
Mitochondrial DNA Alterations in Glioblastoma (GBM)


Abstract
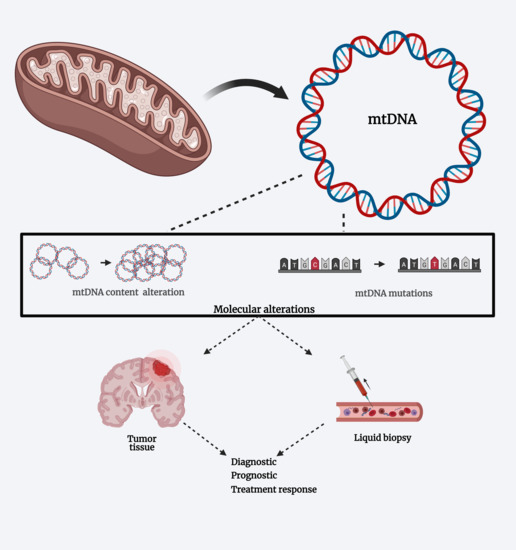
1. Introduction
2. Glioblastoma (GBM)
3. Mitochondria: Genome and Activity
4. Mitochondrial DNA Alterations in Glioblastoma
4.1. Mutations and Polymorphisms
4.2. Variation on MtDNA Content
5. MtDNA Alterations and Liquid Biopsies
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Stetson, L.; Virk, S.M.; Barnholtz-Sloan, J.S. Epidemiology of Gliomas. Cancer Treat. Res. 2015, 163, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Vidone, M.; Clima, R.; Santorsola, M.; Calabrese, C.; Girolimetti, G.; Kurelac, I.; Amato, L.B.; Iommarini, L.; Trevisan, E.; Leone, M.; et al. A comprehensive characterization of mitochondrial DNA mutations in glioblastoma multiforme. Int. J. Biochem. Cell Biol. 2015, 63, 46–54. [Google Scholar] [CrossRef]
- Xiao, A.Y.; Maynard, M.R.; Piett, C.G.; Nagel, Z.D.; Alexander, J.S.; Kevil, C.G.; Berridge, M.V.; Pattillo, C.B.; Rosen, L.R.; Miriyala, S.; et al. Sodium sulfide selectively induces oxidative stress, DNA damage, and mitochondrial dysfunction and radiosensitizes glioblastoma (GBM) cells. Redox Biol. 2019, 26, 101220. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. BioMed Res. Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in gliomas and their vascular microenvironment. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1807, 602–608. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Anni, H.; Dráber, P. Mitochondrial Dysfunction in Gliomas. Semin. Pediatr. Neurol. 2013, 20, 216–227. [Google Scholar] [CrossRef]
- Guntuku, L.; Naidu, V.; Yerra, V.G. Mitochondrial Dysfunction in Gliomas: Pharmacotherapeutic Potential of Natural Compounds. Curr. Neuropharmacol. 2016, 14, 567–583. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Zhang, J.; Li, D.; Qu, F.; Chen, Y.; Li, G.; Jiang, H.; Huang, X.; Yang, H.; Xing, J. Association of leukocyte mitochondrial DNA content with glioma risk: Evidence from a Chinese case-control study. BMC Cancer 2014, 14, 680. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 68, caac.21660. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Weis, S.; Mayr, J.A.; Zimmermann, F.; Geilberger, R.; Sperl, W.; Kofler, B. Alterations of oxidative phosphorylation complexes in astrocytomas. Glia 2014, 62, 514–525. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncology 2016, 18, 16–26. [Google Scholar] [CrossRef]
- Krell, D.; Assoku, M.; Galloway, M.; Mulholland, P.; Tomlinson, I.; Bardella, C. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH Mutations in Glioblastoma. PLoS ONE 2011, 6, e19868. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Rapalino, O.; Gerstner, E.; Chi, A.; Batchelor, T.T.; Cahill, D.P.; Sorensen, A.G.; Rosen, B.R. Detection of oncogenic IDH1 mutations using magnetic resonance spectroscopy of 2-hydroxyglutarate. J. Clin. Investig. 2013, 123, 3659–3663. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef]
- Leather, T.; Jenkinson, M.D.; Das, K.; Poptani, H. Magnetic Resonance Spectroscopy for Detection of 2-Hydroxyglutarate as a Biomarker for IDH Mutation in Gliomas. Metabolites 2017, 7, 29. [Google Scholar] [CrossRef]
