Schrehof, K.; Greber-Platzer, S.; Neumüller, J.; Helmer, H.; et al. Preterm neonates display altered plasmacytoid dendritic cell function and morphology. J. Leukoc. Biol. 2013, 93, 781–788. [Google Scholar] [CrossRef]
- Vély, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F.; et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Collin, M.; Ginhoux, F. Human dendritic cells. Semin. Cell Dev. Biol. 2019, 86, 1–2. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three Markers for Distinct Subsets of Dendritic Cells in Human Peripheral Blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef]
- Haniffa, M.; Shin, A.; Bigley, V.; McGovern, N.; Teo, P.; See, P.; Wasan, P.S.; Wang, X.-N.; Malinarich, F.; Malleret, B.; et al. Human Tissues Contain CD141hi Cross-Presenting Dendritic Cells with Functional Homology to Mouse CD103+ Nonlymphoid Dendritic Cells. Immunity 2012, 37, 60–73. [Google Scholar] [CrossRef]
- Bachem, A.; Guttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Sittig, S.P.; Bakdash, G.; Weiden, J.; Sköld, A.E.; Tel, J.; Figdor, C.G.; de Vries, I.J.M.; Schreibelt, G. A Comparative Study of the T Cell Stimulatory and Polarizing Capacity of Human Primary Blood Dendritic Cell Subsets. Mediat. Inflamm. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Mittag, D.; Proietto, A.I.; Loudovaris, T.; Mannering, S.I.; Vremec, D.; Shortman, K.; Wu, L.; Harrison, L.C. Human Dendritic Cell Subsets from Spleen and Blood Are Similar in Phenotype and Function but Modified by Donor Health Status. J. Immunol. 2011, 186, 6207–6217. [Google Scholar] [CrossRef]
- Nizzoli, G.; Krietsch, J.; Weick, A.; Steinfelder, S.; Facciotti, F.; Gruarin, P.; Bianco, A.; Steckel, B.; Moro, M.; Crosti, M.; et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood 2013, 122, 932–942. [Google Scholar] [CrossRef]
- Segura, E.; Durand, M.; Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ–resident dendritic cells. J. Exp. Med. 2013, 210, 1035–1047. [Google Scholar] [CrossRef]
- Fitzgerald-Bocarsly, P. Human natural interferon-α producing cells. Pharmacol. Ther. 1993, 60, 39–62. [Google Scholar] [CrossRef]
- Gilliet, M.; Cao, W.; Liu, Y.-J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef]
- Hespel, C.; Moser, M. Role of inflammatory dendritic cells in innate and adaptive immunity. Eur. J. Immunol. 2012, 42, 2535–2543. [Google Scholar] [CrossRef]
- Coillard, A.; Segura, E. In vivo Differentiation of Human Monocytes. Front. Immunol. 2019, 10, 1–7. [Google Scholar] [CrossRef]
- Guilliams, M.; Dutertre, C.-A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, 1955–1956. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Yu, H.; Jin, X.; Li, J.; Guo, H.; Shi, Q.; Yin, Z.; Xu, Y.; Wang, X.; Liu, R.; et al. Human Blood CD1c+ Dendritic Cells Encompass CD5high and CD5low Subsets That Differ Significantly in Phenotype, Gene Expression, and Functions. J. Immunol. 2017, 198, 1553–1564. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, P.; Yin, X.; Yin, Z.; Shi, Q.; Cui, Y.; Liu, G.; Wang, S.; Piccaluga, P.P.; Jiang, T.; et al. Human BDCA2+CD123+CD56+ dendritic cells (DC) related to blastic plasmacytoid dendritic cell neoplasm represent a unique myeloid DC subset. Protein Cell 2015, 6, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Szabolcs, P.; Park, K.-D.; Reese, M.; Marti, L.; Broadwater, G.; Kurtzberg, J. Absolute Values of Dendritic Cell Subsets in Bone Marrow, Cord Blood, and Peripheral Blood Enumerated by a Novel Method. Stem Cells 2003, 21, 296–303. [Google Scholar] [CrossRef]
- Pereira, M.I.; Paiva, A. Dendritic Cells in Cord Blood Transplantation: A Review. Stem Cells Int. 2011, 2011, 1–7. [Google Scholar] [CrossRef]
- Encabo, A.; Solves, P.; Carbonell-Uberos, F.; Miñana, M.D. The functional immaturity of dendritic cells can be relevant to increased tolerance associated with cord blood transplantation. Transfusion 2007, 47, 272–279. [Google Scholar] [CrossRef]
- Nguyen, M.; Leuridan, E.; Zhang, T.; De Wit, D.; Willems, F.; Van Damme, P.; Goldman, M.; Goriely, S. Acquisition of Adult-Like TLR4 and TLR9 Responses during the First Year of Life. PLoS ONE 2010, 5, e10407. [Google Scholar] [CrossRef]
