
Mechanisms of Nausea and Vomiting: Current Knowledge and Recent Advances in Intracellular Emetic Signaling Systems


 and
and
Abstract
1. Introduction
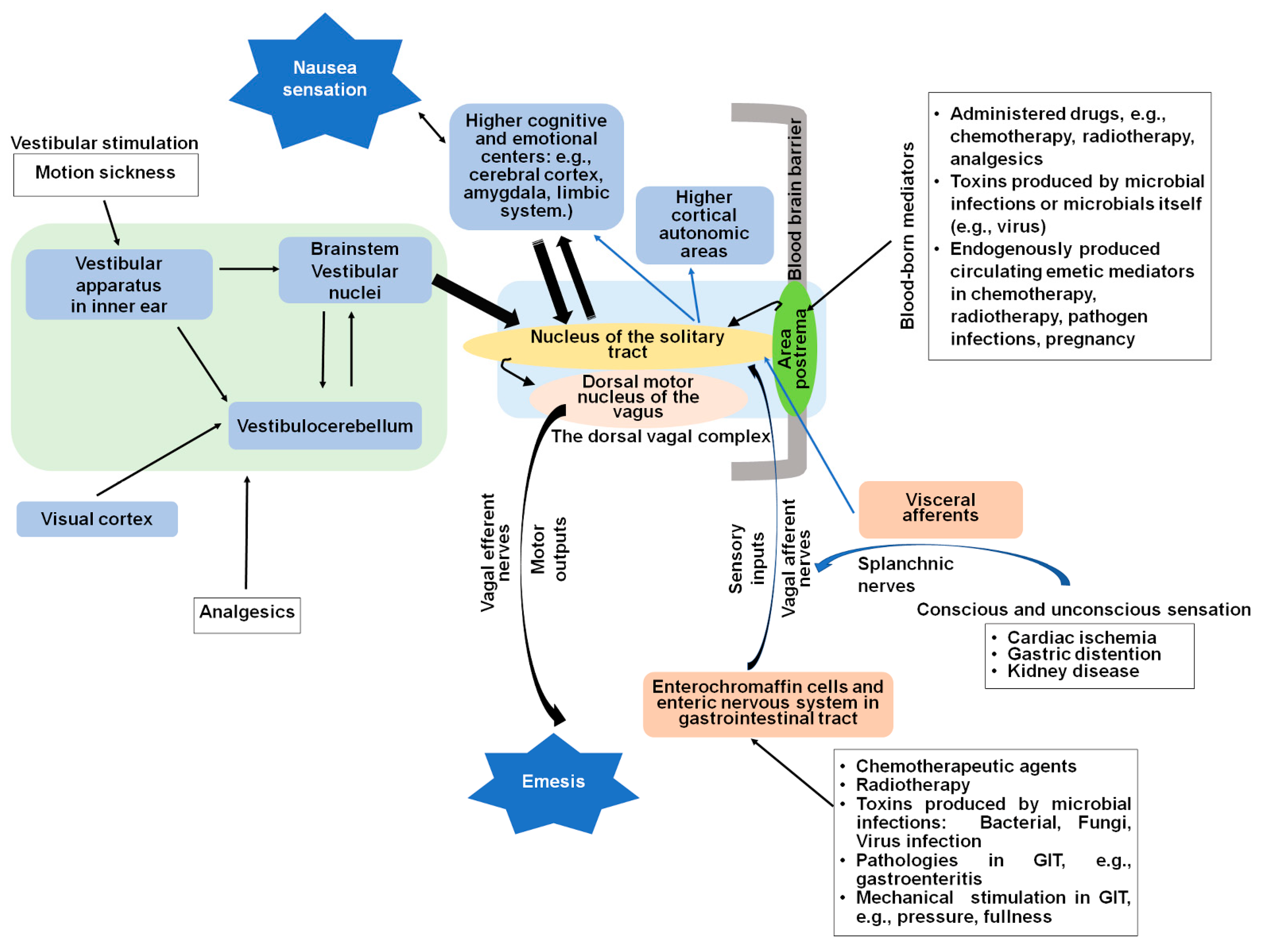
2. Central and Peripheral Sites of Nausea and Vomiting
2.1. The Dorsal Vagal Complex
2.2. Vagal Afferent Pathways Are Involved in Detection of Emetic Stimuli
2.3. The Gastrointestinal Tract
2.4. Nausea Studies: Models, Progress, and Limitations
2.4.1. Human Nausea Studies
2.4.2. Nausea Models in Non-Emetic Species
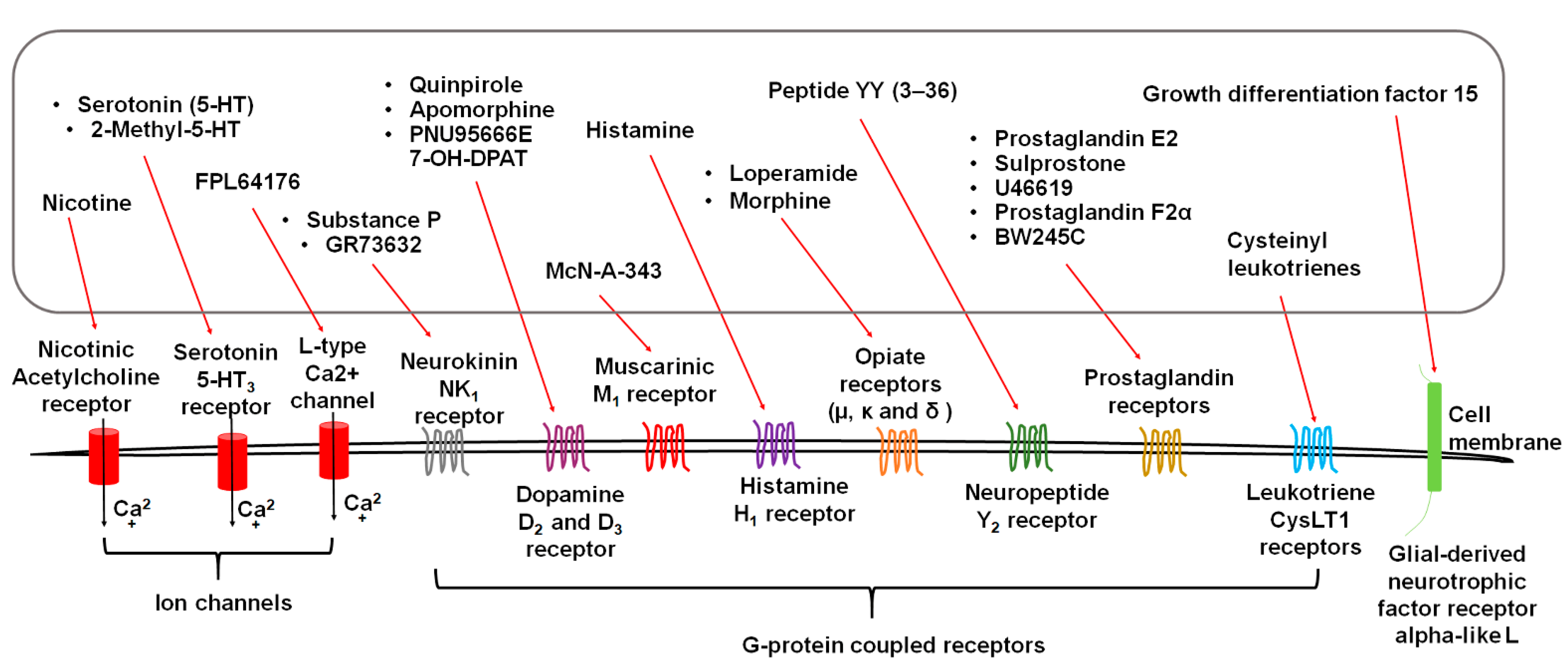
3. Diverse Stimuli Evoke Vomiting via Distinct Receptors
3.1. Serotonin Receptors
3.2. Substance P Neurokinin NK1 Receptor (NK1R)
3.3. Dopamine D2/3 Receptors
3.4. Acetylcholine Receptors
3.5. Histamine H1 Receptor
3.6. Opiate Receptors
3.7. Neuropeptide Y2 Receptors
3.8. GFRAL: Receptor of Growth Differentiation Factor 15 (GDF15)
3.9. Eicosanoid Receptors
3.9.1. Prostaglandins and Receptors
3.9.2. Cysteinyl Leukotrienes and Receptors
4. Physiological Mechanisms of Emesis and Clinical Uses of Antiemetics
4.1. Chemotherapy-Induced Nausea and Vomiting (CINV)
4.2. Radiation-Induced Nausea and Vomiting
4.3. Postoperative Nausea and Vomiting (PONV)
4.4. Cannabinoid Hyperemesis Syndrome
4.5. Motion Sickness: Mechanisms and Treatment
4.6. Emesis Induced by Microbial Infections:
4.6.1. Bacterial Infections and Emesis
4.6.2. Fungal Infections and Emesis
4.6.3. Viral Signaling Mechanisms Responsible for Emesis
4.6.4. Microbiota
5. Intracellular Signal Transduction Systems in Emesis
5.1. Calcium
5.1.1. Emetogens Mobilize Ca2+
5.1.2. Pro- and Antiemetic Effects of L-Type Calcium Channels (LTCC) Agonists and Antagonists
5.1.3. Involvement of Other Ca2+ Channel Modulators in Emesis
5.2. Intracellular Emetic Signals in Diverse Signal Transduction Pathways
5.2.1. Adenylyl Cyclase (cAMP)–Protein Kinase A (PKA)
5.2.2. Extracellular Signal Regulated Kinase 1 and 2 (ERK1/2)
5.2.3. Ca2+/Calmodulin-Dependent Kinase II (CaMKII)
5.2.4. Protein Kinase C (PKC)
5.2.5. Akt/Glycogen Synthase Kinase 3 (GSK-3)
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Gelberg, H. Pathophysiological Mechanisms of Gastrointestinal Toxicity; Elsevier: Amsterdam, The Netherlands, 2018; pp. 139–178. [Google Scholar]
- Singh, P.; Kuo, B. Central aspects of nausea and vomiting in GI disorders. Curr. Treat. Options Gastroenterol. 2016, 14, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Sanger, G.J.; Andrews, P.L. Treatment of nausea and vomiting: Gaps in our knowledge. Auton. Neurosci. 2006, 129, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Dai, M.; Yakushin, S.B.; Cho, C. The neural basis of motion sickness. J. Neurophysiol. 2019, 121, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.D.; Alpers, D.H.; Andrews, P.L.R. Fundamentals of neurogastroenterology. Gut 1999, 45, ii6–ii16. [Google Scholar] [CrossRef] [PubMed]
- Barmack, N.H. Central vestibular system: Vestibular nuclei and posterior cerebellum. Brain Res. Bull. 2003, 60, 511–541. [Google Scholar] [CrossRef]
- Gagliuso, A.H.; Chapman, E.K.; Martinelli, G.P.; Holstein, G.R. Vestibular neurons with direct projections to the solitary nucleus in the rat. J. Neurophysiol. 2019, 122, 512–524. [Google Scholar] [CrossRef]
- Cai, Y.-L.; Ma, W.-L.; Li, M.; Guo, J.-S.; Li, Y.-Q.; Wang, L.-G.; Wang, W.-Z. Glutamatergic vestibular neurons express Fos after vestibular stimulation and project to the NTS and the PBN in rats. Neurosci. Lett. 2007, 417, 132–137. [Google Scholar] [CrossRef]
- Bashashati, M.; McCallum, R.W. Neurochemical mechanisms and pharmacologic strategies in managing nausea and vomiting related to cyclic vomiting syndrome and other gastrointestinal disorders. Eur. J. Pharmacol. 2014, 722, 79–94. [Google Scholar] [CrossRef]
- McKenzie, E.; Chan, D.; Parsafar, S.; Razvi, Y.; McFarlane, T.; Rico, V.; Pasetka, M.; DeAngelis, C.; Chow, E. Evolution of antiemetic studies for radiation-induced nausea and vomiting within an outpatient palliative radiotherapy clinic. Support. Care Cancer 2019, 27, 3245–3252. [Google Scholar] [CrossRef]
- Wickham, R.J. Revisiting the physiology of nausea and vomiting—Challenging the paradigm. Support. Care Cancer 2019, 28, 13–21. [Google Scholar] [CrossRef]
- Navari, R.M. Olanzapine for the prevention and treatment of chronic nausea and chemotherapy-induced nausea and vomiting. Eur. J. Pharmacol. 2014, 722, 180–186. [Google Scholar] [CrossRef]
- Porreca, F.; Ossipov, M.H. Nausea and vomiting side effects with opioid analgesics during treatment of chronic pain: Mechanisms, implications, and management options. Pain Med. 2009, 10, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.S.; Laufer, A. Opioid induced nausea and vomiting. Eur. J. Pharmacol. 2014, 722, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Horn, C.C.; Wallisch, W.J.; Homanics, G.; Williams, J.P. Pathophysiological and neurochemical mechanisms of postoperative nausea and vomiting. Eur. J. Pharmacol. 2014, 722, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Weltens, N.; Iven, J.; Van Oudenhove, L.; Kano, M. The gut-brain axis in health neuroscience: Implications for functional gastrointestinal disorders and appetite regulation. Ann. N. Y. Acad. Sci. 2018, 1428, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Ray, A.P. Evidence for a re-evaluation of the neurochemical and anatomical bases of chemothera-py-induced vomiting. Chem. Rev. 2009, 109, 3158–3199. [Google Scholar] [CrossRef]
- Babic, T.; Browning, K.N. The role of vagal neurocircuits in the regulation of nausea and vomiting. Eur. J. Pharmacol. 2014, 722, 38–47. [Google Scholar] [CrossRef]
- Kenward, H.; Pelligand, L.; Savary-Bataille, K.; Elliott, J. Nausea: Current knowledge of mechanisms, measurement and clinical impact. Veter. J. 2015, 203, 36–43. [Google Scholar] [CrossRef]
- Spiegel, D.R.; Pattison, A.; Lyons, A.; Ansari, U.; McCroskey, A.L.; Luehrs, E.; Barr, L.; Le, S. The role and treatment implications of peripheral and central processing of pain, pruritus, and nausea in heightened somatic awareness: A review. Innov. Clin. Neurosci. 2017, 14, 11–20. [Google Scholar]
- Travagli, R.A.; Hermann, G.E.; Browning, K.N.; Rogers, R.C. Brainstem circuits regulating gastric function. Annu. Rev. Physiol. 2006, 68, 279–305. [Google Scholar] [CrossRef]
- Hasler, W.L. Pathology of emesis: Its autonomic basis. Handb. Clin. Neurol. 2013, 117, 337–352. [Google Scholar] [PubMed]
- Minami, M.; Endo, T.; Hirafuji, M. Role of serotonin in emesis. Nihon Yakurigaku Zasshi 1996, 108, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Diwakarla, S.; Fothergill, L.J.; Fakhry, J.; Callaghan, B.; Furness, J.B. Heterogeneity of enterochromaffin cells within the gastrointestinal tract. Neurogastroenterol. Motil. 2017, 29, e13101. [Google Scholar] [CrossRef]
- Obara, Y.; Machida, T.; Takano, Y.; Shiga, S.; Suzuki, A.; Hamaue, N.; Iizuka, K.; Hirafuji, M. Cisplatin increases the number of enterochromaffin cells containing substance P in rat intestine. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 847–858. [Google Scholar] [CrossRef]
- Chappa, A.K.; Audus, K.L.; Lunte, S.M. Characteristics of substance P transport across the blood–brain barrier. Pharm. Res. 2006, 23, 1201–1208. [Google Scholar] [CrossRef]
- Freed, A.L.; Audus, K.L.; Lunte, S.M. Investigation of substance P transport across the blood-brain barrier. Peptides 2002, 23, 157–165. [Google Scholar] [CrossRef]
- Alcaino, C.; Knutson, K.R.; Treichel, A.J.; Yildiz, G.; Strege, P.R.; Linden, D.R.; Li, J.H.; Leiter, A.B.; Szurszewski, J.H.; Farrugia, G.; et al. A population of gut epithelial enterochromaffin cells is mechanosensitive and requires Piezo2 to convert force into serotonin release. Proc. Natl. Acad. Sci. USA 2018, 115, E7632–E7641. [Google Scholar] [CrossRef]
- Darmani, N.A.; Wang, Y.; Abad, J.; Ray, A.P.; Thrush, G.R.; Ramirez, J. Utilization of the least shrew as a rapid and selective screening model for the antiemetic potential and brain penetration of substance P and NK1 receptor antagonists. Brain Res. 2008, 1214, 58–72. [Google Scholar] [CrossRef]
- Furness, J. Types of neurons in the enteric nervous system. J. Auton. Nerv. Syst. 2000, 81, 87–96. [Google Scholar] [CrossRef]
- Wafai, L.; Taher, M.; Jovanovska, V.; Bornstein, J.C.; Dass, C.R.; Nurgali, K. Effects of oxaliplatin on mouse myenteric neurons and colonic motility. Front. Neurosci. 2013, 7, 30. [Google Scholar] [CrossRef]
- Robinson, A.; Stojanovska, V.; Rahman, A.A.; McQuade, R.M.; Senior, P.V.; Nurgali, K. Effects of oxaliplatin treatment on the enteric glial cells and neurons in the mouse ileum. J. Histochem. Cytochem. 2016, 64, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Vera, G.; Castillo, M.; Cabezos, P.A.; Chiarlone, A.; Martín, M.I.; Gori, A.; Pasquinelli, G.; Barbara, G.; Stanghellini, V.; Corinaldesi, R.; et al. Enteric neuropathy evoked by repeated cisplatin in the rat. Neurogastroenterol. Motil. 2011, 23, 370-e163. [Google Scholar] [CrossRef]