- Lin, A.-P.; Abbas, S.; Kim, S.-W.; Ortega, M.; Bouamar, H.; Escobedo, Y.; Varadarajan, P.; Qin, Y.; Sudderth, J.; Schulz, E.; et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nat. Commun. 2015, 6, 7768. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Guan, K.-L.; Xiong, Y. Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Wang, J.; Su, H.-K.; Zhao, H.-F.; Chen, Z.-P.; To, S.-S.T. Progress in the application of molecular biomarkers in gliomas. Biochem. Biophys. Res. Commun. 2015, 465, 1–4. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Teo, W.-Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.-Y.; Lau, C.C.; Yang, H.; Park, J.; Kang, S.-G.; Li, X.; et al. Relevance of a TCGA-derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci. Rep. 2019, 9, 7442. [Google Scholar] [CrossRef]
- Orzan, F.; Pagani, F.; Cominelli, M.; Triggiani, L.; Calza, S.; De Bacco, F.; Medicina, D.; Balzarini, P.; Panciani, P.P.; Liserre, R.; et al. A simplified integrated molecular and immunohistochemistry-based algorithm allows high accuracy prediction of glioblastoma transcriptional subtypes. Lab. Investig. 2020, 100, 1330–1344. [Google Scholar] [CrossRef]
- Madurga, R.; García-Romero, N.; Jiménez, B.; Collazo, A.; Pérez-Rodríguez, F.; Hernández-Laín, A.; Fernández-Carballal, C.; Prat-Acín, R.; Zanin, M.; Menasalvas, E.; et al. Normal tissue content impact on the GBM molecular classification. Briefings Bioinform. 2020, bbaa129. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef]
- Keatley, K.; Stromei-Cleroux, S.; Wiltshire, T.; Rajala, N.; Burton, G.; Holt, W.V.; Littlewood, D.T.J.; Briscoe, A.G.; Jung, J.; Ashkan, K.; et al. Integrated Approach Reveals Role of Mitochondrial Germ-Line Mutation F18L in Respiratory Chain, Oxidative Alterations, Drug Sensitivity, and Patient Prognosis in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 3364. [Google Scholar] [CrossRef]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current Trends in Targeted Therapies for Glioblastoma Multiforme. Neurol. Res. Int. 2012, 2012, 1–13. [Google Scholar] [CrossRef]
- Iranmanesh, Y.; Jiang, B.; Favour, O.C.; Dou, Z.; Wu, J.; Li, J.; Sun, C. Mitochondria’s Role in the Maintenance of Cancer Stem Cells in Glioblastoma. Front. Oncol. 2021, 11, 101. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. The Warburg effect and mitochondrial stability in cancer cells. Mol. Asp. Med. 2010, 31, 60–74. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer Cell Metabolism: One Hallmark, Many Faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 1–10. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Kirches, E. MtDNA As a Cancer Marker: A Finally Closed Chapter? Curr. Genom. 2017, 18, 255–267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anderson, S.E.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.; Grant, R.; Lewis, S.C.; Whittle, P.I.R. Randomized Phase III controlled trials of therapy in malignant glioma: Where are we after 40 years? Br. J. Neurosurg. 2008, 22, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; John, J.C.S. Modulation of mitochondrial DNA copy number in a model of glioblastoma induces changes to DNA methylation and gene expression of the nuclear genome in tumours. Epigenet. Chromatin 2018, 11, 1–18. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, Y.; Gao, K.; Yang, Q.; Shi, B.; Hou, P.; Ji, M. High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am. J. Cancer Res. 2015, 5, 1207–1216. [Google Scholar]