- De Wit, D.; Olislagers, V.; Goriely, S.; Vermeulen, F.; Wagner, H.; Goldman, M.; Willems, F. Blood plasmacytoid dendritic cell responses to CpG oligodeoxynucleotides are impaired in human newborns. Blood 2004, 103, 1030–1032. [Google Scholar] [CrossRef]
- Papaioannou, N.E.; Pasztoi, M.; Schraml, B. Understanding the Functional Properties of Neonatal Dendritic Cells: A Doorway to Enhance Vaccine Effectiveness? Front. Immunol. 2019, 9, 3123. [Google Scholar] [CrossRef] [PubMed]
- Dakic, A.; Shao, Q.-x.; D’Amico, A.; O’Keefe, M.; Chen, W.-f.; Shortman, K.; Wu, L. Development of the Dendritic Cell System during Mouse Ontogeny. J. Immunol. 2004, 172, 1018–1027. [Google Scholar] [CrossRef]
- Torres, D.; Kohler, A.; Delbauve, S.; Caminschi, I.; Lahoud, M.H.; Shortman, K.; Flamand, V. IL-12p40/IL-10 Producing preCD8α/Clec9A+ Dendritic Cells Are Induced in Neonates upon Listeria monocytogenes Infection. PLoS Pathog. 2016, 12, e1005561. [Google Scholar] [CrossRef]
- Adkins, B.; Du, R.Q. Newborn mice develop balanced Th1/Th2 primary effector responses in vivo but are biased to Th2 secondary responses. J. Immunol. 1998, 160, 4217–4224. [Google Scholar]
- Naderi, N.; Moazzeni, S.; Pourfathollah, A.; Alimoghaddam, K. High Expression of Fas Ligand on Cord Blood Dendritic Cells: A Possible Immunoregulatory Mechanism after Cord Blood Transplantation. Transplant. Proc. 2011, 43, 3913–3919. [Google Scholar] [CrossRef]
- Niederwieser, D.; Herold, M.; Woloszczuk, W.; Aulitzky, W.; Meister, B.; Tilg, H.; Gastl, G.; Bowden, R.; Huber, C. Endogenous IFN-γ during human bone marrow transplantation. Analysis of serum levels of interferon and interferon-dependent secondary messages. Transplantation 1990, 50, 620–625. [Google Scholar] [CrossRef]
- Kim, S.K.; Yun, C.-H.; Han, S.H. Dendritic Cells Differentiated from Human Umbilical Cord Blood-Derived Monocytes Exhibit Tolerogenic Characteristics. Stem Cells Dev. 2015, 24, 2796–2807. [Google Scholar] [CrossRef]
- Goriely, S.; Vincart, B.; Stordeur, P.; Vekemans, J.; Willems, F.; Goldman, M.; De Wit, D. Deficient IL-12(p35) Gene Expression by Dendritic Cells Derived from Neonatal Monocytes. J. Immunol. 2001, 166, 2141–2146. [Google Scholar] [CrossRef] [PubMed]
- Salio, M.; Dulphy, N.; Renneson, J.; Herbert, M.; McMichael, A.; Marchant, A.; Cerundolo, V. Efficient priming of antigen-specific cytotoxic T lymphocytes by human cord blood dendritic cells. Int. Immunol. 2003, 15, 1265–1273. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eijnden, S.V.; Goriely, S.; De Wit, D.; Goldman, M.; Willems, F. Preferential production of the IL-12(p40)/IL-23(p19) heterodimer by dendritic cells from human newborns. Eur. J. Immunol. 2005, 36, 21–26. [Google Scholar] [CrossRef]
- Bettelli, E.; Oukka, M.; Kuchroo, V.K. TH-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007, 8, 345–350. [Google Scholar] [CrossRef]
- Anh, B.V.; Thao, C.T.; Cuong, P.T.; Thuy, N.T.T.; Diem, H.H.; Khanh, B.T.V.; Hue, B.T.H.; Uyen, T.T.T.; Tu, N.D.; Hoai, T.T.T.; et al. Vγ9γδ T Cell Induction by Human Umbilical Cord Blood Monocytes-Derived, Interferon-α-Stimulated Dendritic Cells. Cancer Control 2020, 27. [Google Scholar] [CrossRef]
- Schmid, D.; Park, C.G.; Hartl, C.A.; Subedi, N.; Cartwright, A.N.; Puerto, R.B.; Zheng, Y.; Maiarana, J.; Freeman, G.J.; Wucherpfennig, K.W.; et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Plüddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 1–18. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Rey-Giraud, F.; Hafner, M.; Ries, C.H. In Vitro Generation of Monocyte-Derived Macrophages under Serum-Free Conditions Improves Their Tumor Promoting Functions. PLoS ONE 2012, 7, e42656. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Locati, M. New vistas on macrophage differentiation and activation. Eur. J. Immunol. 2007, 37, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Corliss, B.A.; Azimi, M.S.; Munson, J.M.; Peirce, S.M.; Murfee, W.L. Macrophages: An Inflammatory Link between Angiogenesis and Lymphangiogenesis. Microcirculation 2016, 23, 95–121. [Google Scholar] [CrossRef]
