
Regulation of ClC-2 Chloride Channel Proteostasis by Molecular Chaperones: Correction of Leukodystrophy-Associated Defect


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
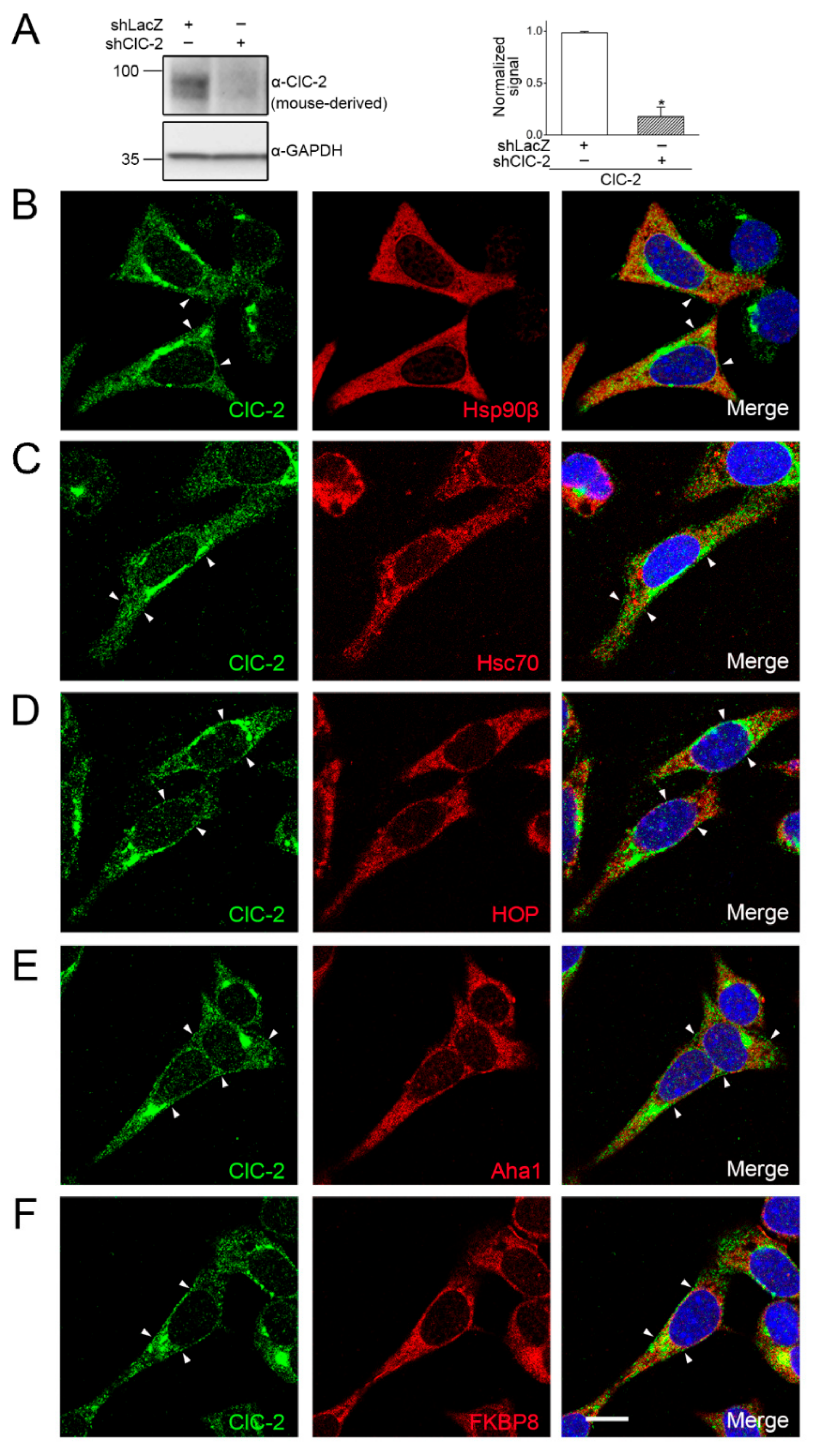
2.1. Association of Endogenous ClC-2 with Chaperones and Co-Chaperones in Native Tissue
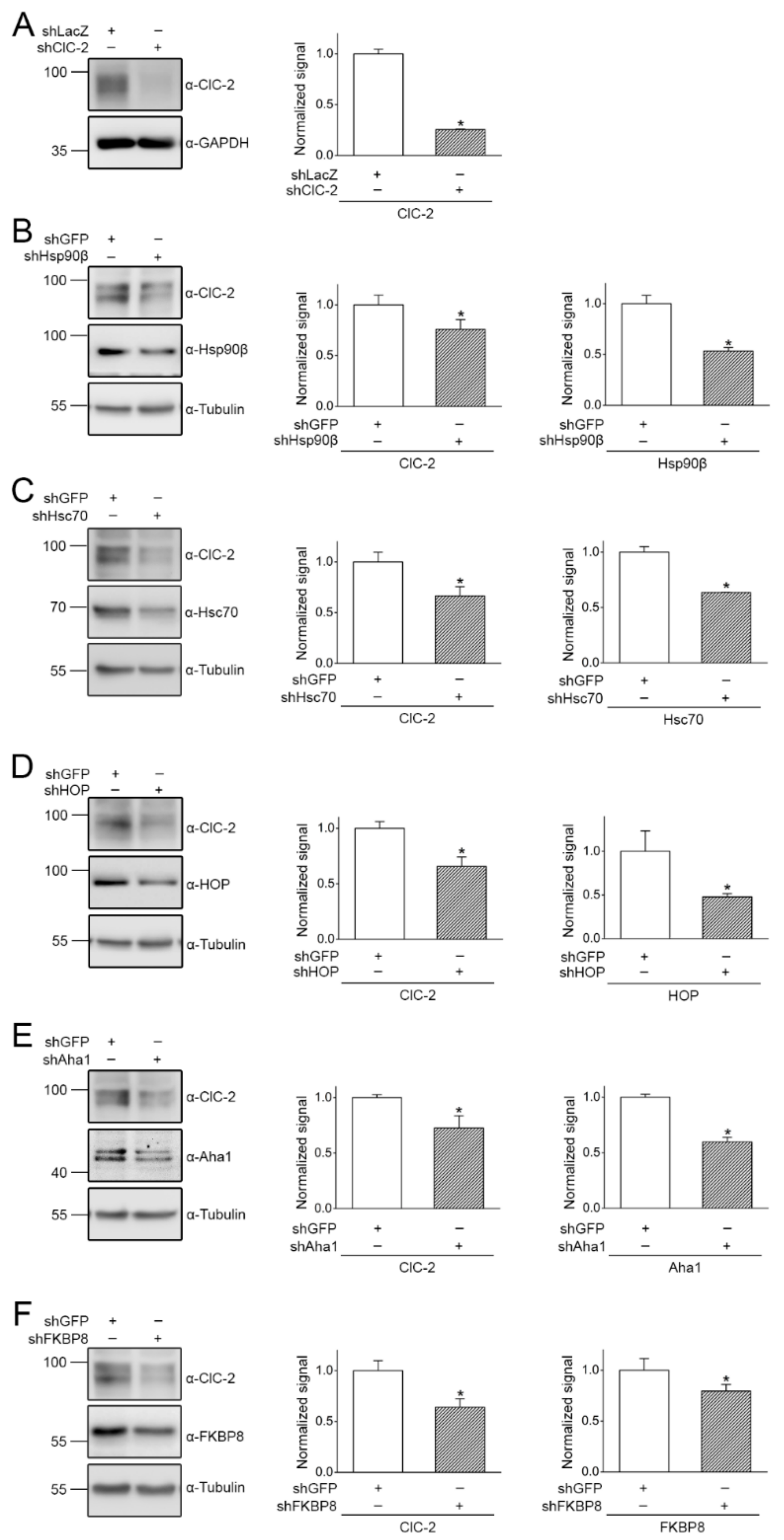
2.2. Regulation of ClC-2 Protein Expression by Chaperones and Co-Chaperones
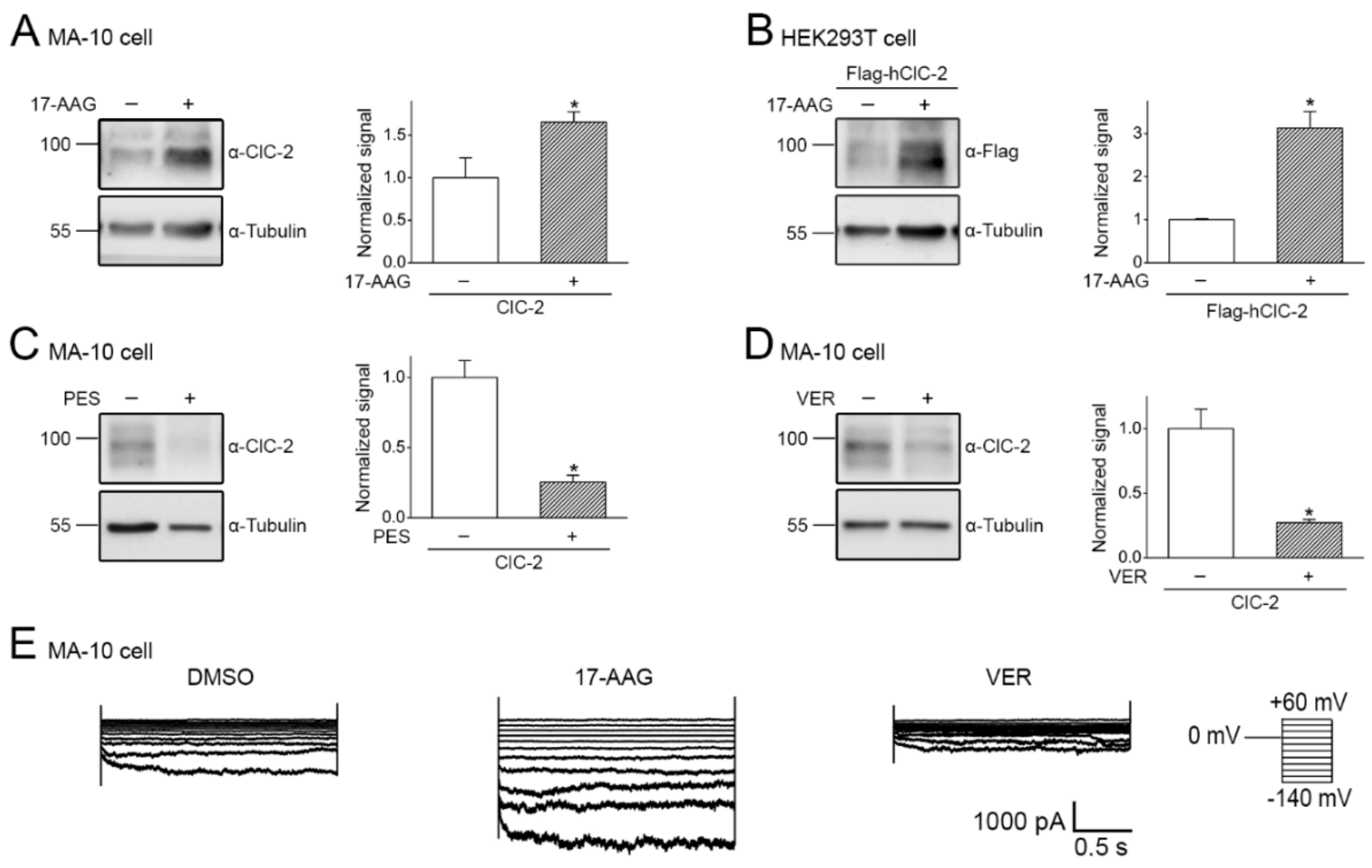
2.3. Pharmacological Promotion of ClC-2 Proteostasis with Hsp90-Targeting Small Molecule
2.4. Correction of Disease-Related Defective ClC-2 Expression by 17-AAG
3. Discussion
4. Materials and Methods
4.1. cDNA Constructs
4.2. Preparation of Animal Samples for Co-Immunoprecipitation
4.3. Cell Culture and DNA Transfection
4.4. RNA Interference
4.5. Immunoblotting
4.6. Immunofluorescence
4.7. Electrophysiology
4.8. Cell Surface Biotinylation
4.9. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 17-AAG | 17-allylamino-17-demethoxygeldanamycin |
| Aha1 | activator of Hsp90 ATPase homolog 1 |
| CHIP | carboxyl terminus Hsc70-interacting protein |
| Cl− | chloride |
| CHO | Chinese hamster ovary |
| CRBN | cereblon |
| CUL | cullin |
| DDB | damage-specific DNA binding protein |
| D-PBS | Dulbecco’s phosphate buffered saline |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| DTT | dithiothreitol |
| ER | endoplasmic reticulum |
| Erg | Ether-à-go-go-related gene |
| FKBP8 | FK506-binding protein 8 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HEK | human embryonic kidney |
| HOP | Hsp70/Hsp90 organizing protein |
| Hsc70 | heat shock cognate protein 70 |
| Hsp90β | heat shock protein 90β |
| IP | immunoprecipitation |
| KV4.3 | voltage-gated potassium channel subtype 4.3 |
| PBS | Phosphate buffered saline |
| PES | 2-phenylethynesulfonamide |
| PMSF | phenylmethylsulfonyl fluoride |
| WT | wild-type |
References
- Cid, L.P.; Montrose-Rafizadeh, C.; Smith, D.I.; Guggino, W.B.; Cutting, G.R. Cloning of a putative human voltage-gated chloride channel (CIC-2) cDNA widely expressed in human tissues. Hum. Mol. Genet. 1995, 4, 407–413. [Google Scholar] [CrossRef]