- Miller, A.D. Central mechanisms of vomiting. Dig. Dis. Sci. 1999, 44, 39S–43S. [Google Scholar] [PubMed]
- Boulet, N.P.; Cloutier, C.J.; Ossenkopp, K.-P.; Kavaliers, M. Oxytocin, social factors, and the expression of conditioned disgust (anticipatory nausea) in male rats. Behav. Pharmacol. 2016, 27, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Napadow, V.; Sheehan, J.D.; Kim, J.; LaCount, L.T.; Park, K.; Kaptchuk, T.J.; Rosen, B.R.; Kuo, B. The brain circuitry underlying the temporal evolution of nausea in humans. Cereb. Cortex 2012, 23, 806–813. [Google Scholar] [CrossRef]
- Horn, C.C. The medical implications of gastrointestinal vagal afferent pathways in nausea and vomiting. Curr. Pharm. Des. 2014, 20, 2703–2712. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A. Serotonin 5-HT 3 receptor antagonists prevent cisplatin-induced emesis in Cryptotis parva: A new experimental model of emesis. J. Neural Transm. 1998, 105, 1143–1154. [Google Scholar] [CrossRef]
- Horn, C.C.; Kimball, B.A.; Wang, H.; Kaus, J.; Dienel, S.; Nagy, A.; Gathright, G.R.; Yates, B.J.; Andrews, P.L.R. Why can’t rodents vomit? A comparative behavioral, anatomical, and physiological study. PLoS ONE 2013, 8, e60537. [Google Scholar] [CrossRef]
- Rudd, J.; Yamamoto, K.; Yamatodani, A.; Takeda, N. Differential action of ondansetron and dexamethasone to modify cisplatin-induced acute and delayed kaolin consumption (“pica”) in rats. Eur. J. Pharmacol. 2002, 454, 47–52. [Google Scholar] [CrossRef]
- Takeda, N.; Hasegawa, S.; Morita, M.; Matsunaga, T. Pica in rats is analogous to emesis: An animal model in emesis research. Pharmacol. Biochem. Behav. 1993, 45, 817–821. [Google Scholar] [CrossRef]
- Yamamoto, K.; Matsunaga, S.; Matsui, M.; Takeda, N.; Yamatodani, A. Pica in mice as a new model for the study of emesis. Methods Find. Exp. Clin. Pharmacol. 2002, 24, 135. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeda, N.; Yamatodani, A. Establishment of an animal model for radiation-induced vomiting in rats using pica. J. Radiat. Res. 2002, 43, 135. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Limebeer, C.L.; Hall, G.; Parker, L.A. Exposure to a lithium-paired context elicits gaping in rats: A model of anticipatory nausea. Physiol. Behav. 2006, 88, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.A.; Limebeer, C.L. Conditioned gaping in rats: A selective measure of nausea. Auton. Neurosci. 2006, 129, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.A. Conditioned flavor avoidance and conditioned gaping: Rat models of conditioned nausea. Eur. J. Pharmacol. 2014, 722, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.D.; Lu, Z.; Rudd, J.A. Looking beyond 5-HT3 receptors: A review of the wider role of serotonin in the pharmacology of nausea and vomiting. Eur. J. Pharmacol. 2014, 722, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Cheng, J.; Yin, J.; Yang, Y.; Guo, J.; Zhang, W.; Xie, B.; Lu, H.; Hao, D. Effects of sacral nerve electrical stimulation on 5-HT and 5-HT3AR/5-HT4R levels in the colon and sacral cord of acute spinal cord injury rat models. Mol. Med. Rep. 2020, 22, 763–773. [Google Scholar] [CrossRef]
- Faerber, L.; Drechsler, S.; Ladenburger, S.; Gschaidmeier, H.; Fischer, W. The neuronal 5-HT3 receptor network after 20 years of research—Evolving concepts in management of pain and inflammation. Eur. J. Pharmacol. 2007, 560, 1–8. [Google Scholar] [CrossRef]
- Aapro, M.; Scotté, F.; Escobar, Y.; Celio, L.; Berman, R.; Franceschetti, A.; Bell, D.; Jordan, K. Practice Patterns for prevention of chemotherapy-induced nausea and vomiting and antiemetic guideline adherence based on real-world prescribing data. Oncology 2021. [Google Scholar] [CrossRef]
- Theriot, J.; Wermuth, H.R.; Ashurst, J.V. Antiemetic serotonin-5-HT3 receptor blockers. In StatPearls; Statpearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kovac, J.R.; Pan, M.; Arent, S.; Lipshultz, L.I. Dietary adjuncts for improving testosterone levels in hypogonadal males. Am. J. Men’s Health 2016, 10, NP109–NP117. [Google Scholar] [CrossRef]
- Jansen, I.; Alafaci, C.; McCulloch, J.; Uddman, R.; Edvinsson, L. Tachykinins (substance P, neurokinin A, neuropeptide K, and neurokinin B) in the cerebral circulation: Vas-omotor responses in vitro and in situ. J. Cereb. Blood Flow Metab. 1991, 11, 567–575. [Google Scholar] [CrossRef]
- Carpenter, D.O.; Briggs, D.B.; Strominger, N. Peptide-induced emesis in dogs. Behav. Brain Res. 1983, 11, 277–281. [Google Scholar] [CrossRef]
- Wu, M.; Harding, R.K.; Hugenholtz, H.; Kucharczyk, J. Emetic effects of centrally administered angiotensin II, arginine vasopressin and neurotensin in the dog. Peptides 1985, 6, 173–175. [Google Scholar] [CrossRef]
- Saito, R.; Takano, Y.; Kamiya, H.-O. Roles of substance P and NK1 receptor in the brainstem in the development of emesis. J. Pharmacol. Sci. 2003, 91, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.; Briggs, D.B.; Strominger, N. Behavioral and electrophysiological studies of peptide-induced emesis in dogs. Fed. Proc. 1984, 43, 2952–2954. [Google Scholar]
- Ray, A.P.; Chebolu, S.; Ramirez, J.; Darmani, N.A. Ablation of least shrew central neurokinin NK₁ receptors reduces GR73632-induced vomiting. Behav. Neurosci. 2009, 123, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Preuss, C.V. Antiemetic neurokinin-1 receptor blockers. In StatPearls; Statpearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hesketh, P.J.; Bohlke, K.; Lyman, G.H.; Basch, E.; Chesney, M.; Clark-Snow, R.A.; Danso, M.A.; Jordan, K.; Somerfield, M.R.; Kris, M.G. Antiemetics: American Society of Clinical Oncology focused guideline update. J. Clin. Oncol. 2016, 34, 381–386. [Google Scholar] [CrossRef]
- Andrews, R.J. The role of tachykinins and the tachykinin NK1 receptor in nausea and emesis. In Handbook of Experimental Pharmacology; Hofmann, F.B., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 359–440. [Google Scholar]
- Weibel, S.; Rücker, G.; Eberhart, L.H.; Pace, N.L.; Hartl, H.M.; Jordan, O.L.; Mayer, D.; Riemer, M.; Schaefer, M.S.; Raj, D.; et al. Drugs for preventing postoperative nausea and vomiting in adults after general anaesthesia: A network meta-analysis. Cochrane Database Syst. Rev. 2020, 10, CD012859. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.J.; Belani, K.G.; Bergese, S.; Chung, F.; Diemunsch, P.; Habib, A.S.; Jin, Z.; Kovac, A.L.; Meyer, T.A.; Urman, R.D.; et al. Fourth consensus guidelines for the management of postoperative nausea and vomiting. Anesthesia Analg. 2020, 131, 411–448. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gainetdinov, R.R.; Gurevich, V.V. G protein-coupled receptor kinases as regulators of dopamine receptor func-tions. Pharmacol. Res. 2016, 111, 1–16. [Google Scholar] [CrossRef]
- Belkacemi, L.; Darmani, N.A. Dopamine receptors in emesis: Molecular mechanisms and potential therapeutic function. Pharmacol. Res. 2020, 161, 105124. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Borison, H.L. A New Concept of organization of the central emetic mechanism: Recent studies on the sites of action of apomorphine, copper sulfate and cardiac glycosides. Gastroenterology 1952, 22, 1–12. [Google Scholar] [CrossRef]
- Darmani, N.A.; Crim, J.L. Delta-9-tetrahydrocannabinol differentially suppresses emesis versus enhanced locomotor ac-tivity produced by chemically diverse dopamine D2/D3 receptor agonists in the least shrew (Cryptotis parva). Pharmacol. Biochem. Behav. 2005, 80, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Zhao, W.; Ahmad, B. The role of D 2 and D 3 dopamine receptors in the mediation of emesis in Cryptotis parva (the least shrew). J. Neural Transm. 1999, 106, 1045–1061. [Google Scholar] [CrossRef]
- Yoshida, N.; Yoshikawa, T.; Hosoki, K. A dopamine D3 receptor agonist, 7-OH-DPAT, causes vomiting in the dog. Life Sci. 1995, 57, PL347–PL350. [Google Scholar] [CrossRef]
- Osinski, M.A.; Uchic, M.E.; Seifert, T.; Shaughnessy, T.K.; Miller, L.N.; Nakane, M.; Cox, B.F.; Brioni, J.D.; Moreland, R.B. Dopamine D, but not D receptor agonists are emetogenic in ferrets. Pharmacol. Biochem. Behav. 2005, 81, 211–219. [Google Scholar] [CrossRef]
- Darmani, N.A.; Crim, J.L.; Janoyan, J.J.; Abad, J.; Ramirez, J. A re-evaluation of the neurotransmitter basis of chemotherapy-induced immediate and delayed vom-iting: Evidence from the least shrew. Brain Res. 2009, 1248, 40–58. [Google Scholar] [CrossRef]
- Veyrat-Follet, C.; Farinotti, R.; Palmer, J.L. Physiology of chemotherapy-induced emesis and antiemetic therapy. Drugs 1997, 53, 206–234. [Google Scholar] [CrossRef]
- Michal, P.; El-Fakahany, E.E.; Doležal, V. Changes in membrane cholesterol differentially influence preferential and non-preferential signaling of the M1 and M3 muscarinic acetylcholine receptors. Neurochem. Res. 2014, 40, 2068–2077. [Google Scholar] [CrossRef]
- Ilyaskina, O.S.; Lemoine, H.; Bünemann, M. Lifetime of muscarinic receptor–G-protein complexes determines coupling efficiency and G-protein subtype selectivity. Proc. Natl. Acad. Sci. USA 2018, 115, 5016–5021. [Google Scholar] [CrossRef]