- De Paoli, F.; Staels, B.; Chinetti, G. Macrophage Phenotypes and Their Modulation in Atherosclerosis. Circ. J. 2014, 78, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeilli, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-Like Phenotype is Independent of Interleukin-4 Receptor Alpha (IL-4Rα) Signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Haskó, G.; Pacher, P.; Deitch, E.A.; Vizi, E.S. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol. Ther. 2007, 113, 264–275. [Google Scholar] [CrossRef]
- Wolfs, I.M.J.; Stoger, L.; Goossens, P.; Pottgens, C.; Gijbels, M.J.J.; Wijnands, E.; van der Vorst, E.P.C.; van Gorp, P.; Beckers, L.; Engel, D.; et al. Reprogramming macrophages to an anti-inflammatory phenotype by helminth antigens reduces murine atherosclerosis. FASEB J. 2014, 28, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Hörhold, F.; Eisel, D.; Oswald, M.; Kolte, A.; Röll, D.; Osen, W.; Eichmüller, S.B.; König, R. Reprogramming of macrophages employing gene regulatory and metabolic network models. PLoS Comput. Biol. 2020, 16, e1007657. [Google Scholar] [CrossRef]
- Dreschers, S.; Ohl, K.; Lehrke, M.; Möllmann, J.; Denecke, B.; Costa, I.; Vogl, T.; Viemann, D.; Roth, J.; Orlikowsky, T.; et al. Impaired cellular energy metabolism in cord blood macrophages contributes to abortive response toward inflammatory threats. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Gille, C.; Spring, B.; Tewes, L.J.; Löffler, J.; Dannecker, G.E.; Hoffmann, M.K.; Eichner, M.; Poets, C.F.; Orlikowsky, T.W. Diminished Response to Interleukin-10 and Reduced Antibody-Dependent Cellular Cytotoxicity of Cord Blood Monocyte-Derived Macrophages. Pediatr. Res. 2006, 60, 152–157. [Google Scholar] [CrossRef][Green Version]
- Dreschers, S.; Ohl, K.; Schulte, N.; Tenbrock, K.; Orlikowsky, T.W. Impaired functional capacity of polarised neonatal macrophages. Sci. Rep. 2020, 10, 624. [Google Scholar] [CrossRef] [PubMed]
- Kraft, J.D.; Horzempa, J.; Davis, C.; Jung, J.-Y.; Peña, M.M.O.; Robinson, C. Neonatal macrophages express elevated levels of interleukin-27 that oppose immune responses. J. Immunol. 2013, 139, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Nau, G.J. Interleukin-12 and Interleukin-27 Regulate Macrophage Control of Mycobacterium tuberculosis. J. Infect. Dis. 2008, 198, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, S.; Pirr, S.; Fehlhaber, B.; Mellinger, L.; Burgmann, J.; Busse, M.; Ginzel, M.; Friesenhagen, J.; von Kockritz-Blickwede, M.; Ulas, T.; et al. In neonates S100A8/S100A9 alarmins prevent the expansion of a specific inflammatory monocyte population promoting septic shock. FASEB J. 2017, 31, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Rebmann, V.; LeMaoult, J.; Horn, P.A.; Carosella, E.D.; Alegre, E. The immunosuppressive molecule HLA-G and its clinical implications. Crit. Rev. Clin. Lab. Sci. 2012, 49, 63–84. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.-H. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front. Immunol. 2018, 9, 2164. [Google Scholar] [CrossRef]
- Lee, C.-L.; Guo, Y.; So, K.-H.; Vijayan, M.; Guo, Y.; Wong, V.H.; Yao, Y.; Lee, K.-F.; Chiu, P.C.; Yeung, W.S. Soluble human leukocyte antigen G5 polarizes differentiation of macrophages toward a decidual macrophage-like phenotype. Hum. Reprod. 2015, 30, 2263–2274. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Rajewsky, K.; Förster, I.; Cumano, A. Evolutionary and somatic selection of the antibody repertoire in the mouse. Science 1987, 238, 1088–1094. [Google Scholar] [CrossRef]
- Li, Z.; Woo, C.J.; Iglesias-Ussel, M.D.; Ronai, D.; Scharff, M.D. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004, 18, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Burrows, P.D.; Wang, J.-Y. B Cell Development and Maturation. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2020; Volume 1254, pp. 1–22. [Google Scholar]
- Murre, C. ‘Big Bang’ of B-cell development revealed. Genes Dev. 2018, 32, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Allen, C.D. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef] [PubMed]