- Thiemann, A.; Grunder, S.; Pusch, M.; Jentsch, T.J. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature 1992, 356, 57–60. [Google Scholar] [CrossRef]
- Sik, A.; Smith, R.L.; Freund, T.F. Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience 2000, 101, 51–65. [Google Scholar] [CrossRef]
- Gyomorey, K.; Yeger, H.; Ackerley, C.; Garami, E.; Bear, C.E. Expression of the chloride channel ClC-2 in the murine small intestine epithelium. Am. J. Physiol. Cell Physiol. 2000, 279, C1787–C1794. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Grunder, S.; Thiemann, A.; Pusch, M.; Jentsch, T.J. Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature 1992, 360, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.E.; Jentsch, T.J. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 1997, 16, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Nehrke, K.; Arreola, J.; Nguyen, H.-V.; Pilato, J.; Richardson, L.; Okunade, G.; Baggs, R.; Shull, G.E.; Melvin, J.E. Loss of hyperpolarization-activated Cl− current in salivary acinar cells from Clcn2 knockout mice. J. Biol. Chem. 2002, 277, 23604–23611. [Google Scholar] [CrossRef]
- Bösl, M.R.; Stein, V.; Hübner, C.; Zdebik, A.A.; Jordt, S.-E.; Mukhopadhyay, A.K.; Davidoff, M.S.; Holstein, A.-F.; Jentsch, T.J. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl− channel disruption. EMBO J. 2001, 20, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Blanz, J.; Schweizer, M.; Auberson, M.; Maier, H.; Muenscher, A.; Hubner, C.A.; Jentsch, T.J. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci. 2007, 27, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Bugiani, M.; Dupuits, C.; Galanaud, D.; Touitou, V.; Postma, N.; van Berkel, C.; Polder, E.; Tollard, E.; Darios, F.; et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: An observational analytical study. Lancet Neurol. 2013, 12, 659–668. [Google Scholar] [CrossRef]
- Guo, Z.; Lu, T.; Peng, L.; Cheng, H.; Peng, F.; Li, J.; Lu, Z.; Chen, S.; Qiu, W. CLCN2-related leukoencephalopathy: A case report and review of the literature. BMC Neurol. 2019, 19, 156. [Google Scholar]