- Li, Y.; Hai, S.; Zhou, Y. Cholinesterase inhibitors for rarer dementias associated with neurological conditions. Cochrane Database Syst. Rev. 2015, 009444. [Google Scholar] [CrossRef]
- Beleslin, D.; Krstić, S. Further studies on nicotine-induced emesis: Nicotinic mediation in area postrema. Physiol. Behav. 1987, 39, 681–686. [Google Scholar] [CrossRef]
- Beleslin, D.B.; Nedelkovski, V. Emesis induced by 4-(m-chlorophenylcarbamoyloxy)-2-butynyltrimethylammonium chloride (McN-A-343): Evidence for a predominant central muscarinic M1 mediation. Neuropharmacology 1988, 27, 949–956. [Google Scholar] [CrossRef]
- Darmani, N.A.; Zhong, W.; Chebolu, S.; Vaezi, M.; Alkam, T. Broad-spectrum antiemetic potential of the L-type calcium channel antagonist nifedipine and evidence for its additive antiemetic interaction with the 5-HT3 receptor antagonist palonosetron in the least shrew (Cryptotis parva). Eur. J. Pharmacol. 2014, 722, 2–12. [Google Scholar] [CrossRef]
- Alt, A.; Pendri, A.; Bertekap, R.L.; Li, G.; Benitex, Y.; Nophsker, M.; Rockwell, K.L.; Burford, N.T.; Sum, C.S.; Chen, J.; et al. Evidence for classical cholinergic toxicity associated with selective activation of M1 muscarinic receptors. J. Pharmacol. Exp. Ther. 2015, 356, 293–304. [Google Scholar] [CrossRef]
- Pergolizzi, J.V.; Philip, B.K.; Leslie, J.B.; Taylor, R.; Raffa, R.B. Perspectives on transdermal scopolamine for the treatment of postoperative nausea and vomiting. J. Clin. Anesthesia 2012, 24, 334–345. [Google Scholar] [CrossRef]
- Takeda, N.; Morita, M.; Hasegawa, S.; Horii, A.; Kubo, T.; Matsunaga, T. Neuropharmacology of motion sickness and emesis. A review. Acta Oto-Laryngol. 1993, 501, 10–15. [Google Scholar] [CrossRef]
- Parsons, M.; Ganellin, C.R. Histamine and its receptors. Br. J. Pharmacol. 2006, 147, S127–S135. [Google Scholar] [CrossRef]
- Chen, M.-M.; Xu, L.-H.; Chang, L.; Yin, P.; Jiang, Z.-L. Reduction of motion sickness through targeting histamine N-methyltransferase in the dorsal vagal complex of the brain. J. Pharmacol. Exp. Ther. 2018, 364, 367–376. [Google Scholar] [CrossRef]
- Bhargava, K.P.; Dixit, K.S. Role of the chemoreceptor trigger zone in histamine-induced emesis. Br. J. Pharmacol. 1968, 34, 508–513. [Google Scholar] [CrossRef][Green Version]
- Deshetty, U.M.; Tamatam, A.; Bhattacharjee, M.; Perumal, E.; Natarajan, G.; Khanum, F. Ameliorative effect of hesperidin against motion sickness by modulating histamine and histamine H1 receptor expression. Neurochem. Res. 2019, 45, 371–384. [Google Scholar] [CrossRef]
- Sato, G.; Uno, A.; Horii, A.; Umehara, H.; Kitamura, Y.; Sekine, K.; Tamura, K.; Fukui, H.; Takeda, N. Effects of hypergravity on histamine H1 receptor mRNA expression in hypothalamus and brainstem of rats: Implications for development of motion sickness. Acta Oto-Laryngol. 2009, 129, 45–51. [Google Scholar] [CrossRef]
- Uno, A.; Takeda, N.; Horii, A.; Morita, M.; Yamamoto, Y.; Yamatodani, A.; Kubo, T. Histamine release from the hypothalamus induced by gravity change in rats and space motion sickness. Physiol. Behav. 1997, 61, 883–887. [Google Scholar] [CrossRef]
- Takeda, N.; Morita, M.; Horii, A.; Nishiike, S.; Kitahara, T.; Uno, A. Neural mechanisms of motion sickness. J. Med. Investig. 2001, 48, 44–59. [Google Scholar]
- Tu, L.; Lu, Z.; Dieser, K.; Schmitt, C.; Chan, S.W.; Ngan, M.P.; Andrews, P.L.R.; Nalivaiko, E.; Rudd, J.A. Brain activation by H1 antihistamines challenges conventional view of their mechanism of action in motion sickness: A behavioral, c-fos and physiological study in suncus murinus (house musk shrew). Front. Physiol. 2017, 8, 412. [Google Scholar] [CrossRef]
- Han, N.-R.; Kim, H.-M.; Jeong, H.-J. Pyeongwee-San extract (KMP6): A new anti-allergic effect. J. Pharm. Pharmacol. 2012, 64, 308–316. [Google Scholar] [CrossRef]
- Blancquaert, J.-P.; Lefebvre, R.A.; Willems, J.L. Emetic and antiemetic effects of opioids in the dog. Eur. J. Pharmacol. 1986, 128, 143–150. [Google Scholar] [CrossRef]
- Ren, Z.Y.; Xu, X.; Bao, Y.; He, J.; Shi, L.; Deng, J.; Gao, X.; Tang, H.; Wang, Y.; Lu, L. The impact of genetic variation on sensitivity to opioid analgesics in patients with postoperative pain: A systematic review and meta-analysis. Pain Physician 2015, 18, 131–152. [Google Scholar]
- Ballantyne, G.H. Peptide YY(1-36) and Peptide YY(3-36): Part I. Distribution, release and actions. Obes. Surg. 2006, 16, 651–658. [Google Scholar] [CrossRef]
- Ono, H.K.; Hirose, S.; Narita, K.; Sugiyama, M.; Asano, K.; Hu, D.-L.; Nakane, A. Histamine release from intestinal mast cells induced by staphylococcal enterotoxin A (SEA) evokes vomiting reflex in common marmoset. PLoS Pathog. 2019, 15, e1007803. [Google Scholar] [CrossRef]
- Kanemasa, T.; Matsuzaki, T.; Koike, K.; Hasegawa, M.; Suzuki, T. Preventive effects of naldemedine, peripherally acting mu-opioid receptor antagonist, on mor-phine-induced nausea and vomiting in ferrets. Life Sci. 2020, 257, 118048. [Google Scholar] [CrossRef]
- Guan, J.L.; Wang, Q.P.; Nakai, Y. Electron microscopic observation of mu-opioid receptor in the rat area postrema. Peptides 1999, 20, 873–880. [Google Scholar] [CrossRef]
- Aicher, S.A.; Goldberg, A.; Sharma, S.; Pickel, V.M. mu-Opioid receptors are present in vagal afferents and their dendritic targets in the medial nucleus of the solitary tract. J. Comp. Neurol. 2000, 422, 181–190. [Google Scholar] [CrossRef]
- Bhandari, P.; Bingham, S.; Andrews, P. The neuropharmacology of loperamide-induced emesis in the ferret: The role of the area postrema, vagus, opiate and 5-HT3 receptors. Neuropharmacology 1992, 31, 735–742. [Google Scholar] [CrossRef]
- Barnes, N.; Bunce, K.; Naylor, R.; Rudd, J. The actions of fentanyl to inhibit drug-induced emesis. Neuropharmacology 1991, 30, 1073–1083. [Google Scholar] [CrossRef]
- Johnston, K.D. The potential for mu-opioid receptor agonists to be anti-emetic in humans: A review of clinical data. Acta Anaesthesiol. Scand. 2010, 54, 132–140. [Google Scholar] [CrossRef]
- Ziffert, I.; Kaiser, A.; Babilon, S.; Mörl, K.; Beck-Sickinger, A.G. Unusually persistent Galphai-signaling of the neuropeptide Y2 receptor depletes cellular Gi/o pools and leads to a Gi-refractory state. Cell Commun. Signal. 2020, 18, 49. [Google Scholar] [CrossRef]
- Bonaventure, P.; Nepomuceno, D.; Mazur, C.; Lord, B.; Rudolph, D.A.; Jablonowski, J.A.; Carruthers, N.I.; Lovenberg, T.W. Characterization of N-(1-Acetyl-2,3-dihydro-1H-indol-6-yl)-3-(3-cyano-phenyl)-N-[1-(2-cyclopentyl-ethyl)-piperidin-4yl]acrylamide (JNJ-5207787), a small molecule antagonist of the neuropeptide Y Y2 receptor. J. Pharmacol. Exp. Ther. 2004, 308, 1130–1137. [Google Scholar] [CrossRef]
- Cox, H.M. Neuropeptide Y receptors; antisecretory control of intestinal epithelial function. Auton. Neurosci. 2007, 133, 76–85. [Google Scholar] [CrossRef]
- Domingues, M.F.; De Assis, D.R.; Piovesan, A.R.; Belo, C.A.D.; Da Costa, J.C. Peptide YY (3–36) modulates intracellular calcium through activation of the phosphatidylinositol pathway in hippocampal neurons. Neuropeptides 2018, 67, 1–8. [Google Scholar] [CrossRef]
- Parker, S.L.; Balasubramaniam, A. Neuropeptide Y Y2 receptor in health and disease. Br. J. Pharmacol. 2008, 153, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bates, M.A.; Bursian, S.J.; Flannery, B.; Zhou, H.R.; Link, J.E.; Zhang, H.; Pestka, J.J. Peptide YY3–36 and 5-hydroxytryptamine mediate emesis induction by trichothecene deoxynivalenol (vomi-toxin). Toxicol. Sci. 2013, 133, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Leslie, R.; McDonald, T.; Robertson, H. Autoradiographic localization of peptide YY and neuropeptide Y binding sites in the medulla oblongata. Peptides 1988, 9, 1071–1076. [Google Scholar] [CrossRef]
- Harding, R.; McDonald, T. Identification and characterization of the emetic effects of peptide YY. Peptides 1989, 10, 21–24. [Google Scholar] [CrossRef]
- Shoblock, J.R.; Welty, N.; Nepomuceno, D.; Lord, B.; Aluisio, L.; Fraser, I.; Motley, S.T.; Sutton, S.W.; Morton, K.; Galici, R.; et al. In vitro and in vivo characterization of JNJ-31020028 (N-(4-{4-[2-(diethylamino)-2-oxo-1-phenylethyl]piperazin-1-yl}-3-fluorophenyl)-2- pyridin-3-ylbenzamide), a selective brain penetrant small molecule antagonist of the neuropeptide Y Y(2) receptor. Psychopharmacology 2010, 208, 265–277. [Google Scholar] [CrossRef]
- Niida, A.; Kanematsu-Yamaki, Y.; Asakawa, T.; Ishimura, Y.; Fujita, H.; Matsumiya, K.; Nishizawa, N.; Adachi, Y.; Mochida, T.; Tsuchimori, K.; et al. Antiobesity and emetic effects of a short-length peptide YY analog and its PEGylated and alkylated derivatives. Bioorganic Med. Chem. 2018, 26, 566–572. [Google Scholar] [CrossRef]
- Borner, T.; Shaulson, E.D.; Ghidewon, M.Y.; Barnett, A.B.; Horn, C.C.; Doyle, R.P.; Grill, H.J.; Hayes, M.R.; De Jonghe, B.C. GDF15 induces anorexia through nausea and emesis. Cell Metab. 2020, 31, 351–362.e5. [Google Scholar] [CrossRef]