- Stewart, J.B.; Alaei-Mahabadi, B.; Sabarinathan, R.; Samuelsson, T.; Gorodkin, J.; Gustafsson, C.M.; Larsson, E. Simultaneous DNA and RNA Mapping of Somatic Mitochondrial Mutations across Diverse Human Cancers. PLoS Genet. 2015, 11, e1005333. [Google Scholar] [CrossRef]
- Yusoff, A.A.M.; Nasir, K.N.M.; Haris, K.; Khair, S.Z.N.M.; Ghani, A.R.I.A.; Idris, Z.; Abdullah, J.M. Detection of somatic mutations in the mitochondrial DNA control region D-loop in brain tumors: The first report in Malaysian patients. Oncol. Lett. 2017, 14, 5179–5188. [Google Scholar] [CrossRef]
- DeHaan, C.; Habibi-Nazhad, B.; Yan, E.; Salloum, N.; Parliament, M.; Allalunis-Turner, J. Mutation in mitochondrial complex I ND6 subunit is associated with defective response to hypoxia in human glioma cells. Mol. Cancer 2004, 3, 19. [Google Scholar] [CrossRef]
- Kirches, E.; Krause, G.; Warich-Kirches, M.; Weis, S.; Schneider, T.; Meyer-Puttlitz, B.; Mawrin, C.; Dietzmann, K. High frequency of mitochondrial DNA mutations in glioblastoma multiforme identified by direct sequence comparison to blood samples. Int. J. Cancer 2001, 93, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.; Salas, A.; Gamborino, E.; Sobrido, M.J.; Macaulay, V.; Carracedo, Á. mtDNA mutations in tumors of the central nervous system reflect the neutral evolution of mtDNA in populations. Oncogene 2003, 23, 1314–1320. [Google Scholar] [CrossRef][Green Version]
- Kirches, E.; Michael, M.; Woy, C.; Schneider, T.; Warich-Kirches, M.; Schneider-Stock, R.; Winkler, K.; Wittig, H.; Dietzmann, K. Loss of heteroplasmy in the displacement loop of brain mitochondrial DNA in astrocytic tumors. Genes Chromosom. Cancer 1999, 26, 80–83. [Google Scholar] [CrossRef]
- Altafi, D.; Sadeghi, S.; Hojatian, H.; Afra, M.T.; Kar, S.P.; Gorji, M.; Houshmand, M. Mitochondrial Polymorphisms, in The D-Loop Area, Are Associated with Brain Tumors. Cell J 2019, 21, 350–356. [Google Scholar]
- Yeung, K.Y.; Dickinson, A.; Donoghue, J.F.; Polekhina, G.; White, S.J.; Grammatopoulos, D.K.; McKenzie, M.; Johns, T.G.; John, J.C.S. The identification of mitochondrial DNA variants in glioblastoma multiforme. Acta Neuropathol. Commun. 2014, 2, 1. [Google Scholar] [CrossRef]
- Montanini, L.; Regna-Gladin, C.; Eoli, M.; Albarosa, R.; Carrara, F.; Zeviani, M.; Bruzzone, M.G.; Broggi, G.; Boiardi, A.; Finocchiaro, G. Instability of mitochondrial DNA and MRI and clinical correlations in malignant gliomas. J. Neuro-Oncol. 2005, 74, 87–90. [Google Scholar] [CrossRef]
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; Seidman, J.G.; et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091. [Google Scholar] [CrossRef]
- Lloyd, R.E.; Keatley, K.; Littlewood, D.T.J.; Meunier, B.; Holt, W.V.; An, Q.; Higgins, S.C.; Polyzoidis, S.; Stephenson, K.F.; Ashkan, K.; et al. Identification and functional prediction of mitochondrial complex III and IV mutations associated with glioblastoma. Neuro-Oncology 2015, 17, 942–952. [Google Scholar] [CrossRef]
- Shen, J.; Song, R.; Lu, Z.; Zhao, H. Mitochondrial DNA copy number in whole blood and glioma risk: A case control study. Mol. Carcinog. 2016, 55, 2089–2094. [Google Scholar] [CrossRef]
- Song, Z.; Laleve, A.; Vallières, C.; McGeehan, J.E.; Lloyd, R.E.I.; Meunier, B. Human Mitochondrial Cytochrome b Variants Studied in Yeast: Not All Are Silent Polymorphisms. Hum. Mutat. 2016, 37, 933–941. [Google Scholar] [CrossRef]