- Gaitán-Peñas, H.; Apaja, P.M.; Arnedo, T.; Castellanos, A.; Elorza-Vidal, X.; Soto, D.; Gasull, X.; Lukacs, G.L.; Estévez, R. Leukoencephalopathy-causing CLCN2 mutations are associated with impaired Cl− channel function and trafficking. J. Physiol. 2017, 595, 6993–7008. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, D.; Pareyson, D.; Savoiardo, M.; Farina, L.; Ciano, C.; Caldarazzo, S.; Sagnelli, A.; Bonato, S.; Nava, S.; Bresolin, N.; et al. Subclinical leukodystrophy and infertility in a man with a novel homozygous CLCN2 mutation. Neurology 2014, 83, 1217–1218. [Google Scholar] [CrossRef] [PubMed]
- Hoegg-Beiler, M.B.; Sirisi, S.; Orozco, I.J.; Ferrer, I.; Hohensee, S.; Auberson, M.; Gödde, K.; Vilches, C.; De Heredia, M.L.; Nunes, V.; et al. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat. Commun. 2014, 5, 3475. [Google Scholar] [CrossRef] [PubMed]
- Goppner, C.; Soria, A.H.; Hoegg-Beiler, M.B.; Jentsch, T.J. Cellular basis of ClC-2 Cl− channel-related brain and testis pathologies. J. Biol. Chem. 2021, 296, 100074. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Koshimizu, E.; Miyatake, S.; Matsumoto, N.; Imamura, A. A novel homozygous mutation of CLCN2 in a patient with characteristic brain MRI images—A first case of CLCN2-related leukoencephalopathy in Japan. Brain Dev. 2019, 41, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Zeydan, B.; Uygunoglu, U.; Altintas, A.; Saip, S.; Siva, A.; Abbink, T.E.M.; van der Knaap, M.S.; Yalcinkaya, C. Identification of 3 Novel Patients with CLCN2-Related Leukoencephalopathy due to CLCN2 Mutations. Eur. Neurol. 2017, 78, 125–127. [Google Scholar] [CrossRef]
- Giorgio, E.; Vaula, G.; Benna, P.; Buono, N.L.; Eandi, C.M.; Dino, D.; Mancini, C.; Cavalieri, S.; Di Gregorio, E.; Pozzi, E.; et al. A novel homozygous change of CLCN2 (p.His590Pro) is associated with a subclinical form of leukoencephalopathy with ataxia (LKPAT). J. Neurol. Neurosurg. Psychiatry 2017, 88, 894–896. [Google Scholar] [CrossRef]
- Fu, S.J.; Hu, M.C.; Peng, Y.J.; Fang, H.Y.; Hsiao, C.T.; Chen, T.Y.; Jeng, C.J.; Tang, C.Y. CUL4-DDB1-CRBN E3 Ubiquitin Ligase Regulates Proteostasis of ClC-2 Chloride Channels: Implication for Aldosteronism and Leukodystrophy. Cells 2020, 9, 1332. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Bagola, K.; Mehnert, M.; Jarosch, E.; Sommer, T. Protein dislocation from the ER. Biochim. Biophys. Acta 2011, 1808, 925–936. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef]