- Stein, K.E. Thymus-Independent and Thymus-Dependent Responses to Polysaccharide Antigens. J. Infect. Dis. 1992, 165, S49–S52. [Google Scholar] [CrossRef] [PubMed]
- Crampton, S.P.; Voynova, E.; Bolland, S. Innate pathways to B-cell activation and tolerance. Ann. N.Y. Acad. Sci. 2010, 1183, 58–68. [Google Scholar] [CrossRef]
- Kräutler, N.J.; Suan, D.; Butt, D.; Bourne, K.; Hermes, J.R.; Chan, T.D.; Sundling, C.; Kaplan, W.; Schofield, P.; Jackson, J.; et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J. Exp. Med. 2017, 214, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Weisel, F.; Shlomchik, M. Memory B Cells of Mice and Humans. Annu. Rev. Immunol. 2017, 35, 255–284. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- He, J.-S.; Narayanan, S.; Subramaniam, S.; Ho, W.Q.; Lafaille, J.J.; Curotto de Lafaille, M.A. Biology of IgE Production: IgE Cell Differentiation and the Memory of IgE Responses. In IgE Antibodies Generation Function; Spinger: Berlin/Heidelberg, Germany, 2015; Volume 388, pp. 1–19. [Google Scholar]
- Siegrist, C.-A.; Aspinall, R. B-cell responses to vaccination at the extremes of age. Nat. Rev. Immunol. 2009, 9, 185–194. [Google Scholar] [CrossRef]
- Zinkernagel, R.M. Maternal Antibodies, Childhood Infections, and Autoimmune Diseases. N. Engl. J. Med. 2001, 345, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.F.; Karron, R.A.; Belshe, R.B.; Thompson, J.; Crowe, J.E., Jr.; Boyce, T.G.; Halburnt, L.L.; Reed, G.W.; Whitehead, S.S.; Anderson, E.L.; et al. Evaluation of a Live, Cold-Passaged, Temperature-Sensitive, Respiratory Syncytial Virus Vaccine Candidate in Infancy. J. Infect. Dis. 2000, 182, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.J.; Perrett, K.P.; Beverley, P. Maintaining protection against invasive bacteria with protein–polysaccharide conjugate vaccines. Nat. Rev. Immunol. 2009, 9, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Quinello, C.; Silveira-Lessa, A.L.; Ceccon, M.E.J.R.; Cianciarullo, M.A.; Carneiro-Sampaio, M.; Palmeira, P. Phenotypic Differences in Leucocyte Populations among Healthy Preterm and Full-Term Newborns. Scand. J. Immunol. 2014, 80, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Morbach, H.; Eichhorn, E.M.; Liese, J.G.; Girschick, H.J. Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 2010, 162, 271–279. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Franklin, S.W.; Zegers, B.J.; Rijkers, G.T. Expression and Functional Characteristics of the Complement Receptor Type 2 on Adult and Neonatal B Lymphocytes. Clin. Immunol. Immunopathol. 1993, 69, 1–8. [Google Scholar] [CrossRef]
- Kaur, K.; Chowdhury, S.; Greenspan, N.S.; Schreiber, J.R. Decreased expression of tumor necrosis factor family receptors involved in humoral immune responses in preterm neonates. Blood 2007, 110, 2948–2954. [Google Scholar] [CrossRef] [PubMed]
- Viemann, D.; Schlenke, P.; Hammers, H.-J.; Kirchner, H.; Kruse, A. Differential expression of the B cell-restricted molecule CD22 on neonatal B lymphocytes depending upon antigen stimulation. Eur. J. Immunol. 2000, 30, 550–559. [Google Scholar] [CrossRef]
- Tasker, L.; Marshall-Clarke, S. Functional responses of human neonatal B lymphocytes to antigen receptor cross-linking and CpG DNA. Clin. Exp. Immunol. 2003, 134, 409–419. [Google Scholar] [CrossRef]
- Glaesener, S.; Jaenke, C.; Habener, A.; Geffers, R.; Hagendorff, P.; Witzlau, K.; Imelmann, E.; Krueger, A.; Meyer-Bahlburg, A. Decreased production of class-switched antibodies in neonatal B cells is associated with increased expression of miR-181b. PLoS ONE 2018, 13, e0192230. [Google Scholar] [CrossRef] [PubMed]
- Adkins, B.; Leclerc, C.; Marshall-Clarke, S. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 2004, 4, 553–564. [Google Scholar] [CrossRef]
- Meyer-Bahlburg, A.; Andrews, S.F.; Yu, K.O.; Porcelli, S.A.; Rawlings, D.J. Characterization of a late transitional B cell population highly sensitive to BAFF-mediated homeostatic proliferation. J. Exp. Med. 2008, 205, 155–168. [Google Scholar] [CrossRef]