- Griguer, C.E.; Cantor, A.B.; Fathallah-Shaykh, H.M.; Gillespie, G.Y.; Gordon, A.S.; Markert, J.M.; Radovanovic, I.; Clement-Schatlo, V.; Shannon, C.N.; Oliva, C.R. Prognostic Relevance of Cytochrome c Oxidase in Primary Glioblastoma Multiforme. PLoS ONE 2013, 8, e61035. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Guerra, F.; Cormio, G.; Pesce, V.; Fracasso, F.; Loizzi, V.; Resta, L.; Putignano, G.; Cantatore, P.; Selvaggi, L.E.; et al. Mitochondrial DNA content and mass increase in progression from normal to hyperplastic to cancer endometrium. BMC Res. Notes 2012, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.T.Y.; Cain, J.E.; Cuddihy, A.; Johnson, J.; Dickinson, A.; Yeung, K.-Y.; Kumar, B.; Johns, T.G.; Watkins, D.N.; Spencer, A.; et al. Mitochondrial DNA plasticity is an essential inducer of tumorigenesis. Cell Death Discov. 2016, 2, 16016. [Google Scholar] [CrossRef]
- Dardaud, L.-M.; Bris, C.; Desquiret-Dumas, V.; Boisselier, B.; Tabouret, E.; Mokhtari, K.; Figarella-Branger, D.; Rousseau, A.; Procaccio, V. High mitochondrial DNA copy number is associated with longer survival in young patients with glioblastoma. Neuro-Oncology 2019, 21, 1084–1085. [Google Scholar] [CrossRef]
- Dickinson, A.M.; Yeung, K.Y.; Donoghue, J.P.; Baker, M.J.; Kelly, R.D.; McKenzie, M.; Johns, T.G.; John, J.C.S. The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ. 2013, 20, 1644–1653. [Google Scholar] [CrossRef]
- Sravya, P.; Nimbalkar, V.P.; Kanuri, N.N.; Sugur, H.; Verma, B.K.; Kundu, P.; Rao, S.; Krishna, A.U.; Somanna, S.; Kondaiah, P.; et al. Low mitochondrial DNA copy number is associated with poor prognosis and treatment resistance in glioblastoma. Mitochondrion 2020, 55, 154–163. [Google Scholar] [CrossRef]
- Saenz-Antoñanzas, A.; Auzmendi-Iriarte, J.; Carrasco-Garcia, E.; Moreno-Cugnon, L.; Ruiz, I.; Villanua, J.; Egaña, L.; Otaegui, D.; Samprón, N.; Matheu, A. Liquid Biopsy in Glioblastoma: Opportunities, Applications and Challenges. Cancers 2019, 11, 950. [Google Scholar] [CrossRef]
- Klekner, Á.; Szivos, L.; Virga, J.; Árkosy, P.; Bognár, L.; Birkó, Z.; Nagy, B. Significance of liquid biopsy in glioblastoma—A review. J. Biotechnol. 2019, 298, 82–87. [Google Scholar] [CrossRef]
- Bark, J.M.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br. J. Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef]
- Sareen, H.; Garrett, C.; Lynch, D.; Powter, B.; Brungs, D.; Cooper, A.; Po, J.; Koh, E.-S.; Vessey, J.Y.; McKechnie, S.; et al. The Role of Liquid Biopsies in Detecting Molecular Tumor Biomarkers in Brain Cancer Patients. Cancers 2020, 12, 1831. [Google Scholar] [CrossRef] [PubMed]
- Birkó, Z.; Nagy, B.; Klekner, Á.; Virga, J. Novel Molecular Markers in Glioblastoma—Benefits of Liquid Biopsy. Int. J. Mol. Sci. 2020, 21, 7522. [Google Scholar] [CrossRef] [PubMed]
- Hallal, S.; Ebrahimkhani, S.; Shivalingam, B.; Graeber, M.B.; Kaufman, K.L.; Buckland, M.E. The emerging clinical potential of circulating extracellular vesicles for non-invasive glioma diagnosis and disease monitoring. Brain Tumor Pathol. 2019, 36, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, M.; Pös, O.; Márton, É.; Buglyó, G.; Soltész, B.; Keserű, J.; Penyige, A.; Szemes, T.; Nagy, B. Circulating Cell-Free Nucleic Acids: Main Characteristics and Clinical Application. Int. J. Mol. Sci. 2020, 21, 6827. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Huang, X.; Zhou, X.; Hu, J.; Li, G.; He, S.; Xing, J. High leukocyte mitochondrial DNA content contributes to poor prognosis in glioma patients through its immunosuppressive effect. Br. J. Cancer 2015, 113, 99–106. [Google Scholar] [CrossRef][Green Version]