- Félétou, M.; Levens, N.R. Neuropeptide Y2 receptors as drug targets for the central regulation of body weight. Curr. Opin. Investig. Drugs 2005, 6, 1002–1011. [Google Scholar]
- Sato, N.; Ogino, Y.; Mashiko, S.; Ando, M. Modulation of neuropeptide Y receptors for the treatment of obesity. Expert Opin. Ther. Patents 2009, 19, 1401–1415. [Google Scholar] [CrossRef]
- Brothers, S.; Wahlestedt, C. Therapeutic potential of neuropeptide Y (NPY) receptor ligands. EMBO Mol. Med. 2010, 2, 429–439. [Google Scholar] [CrossRef]
- Wu, W.; Zhou, H.-R.; Bursian, S.J.; Link, J.E.; Pestka, J.J. Calcium-sensing receptor and transient receptor ankyrin-1 mediate emesis induction by deoxynivalenol (vomitoxin). Toxicol. Sci. 2016, 155, 32–42. [Google Scholar] [CrossRef]
- L’Homme, L.; Sermikli, B.P.; Staels, B.; Piette, J.; Legrand-Poels, S.; Dombrowicz, D. Saturated fatty acids promote GDF15 expression in human macrophages through the PERK/eIF2/CHOP signaling pathway. Nutrients 2020, 12, 3771. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.M.; Kim, H.; Bennett, D.; Calle, R.A.; Collins, S.; Esquejo, R.M.; He, T.; Joaquim, S.; Joyce, A.; Lambert, M.; et al. GDF-15 neutralization alleviates platinum-based chemotherapy-induced emesis, anorexia, and weight loss in mice and nonhuman primates. Cell Metab. 2020, 32, 938–950.e6. [Google Scholar] [CrossRef] [PubMed]
- Petry, C.J.; Ong, K.K.; Burling, K.A.; Barker, P.; Goodburn, S.F.; Perry, J.R.; Acerini, C.L.; Hughes, I.A.; Painter, R.C.; Afink, G.B.; et al. Associations of vomiting and antiemetic use in pregnancy with levels of circulating GDF15 early in the second trimester: A nested case-control study. Welcome Open Res. 2018, 3, 123. [Google Scholar] [CrossRef] [PubMed]
- Fejzo, M.S.; Fasching, P.A.; Schneider, M.O.; Schwitulla, J.; Beckmann, M.W.; Schwenke, E.; MacGibbon, K.W.; Mullin, P.M. Analysis of GDF15 and IGFBP7 in hyperemesis gravidarum support causality. Geburtshilfe Frauenheilkd. 2019, 79, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Fejzo, M.S.; Arzy, D.; Tian, R.; MacGibbon, K.W.; Mullin, P.M. Evidence GDF15 plays a role in familial and recurrent hyperemesis gravidarum. Geburtshilfe Frauenheilkd. 2018, 78, 866–870. [Google Scholar] [CrossRef]
- Fejzo, M.S.; Sazonova, O.V.; Sathirapongsasuti, J.F.; Hallgrímsdóttir, I.B.; Vacic, V.; MacGibbon, K.; Schoenberg, F.P.; Mancuso, N.; Slamon, D.J.; Mullin, P.M. Placenta and appetite genes GDF15 and IGFBP7 are associated with hyperemesis gravidarum. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Fejzo, M.S.; Trovik, J.; Grooten, I.J.; Sridharan, K.; Roseboom, T.J.; Vikanes, Å.; Painter, R.C.; Mullin, P.M. Nausea and vomiting of pregnancy and hyperemesis gravidarum. Nat. Rev. Dis. Prim. 2019, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Andersson-Hall, U.; Joelsson, L.; Svedin, P.; Mallard, C.; Holmäng, A. Growth-differentiation-factor 15 levels in obese and healthy pregnancies: Relation to insulin re-sistance and insulin secretory function. Clin. Endocrinol. 2021. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Moreno, J.J. Eicosanoid receptors: Targets for the treatment of disrupted intestinal epithelial homeostasis. Eur. J. Pharmacol. 2017, 796, 7–19. [Google Scholar] [CrossRef]
- Morteau, O. Prostaglandins and inflammation: The cyclooxygenase controversy. Snake Venoms 2001, 48, 67–81. [Google Scholar] [CrossRef]
- Honda, T.; Kabashima, K. Prostanoids in allergy. Allergol. Int. 2015, 64, 11–16. [Google Scholar] [CrossRef]
- Ke, J.; Yang, Y.; Che, Q.; Jiang, F.; Wang, H.; Chen, Z.; Zhu, M.; Tong, H.; Zhang, H.; Yan, X.; et al. Prostaglandin E2 (PGE2) promotes proliferation and invasion by enhancing SUMO-1 activity via EP4 receptor in endometrial cancer. Tumor Biol. 2016, 37, 12203–12211. [Google Scholar] [CrossRef]
- Gadsby, R.; Barnie-Adshead, A.; Grammatoppoulos, D.; Gadsby, P. Nausea and vomiting in pregnancy: An association between symptoms and maternal prostaglandin E2. Gynecol. Obstet. Investig. 2000, 50, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.M.; Filshie, G.M. Therapeutic abortion using prostaglandin F2alpha. Lancet 1970, 1, 157–159. [Google Scholar] [CrossRef]
- Karim, S.M.M.; Filshie, G.M. Use of prostaglandin E2 for therapeutic abortion. BMJ 1970, 3, 198–200. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kan, K.K.; Ngan, M.P.; Wai, M.K.; Rudd, J.A. Mechanism of the prostanoid TP receptor agonist U46619 for inducing emesis in the ferret. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008, 378, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Kan, K.K.; Jones, R.L.; Ngan, M.-P.; Rudd, J.A.; Wai, M.-K. Emetic action of the prostanoid TP receptor agonist, U46619, in Suncus murinus (house musk shrew). Eur. J. Pharmacol. 2003, 482, 297–304. [Google Scholar] [CrossRef]
- Kan, K.K.; Jones, R.L.; Ngan, M.-P.; Rudd, J.A. Action of prostanoids on the emetic reflex of Suncus murinus (the house musk shrew). Eur. J. Pharmacol. 2003, 477, 247–251. [Google Scholar] [CrossRef]
- Kan, K.K.; Rudd, J.A.; Wai, M.K. Differential action of anti-emetic drugs on defecation and emesis induced by prostaglandin E2 in the ferret. Eur. J. Pharmacol. 2006, 544, 153–159. [Google Scholar] [CrossRef]
- Kan, K.K.; Jones, R.L.; Ngan, M.-P.; Rudd, J.A. Excitatory action of prostanoids on the ferret isolated vagus nerve preparation. Eur. J. Pharmacol. 2004, 491, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kan, K.K.; Wai, M.K.; Jones, R.L.; Rudd, J.A. Role of prostanoid EP3/1 receptors in mechanisms of emesis and defaecation in ferrets. Eur. J. Pharmacol. 2017, 803, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Kan, K.K.; Jones, R.L.; Ngan, M.-P.; A Rudd, J. Actions of prostanoids to induce emesis and defecation in the ferret. Eur. J. Pharmacol. 2002, 453, 299–308. [Google Scholar] [CrossRef]
- Darmani, N.A.; Chebolu, S. The role of endocannabinoids and arachidonic acid metabolites in emesis. In Endocannabinoids: Molecular, Pharmacological, Behavioral and Clinical Features; Bentham Science Publishers: Oak Park, IL, USA, 2013; pp. 25–59. [Google Scholar] [CrossRef]
- Goppelt-Struebe, M. Molecular mechanisms involved in the regulation of prostaglandin biosynthesis by glucocorticoids. Biochem. Pharmacol. 1997, 53, 1389–1395. [Google Scholar] [CrossRef]
- Girod, V.; Dapzol, J.; Bouvier, M.; Grélot, L. The COX inhibitors indomethacin and meloxicam exhibit anti-emetic activity against cisplatin-induced emesis in piglets. Neuropharmacology 2002, 42, 428–436. [Google Scholar] [CrossRef]
- Govindan, R.; McLeod, H.; Mantravadi, P.; Fineberg, N.; Helft, P.; Kesler, K.; Hanna, N.; Stoner, C.; Ansari, R.; Fox, E. Cisplatin, fluorouracil, celecoxib, and RT in resectable esophageal cancer: Preliminary results. Oncology 2004, 18, 18–21. [Google Scholar]
- Chu, C.-C.; Hsing, C.-H.; Shieh, J.-P.; Chien, C.-C.; Ho, C.-M.; Wang, J.-J. The cellular mechanisms of the antiemetic action of dexamethasone and related glucocorticoids against vomiting. Eur. J. Pharmacol. 2014, 722, 48–54. [Google Scholar] [CrossRef]
- Chebolu, S.; Wang, Y.; Ray, A.P.; Darmani, N.A. Pranlukast prevents cysteinyl leukotriene-induced emesis in the least shrew (Cryptotis parva). Eur. J. Pharmacol. 2010, 628, 195–201. [Google Scholar] [CrossRef]
- Jo-Watanabe, A.; Okuno, T.; Yokomizo, T. The role of leukotrienes as potential therapeutic targets in allergic disorders. Int. J. Mol. Sci. 2019, 20, 3580. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yamatodani, A. Involvement of leukotriene pathway in the development of sevoflurane-induced pica in rats. Can. J. Physiol. Pharmacol. 2019, 97, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Bloomquist, E.I.; Kream, R.M. Release of substance P from guinea pig trachea leukotriene D4. Exp. Lung Res. 1990, 16, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Sałat, K. Chemotherapy-induced peripheral neuropathy—Part 2: Focus on the prevention of oxaliplatin-induced neurotoxicity. Pharmacol. Rep. 2020, 72, 508–527. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Endo, T.; Hirafuji, M.; Hamaue, N.; Liu, Y.; Hiroshige, T.; Nemoto, M.; Saito, H.; Yoshioka, M. Pharmacological aspects of anticancer drug-induced emesis with emphasis on serotonin release and vagal nerve activity. Pharmacol. Ther. 2003, 99, 149–165. [Google Scholar] [CrossRef]
- Navari, R.M.; Schwartzberg, L.S. Evolving role of neurokinin 1-receptor antagonists for chemotherapy-induced nausea and vomiting. OncoTargets Ther. 2018, ume 11, 6459–6478. [Google Scholar] [CrossRef]
- Darmani, N.A.; Chebolu, S.; Zhong, W.; Kim, W.D.; Narlesky, M.; Adams, J.; Dong, F. The anti-asthmatic drug pranlukast suppresses the delayed-phase vomiting and reverses intracellular indices of emesis evoked by cisplatin in the least shrew (Cryptotis parva). Eur. J. Pharmacol. 2017, 809, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Torii, Y.; Mutoh, M.; Saito, H.; Matsuki, N. Involvement of free radicals in cisplatin-induced emesis in Suncus murinus. Eur. J. Pharmacol. Environ. Toxicol. Pharmacol. 1993, 248, 131–135. [Google Scholar] [CrossRef]