- Belnoue, E.; Pihlgren, M.; McGaha, T.; Tougne, C.; Rochat, A.-F.; Bossen, C.; Schneider, P.; Huard, B.; Lambert, P.-H.; Siegrist, C.-A. April is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood 2008, 111, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Newport, M.J.; Goetghebuer, T.; Weiss, H.A.; Whittle, H.; Siegrist, C.-A.; Marchant, A. Genetic regulation of immune responses to vaccines in early life. Genes Immun. 2004, 5, 122–129. [Google Scholar] [CrossRef]
- Haas, K.M.; Poe, J.C.; Steeber, D.A.; Tedder, T.F. B-1a and B-1b Cells Exhibit Distinct Developmental Requirements and Have Unique Functional Roles in Innate and Adaptive Immunity to S. pneumoniae. Immunity 2005, 23, 7–18. [Google Scholar] [CrossRef]
- Griffin, D.O.; Holodick, N.E.; Rothstein, T.L. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+CD27+CD43+CD70−. J. Exp. Med. 2011, 208, 67–80. [Google Scholar] [CrossRef]
- Wong, S.-C.; Chew, W.-K.; Tan, J.E.-L.; Melendez, A.J.; Francis, F.; Lam, K.-P. Peritoneal CD5+ B-1 Cells Have Signaling Properties Similar to Tolerant B Cells. J. Biol. Chem. 2002, 277, 30707–30715. [Google Scholar] [CrossRef]
- Kageyama, Y.; Katayama, N. Ontogeny of human B1 cells. Int. J. Hematol. 2020, 111, 628–633. [Google Scholar] [CrossRef]
- Zhong, X.; Gao, W.; Degauque, N.; Bai, C.; Lu, Y.; Kenny, J.; Oukka, M.; Strom, T.B.; Rothstein, T.L. Reciprocal generation of Th1/Th17 and Treg cells by B1 and B2 B cells. Eur. J. Immunol. 2007, 37, 2400–2404. [Google Scholar] [CrossRef]
- Griffin, D.O.; Rothstein, T.L. Human B1 Cell Frequency: Isolation and Analysis of Human B1 Cells. Front. Immunol. 2012, 3, 122. [Google Scholar] [CrossRef]
- Reynaud, C.-A.; Weill, J.-C. Gene profiling of CD11b+ and CD11b− B1 cell subsets reveals potential cell sorting artifacts. J. Exp. Med. 2012, 209, 433–434. [Google Scholar] [CrossRef]
- Cunningham, A.; Flores-Langarica, A.; Bobat, S.; Dominguez Medina, C.C.; Cook, C.N.L.; Ross, E.A.; Lopez-Macias, C.; Hederson, I. B1b Cells Recognize Protective Antigens after Natural Infection and Vaccination. Front. Immunol. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Ghosn, E.E.B.; Yang, Y.; Tung, J.; Herzenberg, L.A. CD11b expression distinguishes sequential stages of peritoneal B-1 development. Proc. Natl. Acad. Sci. USA 2008, 105, 5195–5200. [Google Scholar] [CrossRef]
- Esteve-Solé, A.; Luo, Y.; Vlagea, A.; Deya-Martinez, A.; Yague, J.; Plaza-Martin, A.M.; Juan, M.; Alsina, L. B Regulatory Cells: Players in Pregnancy and Early Life. Int. J. Mol. Sci. 2018, 19, 2099. [Google Scholar] [CrossRef]
- Sarvaria, A.; Basar, R.; Mehta, R.; Shaim, H.; Muftuoglu, M.; Khoder, A.; Sekine, T.; Gokdemir, E.; Kondo, K.; Marin, D.; et al. IL-10+ regulatory B cells are enriched in cord blood and may protect against cGVHD after cord blood transplantation. Blood 2016, 128, 1346–1361. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N.J. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Cupedo, T. Innate Lymphoid Cells: Emerging Insights in Development, Lineage Relationships, and Function. Annu. Rev. Immunol. 2012, 30, 647–675. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 Induces IL-4, IL-5, and IL-13 and Th2-Associated Pathologies In Vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef]
- Wilhelm, C.; Hirota, K.; Stieglitz, B.; Van Snick, J.; Tolaini, M.; Lahl, K.; Sparwasser, T.; Helmby, H.; Stockinger, B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 2011, 12, 1071–1077. [Google Scholar] [CrossRef]
- Monticelli, L.A. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, J. MHCII-Mediated Dialog between Group 2 Innate Lymphoid Cells and CD4+ T Cells Potentiates Type 2 Immunity and Promotes Parasitic Helminth Expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef]