{kind=link}
| Region/Gene | Variation | Type | Amino Acid (aa) Change | Reference |
|---|---|---|---|---|
| D-LOOP | Np61 C→A | Transversion | - | [49] |
| Np64 C→T | Transition | - | [50] | |
| Np71 | G-deletion | - | [49] | |
| Np72 T→C | Transition | - | [51] | |
| Np73 A→G | Transition | - | [44,47,48] | |
| Np146 T→C | Transition | - | [49,52] | |
| Np150 C→T | Transition | - | [53] | |
| Np152 T→C | Transition | - | [49,50] | |
| Np152 A→G | Transition | - | [54] | |
| Np176 | A-deletion | - | [49] | |
| Np185 G→A | Transition | - | [53] | |
| Np185 A→G | Transition | - | [51] | |
| Np186 C→G | Transition | - | [49] | |
| Np189 A→G | Transition | - | [53] | |
| Np194 T→C | Transition | - | [55] | |
| Np195 T→C | Transition | - | [49,50,51,52,54] | |
| Np204 T→C | Transition | - | [53] | |
| Np 204 C→T | Transition | - | [44,46,47,49] | |
| Np207 G→A | Transition | - | [54] | |
| Np249 | A-deletion | - | [49] | |
| Np263 A→G | Transition | - | [44,45,47,49] | |
| Np295 C→T | Transition | - | [51] | |
| Np302 A→C | Transversion | - | [55] | |
| Np303 | CC-insertion | - | [49] | |
| Np303–309 | C-insertion | - | [50] | |
| Np310 T→C | Transition | - | [55] | |
| Np310 (C8TC6)→(C9TC6) | Insertion | - | [51] | |
| Np310 (C7TC6)→(C9TC6) | Insertion | - | [51] | |
| Np311 | CC-insertion | - | [49] | |
| Np311–315 | C-insertion | - | [50] | |
| Np320 C→T | Transition | - | [50] | |
| Np343 C→G | Transversion | - | [50] | |
| Np411 C→G/C | Heteroplasmy | - | [49] | |
| Np476 C→A | Transversion | - | [49] | |
| Np489 T→C | Transition | - | [54] | |
| Np514 (CA)4→(CA)6 | Insertion | - | [51] | |
| Np523–524 | AC-deletion | - | [49] | |
| Np566 C→A | Transversion | - | [49] | |
| Np16069 C→T | Transition | - | [54] | |
| Np16126 T→C | Transition | - | [51,54] | |
| Np16129 G→A | Transition | - | [50] | |
| Np16134 C→T | Transition | - | [51] | |
| Np16145 G→A | Transition | - | [54] | |
| Np16148 C→T | Transition | - | [54] | |
| Np16172 T→C | Transition | - | [54] | |
| Np16186 C→T | Transition | - | [54,55] | |
| Np16189 T→C | Transition | - | [49,50,51,54] | |
| Np16189 (C)10→(C)11 | Insertion | - | [51] | |
| Np16189 (C)10→(C)9 | Deletion | - | [51,52] | |
| Np16189 (C)12→(C)11 | Deletion | - | [51] | |
| Np16193 C→T | Transition | - | [54] | |
| Np16218 C→T | Transition | - | [55] | |
| Np16220 A→C | Tranversion | - | [50] | |
| Np16223 C→T | Transition | - | [54] | |
| Np16224 T→C | Transition | - | [54,55] | |
| Np16234 C→T | Transition | - | [54] | |
| Np16256 C→T | Transition | - | [54] | |
| Np16261 C→T | Transition | - | [49,54] | |
| Np16264 C→T | Transition | - | [50] | |
| Np16265 A→G | Transition | - | [49] | |
| Np16270 C→T | Transition | - | [49,54] | |
| Np16274 G→A | Transition | - | [54] | |
| Np16292 C→T | Transition | - | [54] | |
| Np16293 A→G | Transition | - | [51] | |
| Np16311 T→C | Transition | - | [54,56] | |
| Np16316 A→G | Transition | - | [50] | |
| Np16318 A→C | Tranversion | - | [54] | |
| Np16327 C→T | Transition | - | [54] | |
| Np16336 T→C | Transition | - | [56] | |
| Np16355 C→T | Transition | - | [54] | |
| Np16356 T→C | Transition | - | [49,51] | |
| Np16362 T→C | Transition | - | [54] | |
| Np16381 T→C | Transition | - | [49] | |
| Np16384 G→A | Transition | - | [54] | |