- Alam, J.; Subhan, F.; Ullah, I.; Shahid, M.; Ali, G.; Sewell, R.D.E. Erratum to: Synthetic and natural antioxidants attenuate cisplatin-induced vomiting. BMC Pharmacol. Toxicol. 2017, 18, 9. [Google Scholar] [CrossRef]
- Navari, R.M. Managing nausea and vomiting in patients with cancer: What works. Oncology 2018, 32, 121. [Google Scholar]
- Sutherland, A.; Naessens, K.; Plugge, E.; Ware, L.; Head, K.; Burton, M.J.; Wee, B. Olanzapine for the prevention and treatment of cancer-related nausea and vomiting in adults. Cochrane Database Syst. Rev. 2018, 9, CD012555. [Google Scholar] [CrossRef]
- Feyer, P.; Jahn, F.; Jordan, K. Radiation induced nausea and vomiting. Eur. J. Pharmacol. 2014, 722, 165–171. [Google Scholar] [CrossRef]
- Yee, C.; Drost, L.; Zhang, L.; Wan, B.A.; Ganesh, V.; Tsao, M.; Barnes, E.; Pasetka, M.; DeAngelis, C.; Chow, E. Impact of radiation-induced nausea and vomiting on quality of life. Support. Care Cancer 2018, 26, 3959–3966. [Google Scholar] [CrossRef] [PubMed]
- Rowbottom, L.; McDonald, R.; Turner, A.; Chow, E.; De Angelis, C. An overview of radiation-induced nausea and vomiting. J. Med. Imaging Radiat. Sci. 2016, 47, S29–S38. [Google Scholar] [CrossRef]
- Dennis, K.; De Angelis, C.; Jon, F.; Lauzon, N.; Pasetka, M.; Holden, L.; Barnes, E.; Danjoux, C.; Sahgal, A.; Tsao, M.; et al. Aprepitant and granisetron for the prophylaxis of radiotherapy-induced nausea and vomiting after moder-ately emetogenic radiotherapy for bone metastases: A prospective pilot study. Curr. Oncol. 2014, 21, e760–e767. [Google Scholar] [CrossRef]
- Wong, E.; Pulenzas, N.; Bedard, G.; DeAngelis, C.; Zhang, L.; Tsao, M.; Danjoux, C.; Thavarajah, N.; Lechner, B.; McDonald, R.; et al. Ondansetron rapidly dissolving film for the prophylactic treatment of radiation-induced nausea and vomiting—A pilot study. Curr. Oncol. 2015, 22, 199–210. [Google Scholar] [CrossRef][Green Version]
- Dennis, K.; Ponn, M.; Chow, E. Nausea and vomiting induced by gastrointestinal radiation therapy: Current status and future directions. Curr. Opin. Support Palliat. Care 2015, 9, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Elvir-Lazo, O.L.; White, P.F.; Yumul, R.; Eng, H.C. Management strategies for the treatment and prevention of postoperative/postdischarge nausea and vomiting: An updated review. F1000Research 2020, 9, 983. [Google Scholar] [CrossRef] [PubMed]
- Apfel, C.; Heidrich, F.; Jukar-Rao, S.; Jalota, L.; Hornuss, C.; Whelan, R.; Zhang, K.; Cakmakkaya, O. Evidence-based analysis of risk factors for postoperative nausea and vomiting. Br. J. Anaesth. 2012, 109, 742–753. [Google Scholar] [CrossRef]
- Hase, T.; Hashimoto, T.; Saito, H.; Uchida, Y.; Kato, R.; Tsuruga, K.; Takita, K.; Morimoto, Y. Isoflurane induces c-Fos expression in the area postrema of the rat. J. Anesthesia 2019, 33, 562–566. [Google Scholar] [CrossRef]
- Machu, T.K.; A Harris, R. Alcohols and anesthetics enhance the function of 5-hydroxytryptamine3 receptors expressed in Xenopus laevis oocytes. J. Pharmacol. Exp. Ther. 1994, 271, 898–905. [Google Scholar] [PubMed]
- Kadota, T.; Kakuta, N.; Horikawa, Y.T.; Tsutsumi, R.; Oyama, T.; Tanaka, K.; Tsutsumi, Y.M. Plasma substance P concentrations in patients undergoing general anesthesia: An objective marker associated with postoperative nausea and vomiting. JA Clin. Rep. 2016, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Kanaparthi, A.; Kukura, S.; Slenkovich, N.; AlGhamdi, F.; Shafy, S.Z.; Hakim, M.; Tobias, J.D. Perioperative administration of emend((R)) (Aprepitant) at a tertiary care children’s hospital: A 12-month survey. Clin. Pharmacol. 2019, 11, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Naeem, Z.; Chen, I.L.; Pryor, A.D.; Docimo, S.; Gan, T.J.; Spaniolas, K. Antiemetic prophylaxis and anesthetic approaches to reduce postoperative nausea and vomiting in bariatric surgery patients: A systematic review. Obes. Surg. 2020, 30, 3188–3200. [Google Scholar] [CrossRef]
- Sharkey, K.A.; Darmani, N.A.; Parker, L.A. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur. J. Pharmacol. 2014, 722, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.R.; Gordon, B.K.; Danielson, A.R.; Moulin, A.K. Pharmacologic treatment of cannabinoid hyperemesis syndrome: A systematic review. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 725–734. [Google Scholar] [CrossRef]
- Darmani, N.A.; Johnson, J.C. Central and peripheral mechanisms contribute to the antiemetic actions of del-ta-9-tetrahydrocannabinol against 5-hydroxytryptophan-induced emesis. Eur. J. Pharmacol. 2004, 488, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.M.; Parker, L.A. Cannabinoids as potential treatment for chemotherapy-induced nausea and vomiting. Front. Pharmacol. 2016, 7, 221. [Google Scholar] [CrossRef]
- Darmani, N.A.; Belkacemi, L.; Zhong, W. Delta(9)-THC and related cannabinoids suppress substance P- induced neuro-kinin NK1-receptor-mediated vomiting via activation of cannabinoid CB1 receptor. Eur. J. Pharmacol. 2019, 865, 172806. [Google Scholar] [CrossRef]
- Wang, Y.; Ray, A.P.; McClanahan, B.A.; Darmani, N.A. The antiemetic interaction of Δ9-tetrahydrocannabinol when combined with tropisetron or dexamethasone in the least shrew. Pharmacol. Biochem. Behav. 2009, 91, 367–373. [Google Scholar] [CrossRef]
- Meiri, E.; Jhangiani, H.; Vredenburgh, J.J.; Barbato, L.M.; Carter, F.J.; Yang, H.-M.; Baranowski, V. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr. Med Res. Opin. 2007, 23, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.L.; Agito, M.D. Cannabinoid hyperemesis syndrome: Marijuana is both antiemetic and proemetic. Cleve Clin. J. Med. 2015, 82, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.J.; Desanto, K.; Borgelt, L.; Phillips, K.T.; Monte, A.A. Cannabinoid hyperemesis syndrome: Diagnosis, pathophysiology, and treatment—A systematic review. J. Med. Toxicol. 2017, 13, 71–87. [Google Scholar] [CrossRef]
- DeVuono, M.V.; Parker, L.A. Cannabinoid hyperemesis syndrome: A review of potential mechanisms. Cannabis Cannabinoid Res. 2020, 5, 132–144. [Google Scholar] [CrossRef]
- Smith, T.N.; Walsh, A.; Forest, C.P. Cannabinoid hyperemesis syndrome: An unrecognized cause of nausea and vomiting. JAAPA 2019, 32, 1–5. [Google Scholar] [CrossRef]
- Andrews, P.; Bhandari, P. Resinferatoxin, an ultrapotent capsaicin analogue, has anti-emetic properties in the ferret. Neuropharmacology 1993, 32, 799–806. [Google Scholar] [CrossRef]
- Andrews, P.L.R.; Okada, F.; Woods, A.J.; Hagiwara, H.; Kakaimoto, S.; Toyoda, M.; Matsuki, N. The emetic and anti-emetic effects of the capsaicin analogue resiniferatoxin inSuncus murinus, the house musk shrew. Br. J. Pharmacol. 2000, 130, 1247–1254. [Google Scholar] [CrossRef]
- Darmani, N.A.; Henry, D.A.; Zhong, W.; Chebolu, S. Ultra-low doses of the transient receptor potential vanilloid 1 agonist, resiniferatoxin, prevents vomiting evoked by diverse emetogens in the least shrew (Cryptotis parva). Behav. Pharmacol. 2020, 31, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, K.A.; Cristino, L.; Oland, L.D.; Van Sickle, M.D.; Starowicz, K.; Pittman, Q.J.; Guglielmotti, V.; Davison, J.S.; Di Marzo, V. Arvanil, anandamide and N-arachidonoyl-dopamine (NADA) inhibit emesis through cannabinoid CB1 and vanilloid TRPV1 receptors in the ferret. Eur. J. Neurosci. 2007, 25, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Chebolu, S.; Zhong, W.; Trinh, C.; McClanahan, B.; Brar, R.S. Additive antiemetic efficacy of low-doses of the cannabinoid CB(1/2) receptor agonist Delta(9)-THC with ultralow-doses of the vanilloid TRPV1 receptor agonist resiniferatoxin in the least shrew (Cryptotis parva). Eur. J. Pharmacol. 2014, 722, 147–155. [Google Scholar] [CrossRef]
- Moon, A.M.; Buckley, S.A.; Mark, N.M. Successful treatment of cannabinoid hyperemesis syndrome with topical capsaicin. ACG Case Rep. J. 2018, 5, e3. [Google Scholar] [CrossRef]
- Richards, J.R.; LaPoint, J.M.; Burillo-Putze, G. Cannabinoid hyperemesis syndrome: Potential mechanisms for the benefit of capsaicin and hot water hydrotherapy in treatment. Clin. Toxicol. 2018, 56, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kunde, D.A.; Crawford, A.; Geraghty, D.P. Tachykinin (NK1, NK2 and NK3) receptor, transient receptor potential vanilloid 1 (TRPV1) and early transcription factor, cFOS, mRNA expression in rat tissues following systemic capsaicin treatment. Regul. Pept. 2013, 183, 35–41. [Google Scholar] [CrossRef]
- Parvataneni, S.; Varela, L.; Vemuri-Reddy, S.M.; Maneval, M.L. Emerging role of aprepitant in cannabis hyperemesis syndrome. Cureus 2019, 11, e4825. [Google Scholar] [CrossRef] [PubMed]