- Visan, A. Antigen-presenting ILCs. Nat. Immunol. 2014, 15, 909. [Google Scholar] [CrossRef]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nat. Cell Biol. 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ohne, Y. OMIP-066: Identification of Novel Subpopulations of Human Group 2 Innate Lymphoid Cells in Peripheral Blood. Cytom. Part A 2020, 97, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Bennstein, S.B.; Scherenschlich, N.; Weinhold, S.; Manser, A.R.; Noll, A.; Raba, K.; Kogler, G.; Walter, L.; Uhrberg, M. Transcriptional and functional characterization of neonatal circulating ILCs. Stem Cells Transl. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bennstein, S.B.; Weinhold, S.; Manser, A.R.; Scherenschlich, N.; Noll, A.; Raba, K.; Kogler, G.; Walter, L.; Uhrberg, M. Umbilical cord blood-derived ILC1-like cells constitute a novel precursor for mature KIR+NKG2A− NK cells. eLife 2020, 9, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.R.; Zeldovich, V.B.; Poukchanski, A.; Boothroyd, J.C.; Bakardjiev, A.I. Tissue Barriers of the Human Placenta to Infection with Toxoplasma gondii. Infect. Immun. 2011, 80, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Einenkel, R.; Ehrhardt, J.; Hartmann, K.; Krüger, D.; Muzzio, D.O.; Zygmunt, M. Hormonally controlled ILC antigen presentation potential is reduced during pregnancy. Reproduction 2020, 160, 155–169. [Google Scholar] [CrossRef]
- Riordan, N.H.; Chan, K.; Marleau, A.M.; Ichim, T.E. Cord blood in regenerative medicine: Do we need immune suppression? J. Transl. Med. 2007, 5, 8. [Google Scholar] [CrossRef]
- Sun, M. Effect of Autologous Cord Blood Infusion on Motor Function and Brain Connectivity in Young Children with Cerebral Palsy: A Randomized, Placebo-Controlled Trial. Stem Cells Transl. Med. 2017, 6, 2071–2078. [Google Scholar] [CrossRef]
- Cotten, C.M.; Murtha, A.P.; Goldberg, R.N.; Grotegut, C.A.; Smith, P.B.; Goldstein, R.F.; Fisher, K.A.; Gustafson, K.E.; Waters-Pick, B.; Swamy, G.K.; et al. Feasibility of Autologous Cord Blood Cells for Infants with Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2014, 164, 973–979.e1. [Google Scholar] [CrossRef]
- Cui, X.; Chopp, M.; Shehadah, A.; Zacharek, A.; Kuzmin-Nichols, N.; Sanberg, C.D.; Dai, J.; Zhang, C.; Ueno, Y.; Roberts, C.; et al. Therapeutic Benefit of Treatment of Stroke with Simvastatin and Human Umbilical Cord Blood Cells: Neurogenesis, Synaptic Plasticity, and Axon Growth. Cell Transplant. 2012, 21, 845–856. [Google Scholar] [CrossRef]
- Saha, B. Human umbilical cord blood monocytes, but not adult blood monocytes, rescue brain cells from hypoxic-ischemic injury: Mechanistic and therapeutic implications. PLoS ONE 2019, 14, e0218906. [Google Scholar] [CrossRef]
- Sato, Y.; Tsuji, M. Diverse actions of cord blood cell therapy for hypoxic-ischemic encephalopathy. Pediatr. Int. 2021, 63, 497–503. [Google Scholar] [CrossRef]
- Wennhold, A.; Shimabukuro-Vornhagen, A.; von Bergwelt-Baildon, M. B Cell-Based Cancer Immunotherapy. Transfus. Med. Hemother. 2019, 46, 36–46. [Google Scholar] [CrossRef]
- Cobb, M.; Verneris, M.R. Therapeutic manipulation of innate lymphoid cells. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.A.; Penny, T.R.; Paton, M.C.B.; Sutherland, A.E.; Nekkanti, L.; Yawno, T.; Castillo-Melendez, M.; Fahey, M.C.; Jones, N.M.; Jenkin, G.; et al. Effects of umbilical cord blood cells, and subtypes, to reduce neuroinflammation following perinatal hypoxic-ischemic brain injury. J. Neuro Inflamm. 2018, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Darlington, E. Human Umbilical Cord Blood-Derived Monocytes Improve Cognitive Deficits and Reduce Amyloid-β Pathology in PSAPP Mice. Cell Transplant. 2015, 24, 2237–2250. [Google Scholar] [CrossRef]
- Darlington, D.; Deng, J.; Giunta, B.; Hou, H.; Sanberg, C.D.; Kuzmin-Nichols, N.; Zhou, H.-D.; Mori, T.; Ehrhart, J.; Sanberg, P.R.; et al. Multiple Low-Dose Infusions of Human Umbilical Cord Blood Cells Improve Cognitive Impairments and Reduce Amyloid-β-Associated Neuropathology in Alzheimer Mice. Stem Cells Dev. 2013, 22, 412–421. [Google Scholar] [CrossRef] [PubMed]
- De Haar, C. Generation of a cord blood-derived Wilms Tumor 1 dendritic cell vaccine for AML patients treated with allogeneic cord blood transplantation. Oncoimmunology 2015, 4, e1023973. [Google Scholar] [CrossRef]