| Np16392 G→A | Transition | - | [54] | |
| Np16394 G→A | Transition | - | [54] | |
| Np16405 | A-deletion | - | [49] | |
| Np16438 G→A | Transition | - | [56] | |
| Np16477 G→A | Transition | - | [54] | |
| Np16519 T→C | Transition | - | [49,50,51,54,55] | |
| Np16524 A→G | Transition | - | [56] | |
| Np16526 G→A | Transition | - | [56] | |
| Np16527 C→T | Transition | - | [56] | |
| 16S rRNA | Np1913 G→A | Transition | - | [3] |
| Np1966 G→A | Transition | - | [57] | |
| Np2002 G→A | Transition | - | [55] | |
| Np2130 A→G | Transition | - | [55] | |
| Np2652 G→A | Transition | - | [3] | |
| Np2817 G→A | Transition | - | [55] | |
| Np2976 G→A | Transition | - | [3] | |
| Np3036 G→A | Transition | - | [57] | |
| Np3168 C→T | Transition | - | [55] | |
| tRNAGln | Np4395 A→G | Transition | - | [3] |
| tRNAMet | Np4403 G→A | Transition | - | [3] |
| tRNA | Np596 T→C | Transition | - | [3] |
| tRNA | Np15928 G→A | Transition | - | [54] |
| tRNA | Np15936 T→C | Transition | - | [54] |
| 12S rRNA | Np750 A→G | Transition | - | [50,54] |
| Np1227 G→A | Transition | - | [57] | |
| Np1386 T→C | Transition | - | [55] | |
| Np1415 G→A | Transition | - | [57] | |
| L strand replication origin | Np5752 A→G | Transition | - | [55] |
| ATPase 6 | Np8528 T→C | Transition Nonsynnonymous | Met→Thr (M1T) | [3] |
| Np8603 T→C | Transition Nonsynnonymous | Phe→Ser (F26S) | [3] | |
| Np8632 T→C | Transition Nonsynnonymous | Tyr→His (Y36H) | [3] | |
| Np8668 T→C | Transition Nonsynnonymous | Trp→Arg (W48R) | [3] | |
| Np8860 A→G | Transition Nonsynnonymous | Thr→Ala (T112A) | [50] | |
| Np8919 A→G | Transition Nonsynnonymous | Gln→Gln (Q131Q) | [50] | |
| Np9025 G→A | Transition Nonsynnonymous | Gly→Ser (G167S) | [3] | |
| Np9139 G→A | Transition Nonsynnonymous | Ala→Thr (A205T) | [3] | |
| ATPase 8 | Np8496 T→C | Transition Nonsynnonymous | Met→Thr (M44T) | [3] |
| Np8462 T→C | Transition Nonsynnonymous | Tyr→His (Y33H) | [3] | |
| ND1 | Np3344 T→C | Transition Nonsynnonymous | Ile→Thr (I13T) | [57] |
| Np3357 G→C | Transversion Nonsynnonymous | Met→Ile (M17I) | [3] | |
| Np3538 G→A | Transition Nonsynnonymous | Ala→Thr (A78T) | [3] | |
| Np3631 T→A | Transversion Nonsynnonymous | Ser→Thr (S109T) | [3] | |
| Np3866 T→C | Transition Nonsynnonymous | Ile→Thr (I187T) | [3] | |
| Np4029 C→A | Transversion Nonsynnoymous | Ile→Met (I241M) | [3] | |
| Np4136 A→G | Transition Nonsynnoymous | Tyr→Cys (Y277C) | [3] | |
| ND2 | Np4646 T→C | Transition Synnonymous | Tyr→Tyr (Y59Y) | [51] |
| Np4752 T→C | Transition Nonsynnoymous | Ser→Pro (S95P) | [3] | |
| Np4762 T→C | Transition Nonsynnonymous | Ile→Thr (I98T) | [57] | |
| Np4769 A→G | Transition Synnonymous | Met→Met (M100M) | [50] | |
| Np5198 A→G | Transition Synnonymous | Leu→Leu (L243L) | [51] | |
| Np5263 C→T | Transition Nonsynnoymous | Ala→Val (A265V) | [3] | |
| ND3 | Np10398 A→G | Transition Nonsynnoymous | Thr→Ala (T114A) | [49] |
| ND4L | Np10586 G→A | Transition Synnonymous | Ser→Ser (S39S) | [57] |
| Np10473 C→G | Tranversion Nonsynnoymous | Pro→Ala (P2A) | [55] | |
| ND4 | Np10814 A→C | Tranversion Nonsynnoymous | Lys→Gln (K19Q) | [55] |
| Np10946 | C-insertion | Frameshift | [3] | |
| Np11084 A→G | Transition Nonsynnoymous | Thr→Ala (T109A) | [3] | |
| Np11711 G→A | Transition Nonsynnoymous | Ala→Thr (A318T) | [3] | |
| Np11718 G→A | Transition Nonsynnoymous | Gly→Glu (G320E) | [3] | |