- Claessen, J.H.; Kundrat, L.; Ploegh, H.L. Protein quality control in the ER: Balancing the ubiquitin checkbook. Trends Cell Biol. 2012, 22, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Houck, S.A.; Cyr, D.M. Mechanisms for quality control of misfolded transmembrane proteins. Biochim. Biophys. Acta 2011, 1818, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar]
- Banasavadi-Siddegowda, Y.K.; Mai, J.; Fan, Y.; Bhattacharya, S.; Giovannucci, D.R.; Sanchez, E.R.; Fischer, G.; Wang, X. FKBP38 peptidylprolyl isomerase promotes the folding of cystic fibrosis transmembrane conductance regulator in the endoplasmic reticulum. J. Biol. Chem. 2011, 286, 43071–43080. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Roth, D.M.; Chalfant, M.A.; Youker, R.T.; Matteson, J.; Brodsky, J.L.; Balch, W.E. FK506 binding protein 8 peptidylprolyl isomerase activity manages a late stage of cystic fibrosis transmembrane conductance regulator (CFTR) folding and stability. J. Biol. Chem. 2012, 287, 21914–21925. [Google Scholar] [CrossRef] [PubMed]
- Koulov, A.V.; LaPointe, P.; Lu, B.; Razvi, A.; Coppinger, J.; Dong, M.-Q.; Matteson, J.; Laister, R.; Arrowsmith, C.; Yates, J.R.; et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 2010, 21, 871–884. [Google Scholar]
- Peng, Y.J.; Huang, J.J.; Wu, H.H.; Hsieh, H.Y.; Wu, C.Y.; Chen, S.C.; Chen, T.Y.; Tang, C.Y. Regulation of CLC-1 chloride channel biosynthesis by FKBP8 and Hsp90beta. Sci. Rep. 2016, 6, 32444. [Google Scholar] [CrossRef] [PubMed]
- Hinzpeter, A.; Lipecka, J.; Brouillard, F.; Baudoin-Legros, M.; Dadlez, M.; Edelman, A.; Fritsch, J. Association between Hsp90 and the ClC-2 chloride channel upregulates channel function. Am. J. Physiol. Cell Physiol. 2006, 290, C45–C56. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Shirane, M.; Nakayama, K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 2003, 5, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Nishimura, Y.; Ichimura, T.; Suzuki, K.; Miyamura, T.; Suzuki, T.; Moriishi, K.; Matsuura, Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006, 25, 5015–5025. [Google Scholar] [CrossRef]