- Schmäl, F. Neuronal mechanisms and the treatment of motion sickness. Pharmacology 2013, 91, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.; Sipos, W.; Stark, T.D.; Käser, T.; Knecht, C.; Brunthaler, R.; Saalmüller, A.; Hofmann, T.; Ehling-Schulz, M. First insights into within host translocation of the bacillus cereus toxin cereulide using a porcine model. Front. Microbiol. 2018, 9, 2652. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, R.; Saris, N.-E.L.; Grigoriev, P.A.; Andersson, M.A.; Salkinoja-Salonen, M.S. Ionophoretic properties and mitochondrial effects of cereulide. The emetic toxin of B. cereus. JBIC J. Biol. Inorg. Chem. 1999, 263, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Inprasit, C.; Lin, Y.-W.; Huang, C.-P.; Wu, S.-Y.; Hsieh, C.-L. Targeting TRPV1 to relieve motion sickness symptoms in mice by electroacupuncture and gene deletion. Sci. Rep. 2018, 8, 10365. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Introduction to Pathogens. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.K.; Hirose, S.; Naito, I.; Sato’o, Y.; Asano, K.; Hu, D.L.; Omoe, K.; Nakane, A. The emetic activity of staphylococcal enterotoxins, SEK, SEL, SEM, SEN and SEO in a small emetic animal model, the house musk shrew. Microbiol. Immunol. 2017, 61, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.-L.; Nakane, A. Mechanisms of staphylococcal enterotoxin-induced emesis. Eur. J. Pharmacol. 2014, 722, 95–107. [Google Scholar] [CrossRef]
- Hu, D.-L.; Zhu, G.; Mori, F.; Omoe, K.; Okada, M.; Wakabayashi, K.; Kaneko, S.; Shinagawa, K.; Nakane, A. Staphylococcal enterotoxin induces emesis through increasing serotonin release in intestine and it is downregulated by cannabinoid receptor 1. Cell. Microbiol. 2007, 9, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.-L.; Suga, S.; Omoe, K.; Abe, Y.; Shinagawa, K.; Wakui, M.; Nakane, A. Staphylococcal enterotoxin A modulates intracellular Ca2+ signal pathway in human intestinal epithelial cells. FEBS Lett. 2005, 579, 4407–4412. [Google Scholar] [CrossRef]
- Ehling-Schulz, M.; Fricker, M.; Scherer, S. Identification of emetic toxin producing Bacillus cereus strains by a novel molecular assay. FEMS Microbiol. Lett. 2004, 232, 189–195. [Google Scholar] [CrossRef]
- Agata, N.; Ohta, M.; Mori, M.; Isobe, M. A novel dodecadepsipeptide, cereulide, is an emetic toxin of Bacillus cereus. FEMS Microbiol. Lett. 1995, 129, 17–20. [Google Scholar] [CrossRef]
- Shinagawa, K.; Konuma, H.; Sekita, H.; Sugii, S. Emesis of rhesus monkeys induced by intragastric administration with the HEp-2 vacuolation factor (cereulide) produced by Bacillus cereus. FEMS Microbiol. Lett. 1995, 130, 87–90. [Google Scholar]
- Duffy, T.J. Pharmacology of mushroom poisoning. N. Engl. J. Med. 1973, 289, 379–380. [Google Scholar] [PubMed]
- Bonnet, M.S.; Roux, J.; Mounien, L.; Dallaporta, M.; Troadec, J.-D. Advances in deoxynivalenol toxicity mechanisms: The brain as a target. Toxins 2012, 4, 1120–1138. [Google Scholar] [CrossRef]
- Hagbom, M.; Istrate, C.; Engblom, D.; Karlsson, T.; Rodriguez-Diaz, J.; Buesa, J.; Taylor, J.A.; Loitto, V.-M.; Magnusson, K.-E.; Ahlman, H.; et al. Rotavirus stimulates release of serotonin (5-HT) from human enterochromaffin cells and activates brain structures involved in nausea and vomiting. PLoS Pathog. 2011, 7, e1002115. [Google Scholar] [CrossRef]
- Hagbom, M.; Novak, D.; Ekström, M.; Khalid, Y.; Andersson, M.; Lindh, M.; Nordgren, J.; Svensson, L. Ondansetron treatment reduces rotavirus symptoms—A randomized double-blinded placebo-controlled trial. PLoS ONE 2017, 12, e0186824. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Collinson-Pautz, M.R.; Utama, B.; Estes, M.K. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. mBio 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zeng, C.Q.-Y.; Ball, J.M.; Estes, M.K.; Morris, A.P. The rotavirus enterotoxin NSP4 mobilizes intracellular calcium in human intestinal cells by stimulating phospholipase C-mediated inositol 1,4,5-trisphosphate production. Proc. Natl. Acad. Sci. USA 1997, 94, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Chang-Graham, A.L.; Perry, J.L.; Strtak, A.C.; Ramachandran, N.K.; Criglar, J.M.; Philip, A.A.; Patton, J.T.; Estes, M.K.; Hyser, J.M. Rotavirus calcium dysregulation manifests as dynamic calcium signaling in the cytoplasm and endoplasmic reticulum. Sci. Rep. 2019, 9, 1–20. [Google Scholar] [CrossRef]
- Ray, A.P.; Chebolu, S.; Darmani, N.A. Receptor-selective agonists induce emesis and Fos expression in the brain and enteric nervous system of the least shrew (Cryptotis parva). Pharmacol. Biochem. Behav. 2009, 94, 211–218. [Google Scholar] [CrossRef]
- Hyser, J.M.; Estes, M.K. Pathophysiological consequences of calcium-conducting viroporins. Annu. Rev. Virol. 2015, 2, 473–496. [Google Scholar] [CrossRef]
- Bai, D.; Fang, L.; Xia, S.; Ke, W.; Wang, J.; Wu, X.; Fang, P.; Xiao, S. Porcine deltacoronavirus (PDCoV) modulates calcium influx to favor viral replication. Virology 2020, 539, 38–48. [Google Scholar] [CrossRef]
- Jung, K.; Miyazaki, A.; Saif, L.J. Immunohistochemical detection of the vomiting-inducing monoamine neurotransmitter serotonin and enterochromaffin cells in the intestines of conventional or gnotobiotic (Gn) pigs infected with porcine epidemic diarrhea virus (PEDV) and serum cytokine responses of Gn pigs to acute PEDV infection. Res. Veter. Sci. 2018, 119, 99–108. [Google Scholar] [CrossRef]
- Roy, D.; Khanra, I.; Wang, Z.; Merugu, S.B.; Yunus, F.-U.-N.; Mashausi, D.S.; Li, D. Emerging novel coronavirus is a global threat: Insight in the biology of COVID-19 and its hijacking process of hosts’ cell. Curr. Pharm. Des. 2020, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smyk, W.; Janik, M.K.; Portincasa, P.; Milkiewicz, P.; Lammert, F.; Krawczyk, M. COVID-19: Focus on the lungs but do not forget the gastrointestinal tract. Eur. J. Clin. Investig. 2020, 50, e13276. [Google Scholar] [CrossRef]
- Cataldi, M.; Pignataro, G.; Taglialatela, M. Neurobiology of coronaviruses: Potential relevance for COVID-19. Neurobiol. Dis. 2020, 143, 105007. [Google Scholar] [CrossRef]
- Andrews, P.L.; Cai, W.; Rudd, J.A.; Sanger, G.J. COVID-19, nausea, and vomiting. J. Gastroenterol. Hepatol. 2020, 36, 646–656. [Google Scholar] [CrossRef]
- Modi, S.R.; Collins, J.J.; Relman, D.A. Antibiotics and the gut microbiota. J. Clin. Investig. 2014, 124, 4212–4218. [Google Scholar] [CrossRef]
- Pavia, A.T.; Shipman, L.D.; Wells, J.G.; Puhr, N.D.; Smith, J.D.; McKinley, T.W.; Tauxe, R.V. Epidemiologic evidence that prior antimicrobial exposure decreases resistance to infection by antimicrobial-sensitive salmonella. J. Infect. Dis. 1990, 161, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nat. Cell Biol. 2013, 502, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Masucci, L.; Quaranta, G.; Simonelli, C.; Lopetuso, L.R.; Sanguinetti, M.; Gasbarrini, A.; Cammarota, G. Randomised clinical trial: Faecal microbiota transplantation by colonoscopy plus vancomycin for the treatment of severe refractory Clostridium difficile infection-single versus multiple infusions. Aliment. Pharmacol. Ther. 2018, 48, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.M.; Sanders, E.C. Potential neuroinvasive pathways of SARS-CoV-2: Deciphering the spectrum of neurological deficit seen in coronavirus disease-2019 (COVID-19). J. Med. Virol. 2020, 92, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S. Gastrointestinal symptoms associated with COVID-19: Impact on the gut microbiome. Transl. Res. 2020, 226, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Cai, H.; Shen, Y.; Ni, Q.; Chen, Y.; Hu, S.; Li, J.; Wang, H.; Yu, L.; Huang, H.; et al. Management of corona virus disease-19 (COVID-19): The Zhejiang experience. Zhejiang Da Xue Xue Bao Yi Xue Ban 2020, 49, 147–157. [Google Scholar] [PubMed]
- Zuo, T.; Zhan, H.; Zhang, F.; Liu, Q.; Tso, E.Y.; Lui, G.C.; Chen, N.; Li, A.; Lu, W.; Chan, F.K.; et al. Alterations in fecal fungal microbiome of patients with COVID-19 during time of hospitalization until discharge. Gastroenterology 2020, 159, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Darmani, N.A. Intracellular vomit signals and cascades downstream of emetic receptors: Evidence from the least shrew (Cryptotis parva) model of vomiting. Rem. Open Access 2017, 2, 2. [Google Scholar]
- Zhong, W.; Picca, A.J.; Lee, A.S.; Darmani, N.A. Ca2+ signaling and emesis: Recent progress and new perspectives. Auton. Neurosci. 2017, 202, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.; Miledi, R. A study of synaptic transmission in the absence of nerve impulses. J. Physiol. 1967, 192, 407–436. [Google Scholar] [CrossRef]
- Zuccotti, A.; Clementi, S.; Reinbothe, T.; Torrente, A.; Vandael, D.H.; Pirone, A. Structural and functional differences between L-type calcium channels: Crucial issues for future selective targeting. Trends Pharmacol. Sci. 2011, 32, 366–375. [Google Scholar] [CrossRef]
- Rogers, R.C.; Nasse, J.S.; Hermann, G.E. Live-cell imaging methods for the study of vagal afferents within the nucleus of the solitary tract. J. Neurosci. Methods 2006, 150, 47–58. [Google Scholar] [CrossRef]