- Plantinga, C. Clinical Grade Production of Wilms’ Tumor-1 Loaded Cord Blood-Derived Dendritic Cells to Prevent Relapse in Pediatric AML After Cord Blood Transplantation. Front. Immunol. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Hutten, T.; Thordardottir, S.; Hobo, W.; Hübel, J.; van der Waart, A.; Cany, J.; Dolstra, H.; Hangalapura, B.N. Ex Vivo Generation of Interstitial and Langerhans Cell-like Dendritic Cell Subset–based Vaccines for Hematological Malignancies. J. Immunother. 2014, 37, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.; Buntz, S.; Scotland, P.; Xu, L. A cord blood monocyte–derived cell therapy product accelerates brain remyelination. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Kurtzberg, J. Preclinical characterization of DUOC-01, a cell therapy product derived from banked umbilical cord blood for use as an adjuvant to umbilical cord blood transplantation for treatment of inherited metabolic diseases. Cytotherapy 2015, 17, 803–815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wei, X.-C.; Yang, D.-D.; Han, X.-R.; Zhao, Y.-A.; Li, Y.-C.; Zhang, L.-J.; Wang, J.-J. Bioactivity of umbilical cord blood dendritic cells and anti-leukemia effect. Int. J. Clin. Exp. Med. 2015, 8, 19725–19730. [Google Scholar] [PubMed]
- Than, U.T.T.; Le, H.T.; Hoang, D.H.; Nguyen, X.-H.; Pham, C.T.; Van Bui, K.T.; Bui, H.T.H.; Van Nguyen, P.; Nguyen, T.D.; Do, T.T.H.; et al. Induction of Antitumor Immunity by Exosomes Isolated from Cryopreserved Cord Blood Monocyte-Derived Dendritic Cells. Int. J. Mol. Sci. 2020, 21, 1834. [Google Scholar] [CrossRef]
- Aggarwal, R.; Lu, J.; Kanji, S.; Das, M.; Joseph, M.; Lustberg, M.B.; Ray, A.; Pompili, V.J.; Shapiro, C.L.; Das, H. Human Vγ2Vδ2 T cells limit breast cancer growth by modulating cell survival-, apoptosis-related molecules and microenvironment in tumors. Int. J. Cancer 2013, 133, 2133–2144. [Google Scholar] [CrossRef]
- Womble, T.; Green, S.; Shahaduzzaman, M.; Grieco, J.; Sanberg, P.; Pennypacker, K.; Willing, A. Monocytes are essential for the neuroprotective effect of human cord blood cells following middle cerebral artery occlusion in rat. Mol. Cell. Neurosci. 2014, 59, 76–84. [Google Scholar] [CrossRef]
- Marchetti, V.; Yanes, O.; Aguilar, E.; Wang, M.; Friedlander, D.; Moreno, S.; Storm, K.; Zhan, M.; Naccache, S.; Nemerow, G.; et al. Differential Macrophage Polarization Promotes Tissue Remodeling and Repair in a Model of Ischemic Retinopathy. Sci. Rep. 2011, 1, 76. [Google Scholar] [CrossRef]
- Habib, A. Human Cord Blood Serum-Derived APP α-Secretase Cleavage Activity is Mediated by C1 Complement. Cell Transplant. 2018, 27, 666–676. [Google Scholar] [CrossRef]
- Venkat, P.; Culmone, L.; Chopp, M.; Landschoot-Ward, J.; Wang, F.; Zacharek, A.; Chen, J. HUCBC Treatment Improves Cognitive Outcome in Rats with Vascular Dementia. Front. Aging Neurosci. 2020, 12, 1–14. [Google Scholar] [CrossRef]
- Poltavets, A.S.; Vishnyakova, P.A.; Elchaninov, A.V.; Sukhikh, G.T.; Fatkhudinov, T.K. Macrophage Modification Strategies for Efficient Cell Therapy. Cells 2020, 9, 1535. [Google Scholar] [CrossRef] [PubMed]
- Tracy, E.T.; Zhang, C.Y.; Gentry, T.; Shoulars, K.W.; Kurtzberg, J. Isolation and expansion of oligodendrocyte progenitor cells from cryopreserved human umbilical cord blood. Cytotherapy 2011, 13, 722–729. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, J. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat. Commun. 2018, 9, 4092. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Batra, S.; Jeyaseelan, S. Deletion ofNlrp3Augments Survival during Polymicrobial Sepsis by Decreasing Autophagy and Enhancing Phagocytosis. J. Immunol. 2016, 198, 1253–1262. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nat. Cell Biol. 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- June, H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Beatty, L.; O’Hara, M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol. Ther. 2016, 166, 30–39. [Google Scholar] [CrossRef]
- Ritchie, L. In vivo tracking of macrophage activated killer cells to sites of metastatic ovarian carcinoma. Cancer Immunol. Immunother. 2006, 56, 155–163. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Cunningham, S.; Hackstein, H. Recent Advances in Good Manufacturing Practice-Grade Generation of Dendritic Cells. Transfus. Med. Hemother. 2020, 47, 454–463. [Google Scholar] [CrossRef]