- Rahman, N.A.; Huhtaniemi, I.T. Testicular cell lines. Mol. Cell Endocrinol. 2004, 228, 53–65. [Google Scholar] [CrossRef]
- Zirkin, B.R.; Papadopoulos, V. Leydig cells: Formation, function, and regulation. Biol. Reprod. 2018, 99, 101–111. [Google Scholar] [CrossRef]
- Samant, R.S.; Clarke, P.A.; Workman, P. E3 ubiquitin ligase Cullin-5 modulates multiple molecular and cellular responses to heat shock protein 90 inhibition in human cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, 6834–6839. [Google Scholar] [CrossRef]
- Ehrlich, E.S.; Wang, T.; Luo, K.; Xiao, Z.; Niewiadomska, A.M.; Martinez, T.; Xu, W.; Neckers, L.; Yu, X.-F. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 20330–20335. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hou, Y.; Guo, X.; van der Voet, M.; Boxem, M.; Dixon, J.E.; Chisholm, A.D.; Jin, Y. The EBAX-type Cullin-RING E3 ligase and Hsp90 guard the protein quality of the SAX-3/Robo receptor in developing neurons. Neuron 2013, 79, 903–916. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Samant, R.S.; Clarke, P.A.; Workman, P. The expanding proteome of the molecular chaperone HSP90. Cell Cycle 2012, 11, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Vabulas, R.M.; Macek, B.; Pinkert, S.; Cox, J.; Mann, M.; Hartl, F.U. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol. Cell Proteom. 2012, 11, M111.014654. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Moghaddas Gholami, A.; Kuster, B. Systematic identification of the HSP90 candidate regulated proteome. Mol. Cell Proteom. 2012, 11, M111.016675. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Workman, P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer 2006, 13 (Suppl. 1), S125–S135. [Google Scholar] [CrossRef]
- Chiosis, G.; Rodina, A.; Moulick, K. Emerging Hsp90 inhibitors: From discovery to clinic. Anticancer Agents Med. Chem. 2006, 6, 1–8. [Google Scholar] [CrossRef]