- Rogers, R.C.; Van Meter, M.J.; Hermann, G.E. Tumor necrosis factor potentiates central vagal afferent signaling by mod-ulating ryanodine channels. J. Neurosci. 2006, 26, 12642–12646. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.C.; Hermann, G.E. Tumor necrosis factor activation of vagal afferent terminal calcium is blocked by cannabinoids. J. Neurosci. 2012, 32, 5237–5241. [Google Scholar] [CrossRef]
- Suzuki, Y.; Inoue, T.; Ra, C. L-type Ca2+ channels: A new player in the regulation of Ca2+ signaling, cell activation and cell survival in immune cells. Mol. Immunol. 2010, 47, 640–648. [Google Scholar] [CrossRef]
- Hargreaves, A.C.; Gunthorpe, M.J.; Taylor, C.; Lummis, S.C. Direct inhibition of 5-hydroxytryptamine3 receptors by antagonists of L-type Ca2+ channels. Mol. Pharmacol. 1996, 50, 1284–1294. [Google Scholar]
- Homma, K.; Kitamura, Y.; Ogawa, H.; Oka, K. Serotonin induces the increase in intracellular Ca2+ that enhances neurite outgrowth in PC12 cells via activation of 5-HT3 receptors and voltage-gated calcium channels. J. Neurosci. Res. 2006, 84, 316–325. [Google Scholar] [CrossRef]
- Ronde, P.; Nichols, R.A. 5-HT3 receptors induce rises in cytosolic and nuclear calcium in NG108–15 cells via calci-um-induced calcium release. Cell Calcium 1997, 22, 357–365. [Google Scholar] [CrossRef]
- Takenouchi, T.; Munekata, E. Serotonin increases cytoplasmic Ca2+ concentration in PC12h cells: Effect of tachykinin peptides. Neurosci. Lett. 1998, 246, 141–144. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Hattori, Y.; Houzen, H.; Kanno, M.; Yasuda, K. Histamine H1-receptor-mediated increase in the Ca2+ transient without a change in the Ca2+ current in electrically stimulated guinea-pig atrial myocytes. Br. J. Pharmacol. 1998, 124, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.; Schmitt, L.-I.; Jastrow, H.; Thomale, J.; Kleinschnitz, C.; Hagenacker, T. Cisplatin alters the function and expression of N-type voltage-gated calcium channels in the absence of morphological damage of sensory neurons. Mol. Pain 2017, 13, 1744806917746565. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.; Schmitt, L.-I.; Erkel, M.; Melnikova, M.; Thomale, J.; Hagenacker, T. Cisplatin-induced neuropathic pain is mediated by upregulation of N-type voltage-gated calcium channels in dorsal root ganglion neurons. Exp. Neurol. 2017, 288, 62–74. [Google Scholar] [CrossRef]
- Almirza, W.; Peters, P.; Van Zoelen, E.; Theuvenet, A. Role of Trpc channels, Stim1 and Orai1 in PGF2α-induced calcium signaling in NRK fibroblasts. Cell Calcium 2012, 51, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Chebolu, S.; Darmani, N.A. Intracellular emetic signaling evoked by the L-type Ca2+ channel agonist FPL64176 in the least shrew (Cryptotis parva). Eur. J. Pharmacol. 2018, 834, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Aman, T.K.; Shen, R.-Y.; Haj-Dahmane, S. D2-like dopamine receptors depolarize dorsal raphe serotonin neurons through the activation of nonselective cationic conductance. J. Pharmacol. Exp. Ther. 2006, 320, 376–385. [Google Scholar] [CrossRef]
- Wu, J.; Dougherty, J.J.; Nichols, R.A. Dopamine receptor regulation of Ca2+ levels in individual isolated nerve terminals from rat striatum: Comparison of presynaptic D1-like and D2-like receptors. J. Neurochem. 2006, 98, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.; Correia-de-Sa, P. Protein kinase A and Ca(v)1 (L-Type) channels are common targets to facilitatory adenosine A2A and muscarinic M1 receptors on rat motoneurons. Neurosignals 2005, 14, 262–272. [Google Scholar] [CrossRef]
- Sculptoreanu, A.; Yoshimura, N.; De Groat, W.C.; Somogyi, G.T. Protein kinase C is involved in M1-muscarinic receptor-mediated facilitation of L-type Ca2+ channels in neurons of the major pelvic ganglion of the adult male rat. Neurochem. Res. 2001, 26, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Barajas, M.; Andrade, A.; Hernandez-Hernandez, O.; Felix, R.; Arias-Montaño, J.-A. Histamine-induced Ca2+ entry in human astrocytoma U373 MG cells: Evidence for involvement of store-operated channels. J. Neurosci. Res. 2008, 86, 3456–3468. [Google Scholar] [CrossRef]
- Ono, T.; Inoue, M.; Rashid, M.H.; Sumikawa, K.; Ueda, H. Stimulation of peripheral nociceptor endings by low dose morphine and its signaling mechanism. Neurochem. Int. 2002, 41, 399–407. [Google Scholar] [CrossRef]
- Smart, D.; A Hirst, R.; Hirota, K.; Grandy, D.K.; Lambert, D.G. The effects of recombinant rat μ-opioid receptor activation in CHO cells on phospholipase C, [Ca2+]i and adenylyl cyclase. Br. J. Pharmacol. 1997, 120, 1165–1171. [Google Scholar] [CrossRef]
- Splettstoesser, F.; Florea, A.-M.; Büsselberg, D. IP3 receptor antagonist, 2-APB, attenuates cisplatin induced Ca2+ -influx in HeLa-S3 cells and prevents activation of calpain and induction of apoptosis. Br. J. Pharmacol. 2007, 151, 1176–1186. [Google Scholar] [CrossRef]
- Leo, M.; Schmitt, L.-I.; Küsterarent, P.; Kutritz, A.; Rassaf, T.; Kleinschnitz, C.; Hendgen-Cotta, U.B.; Hagenacker, T. Platinum-based drugs cause mitochondrial dysfunction in cultured dorsal root ganglion neurons. Int. J. Mol. Sci. 2020, 21, 8636. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lagunas, M.J.; Martin, R.; Moreno, J.J.; Ferrer, R. PGE2promotes Ca2+-mediated epithelial barrier disruption through EP1and EP4receptors in Caco-2 cell monolayers. Am. J. Physiol. Physiol. 2010, 299, C324–C334. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Poulain, B. Bacterial toxins and the nervous system: Neurotoxins and multipotential toxins interacting with neuronal cells. Toxins 2010, 2, 683–737. [Google Scholar] [CrossRef] [PubMed]
- Peregrin, A.T. The effects on net fluid transport of noxious stimulation of jejunal mucosa in anaesthetized rats. Acta Physiol. Scand. 1999, 166, 55–64. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yoshimaru, T.; Inoue, T.; Ra, C. Ca v 1.2 L-type Ca2+ channel protects mast cells against activation-induced cell death by preventing mito-chondrial integrity disruption. Mol. Immunol. 2009, 46, 2370–2380. [Google Scholar] [CrossRef]
- Yoshimaru, T.; Suzuki, Y.; Inoue, T.; Ra, C. L-type Ca2+ channels in mast cells: Activation by membrane depolarization and distinct roles in regulating mediator release from store-operated Ca2+ channels. Mol. Immunol. 2009, 46, 1267–1277. [Google Scholar] [CrossRef]
- Lomax, R.B.; Gallego, S.; Novalbos, J.; García, A.G.; Warhurst, G. L-type calcium channels in enterochromaffin cells from guinea pig and human duodenal crypts: An in situ study. Gastroenterology 1999, 117, 1363–1369. [Google Scholar] [CrossRef]
- Yamamoto, K.; Asano, K.; Tasaka, A.; Ogura, Y.; Kim, S.; Ito, Y.; Yamatodani, A. Involvement of substance P in the development of cisplatin-induced acute and delayed pica in rats. Br. J. Pharmacol. 2014, 171, 2888–2899. [Google Scholar] [CrossRef]
- Godfraind, T.; Miller, R.; Wibo, M. Calcium antagonism and calcium entry blockade. Pharmacol. Rev. 1986, 38, 321–416. [Google Scholar]
- Samardzic, R.; Bajcetic, M.; Beleslin, D.B. Opposite effects of ethanol and nitrendipine on nicotine-induced emesis and convulsions. Alcohol 1999, 18, 215–219. [Google Scholar] [CrossRef]
- Van Driessche, A.; Sermijn, E.; Paemeleire, K.; Van Coster, R.; Vogelaers, D. Cyclic vomiting syndrome: Case report and short review of the literature. Acta Clin. Belg. 2012, 67, 123–126. [Google Scholar] [PubMed]
- Kothare, S.V. Efficacy of flunarizine in the prophylaxis of cyclical vomiting syndrome and abdominal migraine. Eur. J. Paediatr. Neurol. 2005, 9, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Garaschuk, O.; Yaari, Y.; Konnerth, A. Release and sequestration of calcium by ryanodine-sensitive stores in rat hippo-campal neurones. J. Physiol. 1997, 502, 13–30. [Google Scholar] [CrossRef]
- Gómez-Viquez, L.; Guerrero-Serna, G.; García, U.; Guerrero-Hernandez, A. SERCA pump optimizes Ca2+ release by a mechanism independent of store filling in smooth muscle cells. Biophys. J. 2003, 85, 370–380. [Google Scholar] [CrossRef]
- Gómez-Viquez, N.L.; Guerrero-Serna, G.; Arvizu, F.; García, U.; Guerrero-Hernandez, A. Inhibition of SERCA pumps induces desynchronized RyR activation in overloaded internal Ca2+ stores in smooth muscle cells. Am. J. Physiol. Physiol. 2010, 298, C1038–C1046. [Google Scholar] [CrossRef]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and Regulation of TRPC Channels in Store-Operated Ca2+ Entry. Curr. Top. Membr. 2013, 71, 149–179. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W. Store-Operated Calcium Channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Parrazal, L.; Fernandez-Ruiz, J.; Toledo, R.; Manzo, J.; Morgado-Valle, C. Inhibition of endoplasmic reticulum Ca2+ ATPase in preBötzinger complex of neonatal rat does not affect respiratory rhythm generation. Neuroscience 2012, 224, 116–124. [Google Scholar] [CrossRef]