| Np11719 G→A | Transition Synnoymous | Gly→Gly (G320G) | [50] | |
| Np11361 T→C | Transition Nonsynnoymous | Met→Thr (M201T) | [55] | |
| Np11512 C→A | Tranversion Nonsynnoymous | Asn→Lys (N251K) | [55] | |
| Np11674 C→T | Transition Synnonymous | Thr→Thr (T305T) | [55] | |
| Np11693 G→A | Transition Nonsynnoymous | Ala→Thr (A312T) | [3] | |
| Np11866 | C-insertion | Frameshift | [3] | |
| Np11928 A→G | Transversion Nonsynnoymous | Asn→Ser (N390S) | [3] | |
| Np11978 T→A | Transition Nonsynnoymous | Ser→Thr (S407T) | [3] | |
| Np12033 A→G | Transition Nonsynnoymous | Asn→Ser (N425S) | [3] | |
| Np12101 T→C | Transition Nonsynnoymous | Ser→Pro (S448P) | [55] | |
| Np12102 C→T | Transition Nonsynnoymous | Ser→Phe (S448F) | [55] | |
| ND5 | Np12418 | A-deletion | Frameshift | [3] |
| Np12936 A→G | Transition Synnonymous | Gln→Gln (Q200Q) | [51] | |
| Np12877 G→C | Transversion Nonsynnoymous | Gly→Arg (G181R) | [55] | |
| Np13061 C→A | Transversion Nonsynnoymous | Pro→Gln (P242Q) | [55] | |
| Np13043 C→T | Transition Nonsynnoymous | Ala→Val (A236V) | [55] | |
| Np13063 G→A | Transition Nonsynnoymous | Val→Ile (V243I) | [3] | |
| Np13112 T→C | Transition Nonsynnoymous | Leu→Ser (L259S) | [3] | |
| Np13124 T→C | Transition Nonsynnoymous | Phe→Ser (F263S) | [51] | |
| Np13135 G→A | Transition Nonsynnoymous | Ala→Thr (A267T) | [3] | |
| Np13468 C→A | Transvertion Nonsynnoymous | Leu→Met (L378M) | [3] | |
| Np13613 T→C | Transition Nonsynnoymous | Met→Thr (M426T) | [3] | |
| Np13835 C→T | Transition Nonsynnoymous | Thr→Met (T500M) | [3] | |
| Np13858 A→G | Transition Nonsynnoymous | Thr→Ala (T508A) | [3] | |
| Np13879 T→C | Transition Nonsynnoymous | Ser→Pro (S515P) | [3] | |
| ND6 | Np14159 C→G | Transversion Nonsynnoymous | Arg→Pro (R172P) | [55] |
| Np14160 G→C | Transversion Nonsynnoymous | Arg→Gly (R172G) | [55] | |
| Np14180 T→C | Transition Nonsynnoymous | Tyr→Cys (Y165C) | [3] | |
| Np14426 C→T | Transition Nonsynnoymous | Gly→Glu (G85E) | [55] | |
| Np14470 T→C | Transition Synnonymous | Gly→Gly (G68G) | [50] | |
| Np14620 C→T | Transition Synnonymous | Gly→Gly (G18G) | [51] | |
| Np14634 T→C | Transition Nonsynnoymous | Met→Val (M14V) | [50] | |
| CYTB | Np14766 C→T | Transition Nonsynnoymous | Thr→Ile (T7I) | [58] |
| Np14793 A→G | Transition Nonsynnoymous | His→Arg (H16R) | [58] | |
| Np14798 C→T | Transition Nonsynnoymous | Phe→Leu (F18L) | [25,45,46,52,53] | |
| Np14814 C→T | Transition Nonsynnoymous | Thr→Ile (T23I) | [3] | |
| Np15042 G→A | Transition Nonsynnoymous | Gly→Glu (G99E) | [3] | |
| Np15071 T→C | Transition Nonsynnoymous | Tyr→His (Y109H) | [3] | |
| Np15168 G→A | Transition | Premature Stop Codon | [3] | |
| Np15048 G→C | Transition Nonsynnoymous | Gly→Ala (G101A) | [58,59] | |
| Np15218 A→G | Transition Nonsynnoymous | Thr→Ala (T158A) | [58] | |
| Np15257 G→A | Transition Nonsynnoymous | Asn→Asp (N171D) | [58,60] | |
| Np15264 C→T | Transition Nonsynnoymous | Pro→Leu (P173L) | [55] | |
| Np15267 C→G | Transversion Nonsynnoymous | Thr→Ser (T174S) | [55] | |
| Np15326 A→G | Transition Nonsynnoymous | Thr→Ala (T194A) | [50,58] | |
| Np15375 G→A | Transition Nonsynnoymous | Gly→Glu (G210E) | [3] | |
| Np15452 C→A | Transversion Nonsynnoymous | Leu→Ile (L236I) | [58] | |
| Np15453 T→C | Transition Nonsynnoymous | Leu→Pro (L236P) | [58] | |
| Np15479 T→C | Transition Nonsynnoymous | Phe→Leu (F245L) | [58] | |