- Jeng, C.J.; Fu, S.J.; You, C.Y.; Peng, Y.J.; Hsiao, C.T.; Chen, T.Y.; Tang, C.Y. Defective Gating and Proteostasis of Human ClC-1 Chloride Channel: Molecular Pathophysiology of Myotonia Congenita. Front. Neurol. 2020, 11, 76. [Google Scholar] [CrossRef]
- Chen, T.Y. Structure and function of clc channels. Annu. Rev. Physiol. 2005, 67, 809–839. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Poet, M.; Fuhrmann, J.C.; Zdebik, A.A. Physiological functions of CLC Cl− channels gleaned from human genetic disease and mouse models. Annu. Rev. Physiol. 2005, 67, 779–807. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Peng, Y.J.; Hu, M.C.; Huang, J.J.; Chien, Y.C.; Wu, J.T.; Chen, T.Y.; Tang, C.Y. The Cullin 4A/B-DDB1-Cereblon E3 Ubiquitin Ligase Complex Mediates the Degradation of CLC-1 Chloride Channels. Sci. Rep. 2015, 5, 10667. [Google Scholar] [CrossRef] [PubMed]
- Stricher, F.; Macri, C.; Ruff, M.; Muller, S. HSPA8/HSC70 chaperone protein: Structure, function, and chemical targeting. Autophagy 2013, 9, 1937–1954. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Daniels, C.K.; Cao, S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C. The role of the cytosolic HSP70 chaperone system in diseases caused by misfolding and aberrant trafficking of ion channels. Dis. Model. Mech. 2014, 7, 319–329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Defranco, D.B. Role of molecular chaperones in subnuclear trafficking of glucocorticoid receptors. Kidney Int. 2000, 57, 1241–1249. [Google Scholar] [CrossRef]
- Picard, D. Chaperoning steroid hormone action. Trends Endocrinol. Metab. 2006, 17, 229–235. [Google Scholar] [CrossRef]
- Cato, L.; Neeb, A.; Brown, M.; Cato, A.C. Control of steroid receptor dynamics and function by genomic actions of the cochaperones p23 and Bag-1L. Nucl. Recept. Signal. 2014, 12, e005. [Google Scholar] [CrossRef]
- Peng, Y.J.; Lee, Y.C.; Fu, S.J.; Chien, Y.C.; Liao, Y.F.; Chen, T.Y.; Jeng, C.J.; Tang, C.Y. FKBP8 Enhances Protein Stability of the CLC-1 Chloride Channel at the Plasma Membrane. Int. J. Mol. Sci. 2018, 19, 3783. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Barriere, H.; Bagdany, M.; Rabeh, W.M.; Du, K.; Hohfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef]