- Michelangeli, F.; East, J.M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 2011, 39, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Solovyova, N.; Verkhratsky, A. Neuronal endoplasmic reticulum acts as a single functional Ca2+ store shared by ryanodine and inositol-1,4,5-trisphosphate receptors as revealed by intra-ER [Ca2+] recordings in single rat sensory neurones. Pflügers Archiv. 2003, 446, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Chebolu, S.; Darmani, N.A. Thapsigargin-induced activation of Ca(2+)-CaMKII-ERK in brainstem contributes to substance P release and induction of emesis in the least shrew. Neuropharmacology 2016, 103, 195–210. [Google Scholar] [CrossRef]
- Zhong, W.; Chebolu, S.; Darmani, N. Intracellular emetic signaling cascades by which the selective neurokinin type 1 receptor (NK1R) agonist GR73632 evokes vomiting in the least shrew (Cryptotis parva). Neurochem. Int. 2019, 122, 106–119. [Google Scholar] [CrossRef]
- Zhong, W.; Hutchinson, T.E.; Chebolu, S.; Darmani, N.A. Serotonin 5-HT3 receptor-mediated vomiting occurs via the activation of Ca2+/CaMKII-dependent ERK1/2 signaling in the least shrew (Cryptotis parva). PLoS ONE 2014, 9, e104718. [Google Scholar] [CrossRef]
- Mori, F.; Pérez-Torres, S.; De Caro, R.; Porzionato, A.; Macchi, V.; Beleta, J.; Gavaldà, A.; Palacios, J.; Mengod, G. The human area postrema and other nuclei related to the emetic reflex express cAMP phosphodiesterases 4B and 4D. J. Chem. Neuroanat. 2010, 40, 36–42. [Google Scholar] [CrossRef]
- Hutchinson, T.E.; Zhong, W.; Chebolu, S.; Wilson, S.M.; Darmani, N.A. L-type calcium channels contribute to 5-HT3-receptor-evoked CaMKIIalpha and ERK activation and induction of emesis in the least shrew (Cryptotis parva). Eur. J. Pharmacol. 2015, 755, 110–118. [Google Scholar] [CrossRef]
- Darmani, N.A.; Zhong, W.; Chebolu, S.; Mercadante, F. Differential and additive suppressive effects of 5-HT3 (palonosetron)- and NK1 (netupitant)-receptor antagonists on cisplatin-induced vomiting and ERK1/2, PKA and PKC activation. Pharmacol. Biochem. Behav. 2015, 131, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Chebolu, S.; Darmani, N.A. Central and peripheral emetic loci contribute to vomiting evoked by the Akt in-hibitor MK-2206 in the least shrew model of emesis. Eur. J. Pharmacol. 2021, 900, 174065. [Google Scholar] [CrossRef]
- Alkam, T.; Chebolu, S.; Darmani, N.A. Cyclophosphamide causes activation of protein kinase A (PKA) in the brainstem of vomiting least shrews (Cryptotis parva). Eur. J. Pharmacol. 2014, 722, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Schernthaner-Reiter, M.H.; Trivellin, G.; Stratakis, C.A. Chaperones, somatotroph tumors and the cyclic AMP (cAMP)-dependent protein kinase (PKA) pathway. Mol. Cell Endocrinol. 2020, 499, 110607. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.; Briggs, D.B.; Knox, A.P.; Strominger, N. Excitation of area postrema neurons by transmitters, peptides, and cyclic nucleotides. J. Neurophysiol. 1988, 59, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Saunders, M.P.; Salisbury, A.J.; Long, L.; O’Byrne, K.J.; Braybrooke, J.P.; Dowsett, M.; Taylor, M.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I study of the novel cyclic AMP (cAMP) analogue 8-chloro-cAMP in patients with cancer: Toxicity, hormonal, and immunological effects. Clin. Cancer Res. 1999, 5, 1682–1689. [Google Scholar]
- Vanmierlo, T.; Creemers, P.; Akkerman, S.; van Duinen, M.; Sambeth, A.; De Vry, J.; Uz, T.; Blokland, A.; Prickaerts, J. The PDE4 inhibitor roflumilast improves memory in rodents at non-emetic doses. Behav. Brain Res. 2016, 303, 26–33. [Google Scholar] [CrossRef]
- Darmani, N.A.; Dey, D.; Chebolu, S.; Amos, B.; Kandpal, R.; Alkam, T. Cisplatin causes over-expression of tachykinin NK(1) receptors and increases ERK1/2- and PKA- phosphorylation during peak immediate- and delayed-phase emesis in the least shrew (Cryptotis parva) brainstem. Eur. J. Pharmacol. 2013, 698, 161–169. [Google Scholar] [CrossRef]
- Kim, M.; Javed, N.H.; Yu, J.G.; Christofi, F.; Cooke, H.J. Mechanical stimulation activates Galphaq signaling pathways and 5-hydroxytryptamine release from human carcinoid BON cells. J. Clin. Investig. 2001, 108, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; Russo, A.; Salio, C. Fos and pERK immunoreactivity in spinal cord slices: Comparative analysis of in vitro models for testing putative antinociceptive molecules. Ann. Anat. 2014, 196, 217–223. [Google Scholar] [CrossRef]
- Khan, I.; Tantray, M.A.; Alam, M.S.; Hamid, H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Ikeda, Y.; Murakami, M.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y. Roles of PI3K/AKT/GSK3 pathway involved in psychiatric illnesses. Diseases 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Shanker, K.-S.; Swales, K.; Decordova, S.; Deyoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A first-time-in-human study of GSK2636771, a phosphoinositide 3 kinase beta-selective inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, S.; A Mayer, I. Management of toxicity to isoform α-specific PI3K inhibitors. Ann. Oncol. 2019, 30 (Suppl. 10), x21–x26. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef]
- Mazzardo-Martins, L.; Martins, D.; Stramosk, J.; Cidral-Filho, F.; Santos, A. Glycogen synthase kinase 3-specific inhibitor AR-A014418 decreases neuropathic pain in mice: Evidence for the mechanisms of action. Neuroscience 2012, 226, 411–420. [Google Scholar] [CrossRef]
- Saraswati, A.P.; Hussaini, S.A.; Krishna, N.H.; Babu, B.N.; Kamal, A. Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions. Eur. J. Med. Chem. 2018, 144, 843–858. [Google Scholar] [CrossRef]
- Walz, A.; Ugolkov, A.; Chandra, S.; Kozikowski, A.; Carneiro, B.A.; O’Halloran, T.V.; Giles, F.J.; Billadeau, D.D.; Mazar, A.P. Molecular pathways: Revisiting glycogen synthase kinase-3beta as a target for the treatment of cancer. Clin. Cancer Res. 2017, 23, 1891–1897. [Google Scholar] [CrossRef]
- Sahin, I.; Eturi, A.; De Souza, A.; Pamarthy, S.; Tavora, F.; Giles, F.J.; Carneiro, B.A. Glycogen synthase kinase-3 beta inhibitors as novel cancer treatments and modulators of antitumor immune responses. Cancer Biol. Ther. 2019, 20, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Darmani, N. The pivotal role of glycogen synthase kinase 3 (GSK-3) in vomiting evoked by specific emetogens in the least shrew (Cryptotis parva). Neurochem. Int. 2020, 132, 104603. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Darmani, N.A. Role of PI3K/Akt/GSK-3 pathway in emesis and potential new antiemetics. J. Cell Signal. 2020, 1, 155–159. [Google Scholar] [PubMed]
- Lawes, I.N. The origin of the vomiting response: A neuroanatomical hypothesis. Can. J. Physiol. Pharmacol. 1990, 68, 254–259. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target Receptor/Protein | Emetogens (Agonist/Inhibitor) | Affected Signaling Molecules | References |
|---|---|---|---|
| Serotonin 5-HT3 receptor | 2-Methyl-5-HT (agonist) | Phosphorylation of CaMKIIα and ERK | Zhong et al., 2014 [276], Hutchinson et al., 2015 [278] |
| Neuroknin NK1 receptor | GR73632 (agonist) | Phosphorylation of CaMKIIα, ERK, Akt, and PKC | Zhong et al., 2019 [275] |
| L-type Ca2+ channel | FPL64176 (agonist) | Phosphorylation of ERK1/2, PKC, and Akt | Zhong et al., 2018 [244] |
| Nonselective | Cisplatin | Phosphorylation of proteins ERK1/2, PKC, and PKA | Darmani et al., 2015 [279] |
| Sarcoplasmic endoplasmic reticulum calcium ATPase | Thapsigargin (inhibitor) | Phosphorylation of CaMKIIα and ERK | Zhong et al., 2016 [274] |
| Akt | MK-2206 (inhibitor) Perifosine (inhibitor) | Phosphorylation of ERK | Zhong et al., 2021 [280] |
| Nonselective | Cyclophosphamide | Tissue level of cAMP, phosphorylation of PKA | Alkam et al., 2014 [281] |
| Phosphodiesterase | Rolipram (inhibitor) | Tissue level of cAMP | Alkam et al., 2014 [281] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, W.; Shahbaz, O.; Teskey, G.; Beever, A.; Kachour, N.; Venketaraman, V.; Darmani, N.A. Mechanisms of Nausea and Vomiting: Current Knowledge and Recent Advances in Intracellular Emetic Signaling Systems. Int. J. Mol. Sci. 2021, 22, 5797. https://doi.org/10.3390/ijms22115797
Zhong W, Shahbaz O, Teskey G, Beever A, Kachour N, Venketaraman V, Darmani NA. Mechanisms of Nausea and Vomiting: Current Knowledge and Recent Advances in Intracellular Emetic Signaling Systems. International Journal of Molecular Sciences. 2021; 22(11):5797. https://doi.org/10.3390/ijms22115797
Chicago/Turabian StyleZhong, Weixia, Omar Shahbaz, Garrett Teskey, Abrianna Beever, Nala Kachour, Vishwanath Venketaraman, and Nissar A. Darmani. 2021. "Mechanisms of Nausea and Vomiting: Current Knowledge and Recent Advances in Intracellular Emetic Signaling Systems" International Journal of Molecular Sciences 22, no. 11: 5797. https://doi.org/10.3390/ijms22115797
APA StyleZhong, W., Shahbaz, O., Teskey, G., Beever, A., Kachour, N., Venketaraman, V., & Darmani, N. A. (2021). Mechanisms of Nausea and Vomiting: Current Knowledge and Recent Advances in Intracellular Emetic Signaling Systems. International Journal of Molecular Sciences, 22(11), 5797. https://doi.org/10.3390/ijms22115797