| Np15500 G→A | Transition Nonsynnoymous | Asp→Asn (D252N) | [58,60] | |
| Np15506 G→A | Transition Nonsynnoymous | Asp→Asn (D254N) | [3] | |
| Np15693 T→C | Transition Nonsynnoymous | Met→Thr (M316T) | [58] | |
| Np15758 A→G | Transition Nonsynnoymous | Ile→Val (I338V) | [58] | |
| Np15773 G→A | Transition Nonsynnoymous | Val→Met (V343M) | [3] | |
| Np15803 G→A | Transition Nonsynnoymous | Val→Met (V353M) | [58] | |
| Np15884 G→C | Transversion Nonsynnoymous | Ala→Pro (A380P) | [54] | |
| COI | Np5999 T→C | Transition Synnonymous | Ala→Ala (A32A) | [51] |
| Np6047 A→G | Transition Synnonymous | Leu→Leu (L48L) | [51] | |
| Np6111 G→A | Transition Nonsynnoymous | Val→Met (V70M) | [3] | |
| Np6180 G→A | Transition Nonsynnoymous | Ala→Thr (A93T) | [3] | |
| Np6237 C→A | Transversion Nonsynnoymous | Leu→Met (L112M) | [58] | |
| Np6267 G→A | Transition Nonsynnoymous | Ala→Thr (A122T) | [58] | |
| Np6422 C→T | Transition Synnonymous | Pro→Pro (P173) | [55] | |
| Np6619 G→A | Transition Nonsynnonymous | Gly→Asp (G239D) | [57] | |
| Np6692 | A-deletion | (M271Ter) | [58] | |
| Np6709 G→A | Transition Nonsynnoymous | Gly→Glu (G269E) | [3] | |
| Np6999 G→A | Transition Nonsynnoymous | Val→Met (V366M) | [55] | |
| Np7080 T→C | Transition Nonsynnoymous | Phe→Leu (F393L) | [3] | |
| Np7153 T→C | Transition Nonsynnoymous | Met→Thr (M417T) | [3] | |
| COII | Np7919 G→A | Transition Nonsynnoymous | Asp→Asn (D112N) | [3] |
| Np7965 T→C | Transition Nonsynnoymous | Phe→Ser (F127S) | [3] | |
| Np7976 G→A | Transition Nonsynnoymous | Gly→Ser (G131S) | [3] | |
| Np8251 G→A | Transition Synnonymous | Gly→Gly (G222G) | [55] | |
| Np8252 C→A | Tranversion Nonsynnoymous | Pro→Thr (P223T) | [55] | |
| COIII | Np9252 T→C | Transition Nonsynnoymous | Trp→Arg (W16R) | [58] |
| Np9276 G→A | Transition Nonsynnoymous | Ala→Thr (A24T) | [3] | |
| Np9280 T→C | Transition Nonsynnoymous | Leu→Pro (L25P) | [3] | |
| Np9300 G→A | Transition Nonsynnoymous | Ala→Thr (A32T) | [58] | |
| Np9438 G→A | Transition Nonsynnoymous | Gly→Ser (G78S) | [3] | |
| Np9469 C→T | Transition Nonsynnoymous | Thr→Ile (T88I) | [58] | |
| Np9477 G→A | Transition Nonsynnoymous | Val→Ile (V91I) | [58] | |
| Np9627 G→A | Transition | Premature stop codon | [3] | |
| Np9655 G→A | Transition Nonsynnonymous | Ser→Asn (S150N) | [57,58] | |
| Np9667 A→G | Transition Nonsynnonymous | Asn→Ser (N154S) | [58] | |
| Np9786 G→A | Transition Nonsynnonymous | Gly→Ser (G194S) | [3] | |
| Np9966 G→A | Transition Nonsynnoymous | Val→Ile (V254I) | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leão Barros, M.B.; Pinheiro, D.d.R.; Borges, B.d.N. Mitochondrial DNA Alterations in Glioblastoma (GBM). Int. J. Mol. Sci. 2021, 22, 5855. https://doi.org/10.3390/ijms22115855
Leão Barros MB, Pinheiro DdR, Borges BdN. Mitochondrial DNA Alterations in Glioblastoma (GBM). International Journal of Molecular Sciences. 2021; 22(11):5855. https://doi.org/10.3390/ijms22115855
Chicago/Turabian StyleLeão Barros, Mariceli Baia, Danilo do Rosário Pinheiro, and Bárbara do Nascimento Borges. 2021. "Mitochondrial DNA Alterations in Glioblastoma (GBM)" International Journal of Molecular Sciences 22, no. 11: 5855. https://doi.org/10.3390/ijms22115855
APA StyleLeão Barros, M. B., Pinheiro, D. d. R., & Borges, B. d. N. (2021). Mitochondrial DNA Alterations in Glioblastoma (GBM). International Journal of Molecular Sciences, 22(11), 5855. https://doi.org/10.3390/ijms22115855