- Pillai, R.N.; Ramalingam, S.S. Heat shock protein 90 inhibitors in non-small-cell lung cancer. Curr. Opin. Oncol. 2014, 26, 159–164. [Google Scholar] [CrossRef]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Jacobs, H.M.; Delmore, J.E.; Buon, L.; Hunter, Z.R.; Monrose, V.; Yu, J.; Smith, P.G.; Richardson, P.G.; Anderson, K.C.; et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol. Cancer Ther. 2012, 11, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nakatani, T.; Kamitani, T. Inhibition of NEDD8-conjugation pathway by novel molecules: Potential approaches to anticancer therapy. Mol. Oncol. 2012, 6, 267–275. [Google Scholar] [CrossRef]
- Brownell, J.E.; Sintchak, M.D.; Gavin, J.M.; Liao, H.; Bruzzese, F.J.; Bump, N.J.; Soucy, T.A.; Milhollen, M.A.; Yang, X.; Burkhardt, A.L.; et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: The NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell 2010, 37, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, S.-J.; Hu, M.-C.; Hsiao, C.-T.; Cheng, A.-T.; Chen, T.-Y.; Jeng, C.-J.; Tang, C.-Y. Regulation of ClC-2 Chloride Channel Proteostasis by Molecular Chaperones: Correction of Leukodystrophy-Associated Defect. Int. J. Mol. Sci. 2021, 22, 5859. https://doi.org/10.3390/ijms22115859
Fu S-J, Hu M-C, Hsiao C-T, Cheng A-T, Chen T-Y, Jeng C-J, Tang C-Y. Regulation of ClC-2 Chloride Channel Proteostasis by Molecular Chaperones: Correction of Leukodystrophy-Associated Defect. International Journal of Molecular Sciences. 2021; 22(11):5859. https://doi.org/10.3390/ijms22115859
Chicago/Turabian StyleFu, Ssu-Ju, Meng-Chun Hu, Cheng-Tsung Hsiao, An-Ting Cheng, Tsung-Yu Chen, Chung-Jiuan Jeng, and Chih-Yung Tang. 2021. "Regulation of ClC-2 Chloride Channel Proteostasis by Molecular Chaperones: Correction of Leukodystrophy-Associated Defect" International Journal of Molecular Sciences 22, no. 11: 5859. https://doi.org/10.3390/ijms22115859
APA StyleFu, S.-J., Hu, M.-C., Hsiao, C.-T., Cheng, A.-T., Chen, T.-Y., Jeng, C.-J., & Tang, C.-Y. (2021). Regulation of ClC-2 Chloride Channel Proteostasis by Molecular Chaperones: Correction of Leukodystrophy-Associated Defect. International Journal of Molecular Sciences, 22(11), 5859. https://doi.org/10.3390/ijms22115859