- Kumar, B.; Kale, V.; Limaye, L. Umbilical cord blood-derived CD11c+ dendritic cells could serve as an alternative allogeneic source of dendritic cells for cancer immunotherapy. Stem Cell Res. Ther. 2015, 6, 184. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.; Rubinstein, P.; Zhang, M.-J.; Stevens, C.; Kurtzberg, J.; Scaradavou, A.; Loberiza, F.R.; E Champlin, R.; Klein, J.P.; Horowitz, M.M.; et al. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: A comparison study. Lancet 2007, 369, 1947–1954. [Google Scholar] [CrossRef]
- Palmerini, S.; Pfannenstiel, V.; Waldmann, A.; Bergs, J.W.J.; Brill, B.; Huenecke, S.; Klingebiel, T.; Rodel, F.; Buchholz, C.J.; Wels, W.S.; et al. A serum-free protocol for the ex vivo expansion of Cytokine-Induced Killer cells using gas-permeable static culture flasks. Cytotherapy 2020, 22, 511–518. [Google Scholar] [CrossRef]
- Buschow, I.; Hoen, E.N.M.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.; Rasopo, G.; Wubbolts, R.; et al. MHC II in Dendritic Cells is Targeted to Lysosomes or T Cell-Induced Exosomes Via Distinct Multivesicular Body Pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Wahlund, C.J.E.; Güclüler, G.; Hiltbrunner, S.; Veerman, R.E.; Näslund, T.I.; Gabrielsson, S. Exosomes from antigen-pulsed dendritic cells induce stronger antigen-specific immune responses than microvesicles in vivo. Sci. Rep. 2017, 7, 17095. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.-P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell Subset | Adult | Cord Blood | References | ||
|---|---|---|---|---|---|
| % | ×103/mL | % | ×103/mL | ||
| Monocytes | 5.28 | 397.85 | 7.91 | 1338.42 | [14] |
| Classical | 74.70 | 302.98 | 85.55 | 1160.95 | |
| Intermediate | 3.41 | 14.16 | 3.22 | 45.4 | |
| Non-classical | 12.00 | 43.33 | 4.81 | 61.25 | |
| Dendritic cells | 0.49 | 37.12 | 0.35 | 56.54 | [14,33,34] |
| mDC | 0.14 | 10.24 | 0.06 | 9.49 | |
| pDC | 0.05 | 3.55 | 0.06 | 8.14 | |
| B cells | 2.41 | 173.99 | 3.10 | 518.94 | [14] |
| Immature | 2.10 | 2.58 | 9.24 | 34.15 | |
| Memory | 22.5 | 31.39 | - | - | |
| Plasmablast | 2.60 | 4.6 | - | - | |
| B1 cells | 3.51 | 6.11 | 1.72 | 9.11 | |
| ILC | [35] | ||||
| ILC1 | 29.00 | 0.468 | [35] | [35] | |
| ILC2 | 31.00 | 0.585 | [35] | [35] | |
| ILC3 | 32.00 | 0.513 | [35] | [35] | |
| Condition | Recipient | UCB-APC | Effect | References |
|---|---|---|---|---|
| Acute ischemic stroke | Human | Monocytes | Ongoing recruitment | NCT02433509 |
| middle cerebral artery occlusion | Rat | CD14+ monocytes |
| [156] |
| Cerebral palsy | Mouse | CD14+ monocytes |
| [160] |
| Alzheimer’s disease | Mouse | CD14+ monocytes |
| [161,162] |
| Pediatric AML | In vitro | CD34+-derived DC electroporated with WT1 mRNA |
| [163,164] |
| Adult AML | In vitro and Mouse | CD34+-derived DC |
| [165] |
| Central nervous system injuries | Mouse | DUOC-01 (CD14+ monocytes) |
| [166,167] |
| Other | Ex vivo | CD34+-derived DC |
| [168] |
| In vitro | Monocyte-derived DC exosomes |
| [169] | |
| In vitro | MoDC |
| [72,170] | |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunningham, S.; Hackstein, H. Cord-Blood-Derived Professional Antigen-Presenting Cells: Functions and Applications in Current and Prospective Cell Therapies. Int. J. Mol. Sci. 2021, 22, 5923. https://doi.org/10.3390/ijms22115923
Cunningham S, Hackstein H. Cord-Blood-Derived Professional Antigen-Presenting Cells: Functions and Applications in Current and Prospective Cell Therapies. International Journal of Molecular Sciences. 2021; 22(11):5923. https://doi.org/10.3390/ijms22115923
Chicago/Turabian StyleCunningham, Sarah, and Holger Hackstein. 2021. "Cord-Blood-Derived Professional Antigen-Presenting Cells: Functions and Applications in Current and Prospective Cell Therapies" International Journal of Molecular Sciences 22, no. 11: 5923. https://doi.org/10.3390/ijms22115923
APA StyleCunningham, S., & Hackstein, H. (2021). Cord-Blood-Derived Professional Antigen-Presenting Cells: Functions and Applications in Current and Prospective Cell Therapies. International Journal of Molecular Sciences, 22(11), 5923. https://doi.org/10.3390/ijms22115923